

Abstract
Mutations in the Autoimmune regulator (Aire) gene result in a clinical phenomenon known as Autoimmune Polyglandular Syndrome Type I (APS1), which classically manifests as a triad of adrenal insufficiency, hypoparathyroidism, and chronic mucocutaneous infections. In addition to this triad, a number of other autoimmune diseases have been observed in APS1 patients including Sjögren's syndrome, vitiligo, alopecia, uveitis, and others. Aire-deficient mice, the animal model for APS1, have highlighted the role of the thymus in the disease process and demonstrated a failure in central tolerance in aire-deficient mice. However, autoantibodies have been observed against multiple organs in both mice and humans, making it unclear what the specific role of B and T cells are in the pathogenesis of disease. Utilizing the aire-deficient mouse as a preclinical model for APS1, we have investigated the relative contribution of specific lymphocyte populations, with the goal of identifying the cell populations which may be targeted for rational therapeutic design. Here we show that T cells are indispensable to the breakdown of self-tolerance, in contrast to B cells which play a more limited role in autoimmunity. Th1 polarized CD4+ T cells, in particular, are major contributors to the autoimmune response. With this knowledge, we go on to utilize therapies targeted at T cells to investigate their ability to modulate disease in vivo. Depletion of CD4+ T cells using a neutralizing antibody ameliorated the disease process. Thus, therapies targeted specifically at the CD4+ T cell subset may help control autoimmune disease in patients with APS1.
Keywords: Autoimmunity, Tolerance, T cells, Thymus
Introduction
Autoimmune Polyglandular Syndrome Type 1 (APS1) is a monogenic autoimmune disease that is inherited in an autosomal recessive pattern (1). Individuals with this disease present with a clinical triad including adrenal insufficiency, chronic mucocutaneous infections, and hypoparathyroidism. In addition to these hallmarks of disease, patients frequently have other autoimmune manifestations including type 1 diabetes, Sjögren's syndrome, vitiligo, keratitis, alopecia, gastritis, and other syndromes believed to be of an autoimmune etiology (2).
The basis for such a strong predisposition to autoimmunity was genetically mapped by two independent groups, resulting in the identification of the Autoimmune regulator (Aire) gene (3, 4). The Aire protein, which bears strong resemblance to a transcription factor and has been shown to localize to nuclear speckles (5), is expressed in a subset of medullary thymic epithelial cells (mTECs) that are associated with negative selection of developing thymocytes. Within mTECs, Aire controls the promiscuous expression of many peripheral autoantigens through mechanisms that are not completely understood (6). The absence of Aire expression results in an inability to remove autoreactive thymocytes from the immune repertoire, ultimately resulting in autoimmune disease against multiple tissues (7).
Despite the evidence suggesting the thymus as the key to the initiation of the disease process, multiple cells could play a role in the autoimmunity that eventually ensues and tissue destruction may be mediated by cell types other than T cells. In APS1 patients and aire-deficient mice, autoantibodies recognizing several organ-specific autoantigens have been identified including insulin, glutamic acid decarboxylase, cytochrome P450, 21-hydroxylase (8), and more recently tudor-domain containing protein 6 in humans (9) as well as interphotoreceptor retinoid binding protein (7), fodrin (10), pancreas specific protein disulfide isomerase (11), and mucin-6 (12) in aire-deficient mice It is unclear, however, whether or not these autoantibodies are pathogenic and what role they, or the B cells that produce them, may play in the progression of disease.
Aire-deficient mice remain the best tool available to study this unique process and mimic the human disease in many ways. Due in part to the difficulties in studying human patients and their relative rarity in clinical medicine, little is known about the specific contribution of different cell types in disease pathogenesis and progression. To further understand the role that the immune system plays in aire-mediated autoimmunity, we performed a detailed analysis of lymphocyte function within aire-deficient mice and bred the aire mutation onto several genetic backgrounds including mice deficient for T and B cells. Here, we present the results of these studies on the relative role of T and B cells in mediating disease and demonstrate that T cells are indispensable to the disease process, whereas B cells play a more limited role in autoimmunity. Therapies targeting CD4+ T cells ameliorated autoimmunity, supporting these genetic and adoptive transter studies and suggesting a clinically relevant avenue of therapeutic exploration.
Materials and Methods
Mice
Aire-deficient mice were generated as previously described (6) and were backcrossed into the C57BL/6 and NOD Lt/J backgrounds greater than 10 generations. IgH-deficient (13), STAT4-deficient (14), and STAT6-deficient (15) on the NOD background and CIITA-deficient mice (16) on the C57/BL6 background were purchased from Jackson Labs and bred to mice in our facility. All mice were housed in a pathogen-free barrier facility at UCSF. Experiments complied with the Animal Welfare Act and NIH guidelines for the ethical care and use of animals in biomedical research and were approved by the UCSF Animal Care and Use Committee.
Antibodies
All antibodies used for flow cytometry (anti-CD4 [RM4-5], CD8 [56-6.7], CD45 [30-F11], IL-4 [11B11], Il-10 [JES5-16E3], IL-17 [TC11-18H10], IFN-γ [XMG1.2] and isotype controls) were purchased from BD Biosciences. The anti-CD4 antibody GK1.5 and anti-CD8 antibody YTS-169.4 used for depletion experiments were gifts from Dr. Jeff Bluestone.
Histology
Organs from mice were harvested, fixed overnight in 10% formalin, embedded in paraffin, sectioned, and stained for hematoxylin and eosin. Tissue sections were scored on a grading system from 0 to 4, where 0 was no indication of immune infiltrate, 1 was a tissue that was 1-25% infiltrated, 2 was a tissue that was 26-50% infiltrated, 3 was a tissue that was 51-75% infiltrated, and 4 was a tissue that was greater than 76% infiltrated.
Immunostaining
Immune cell subtypes were visualized by immunohistochemistry using antibodies specific for CD4, CD8, and IgD (BD Pharmingen) and a DAB staining kit (Vector Labs).
Adoptive transfer
Cervical lymph node cells and splenocytes were harvested and CD4+ or CD8+ T cells were depleted using complement. Cell populations (5 × 106 CD4+ and CD8+, CD4+ depleted, or CD8+ depleted) were injected I.V. into scid.NOD mice. On days 0, 5, 19, and 33 animals were treated with 0.5mg/mouse of anti-CD4 (GK1.5, CD4+ depleted) or anti-CD8 (YTS169.4, CD8 depleted) injected IP to remove residual CD4+ or CD8+ T cells (17). Animals were aged 40 days post-transfer then sacrificed and analyzed as described.
Sera transfers
Wild-type or immunodeficient animals were injected i.p. with 150 microliters of wild-type or aire-deficient sera on day 0 followed by a repeat injection of 100 microliters on day 2. Animals were weighed three times a week. Two weeks following the initial injection, animals were sacrificed and analyzed as described above. For repeat administration, wild-type animals were injected i.p. with 75 microliters of wild-type or aire-deficient every day, for 10 days (a total of 750ul of sera). Animals were sacrificed and analyzed as described above at day 14.
Flow cytometry
Salivary glands were incubated in 2mg/ml collagenase D in DMEM supplemented with 10ug DNAse I for 40 minutes. The remaining tissue was dispersed by vortexing and filtered through nylon mesh. Cells were placed in culture in DMEM complete media with 10% fetal calf serum with Golgi-Stop (Becton Dickinson) and stimulated with 10 ng/ml phorbol 12-myristate 13-acetate (PMA) and 0.5uM ionomycin (Sigma Aldrich) for 4 hours at 37°C. After the incubation, cells were surface stained with antibodies specific for CD4, CD8, and CD45 to gate lymphocytes, then permeabilized and stained with antibodies specific for IL-4, IL-10, IL-17, interferon-gamma, or isotype control. Cells were analyzed on a LSRII flow cytometer (Becton Dickinson).
For lymph node and splenic cell preparations, cervical lymph nodes or spleens were removed and dispersed by mechanical separation. Cells were then stimulated and analyzed as described above.
Anti-CD4 administration
Animals were given 500 micrograms of anti-CD4 (GK1.5) or rat isotype control (Jackson Immunoresearch) via intraperitoneal injection once a week starting at age day 21. Animals were sacrificed on day 49. Histology was analyzed as described.
Statistics
Data was analyzed using Mann Whitney non-parametric test with Prism software (Graphpad, San Diego, CA).
Results
T cells are required for the disease process
The relative contribution of T and B cells to autoimmunity varies in different model systems. For example, in the NOD mouse model of diabetes, the presence of B cells and autoantibodies are critical to both insulitis and overt diabetes (18). Similarly, T cells are known to be required for islet infiltration and destruction (19). While high titer autoantibodies are present in aire-deficient mice and target both known and unknown organ-specific autoantigens (6, 7), it's unclear whether or not these autoantibodies play a pathogenic role. Similarly, previous experiments in our lab and others have determined that autoreactive T cells escape the thymus in aire-deficient animals (7, 20, 21), but it is not clear whether the ultimate destruction of a target organ is entirely T cell mediated. To determine the relative contribution of T and B cells to the immune damage observed in aire-deficient mice, we bred aire-deficient animals on the NOD background to IgH-deficient (18) or TCRα-deficient (22) animals. The resulting mice lacked both aire and either B or T cells, respectively (aire/IgH double knock out [DKO] or aire/TCRα DKO). As has been previously observed, aire-deficient mice on the NOD background undergo a spontaneous wasting disease that results in death (20). In contrast, all aire/TCRα DKO mice were healthy and their weight remained stable as far out as 40 weeks of age (data not shown), a time at which no aire-deficient or aire/IgH deficient animal survived. To further investigate if this observation reflected a lack of active disease in these animals, mice were sacrificed at 6-10 weeks of age (aire single knockout, aire/IgH DKO) or 25 weeks of age (aire/TCRα DKO) and their tissues were analyzed for the accumulation of lymphocytes. Aire/IgH DKO mice exhibited a distinct mononuclear cell infiltrate that mirrored what we observed in aire-deficient mice (retina and pancreas shown in figure 1A & B respectively, remaining organs depicted in figure 1C & 1D). Aire/TCRα DKO animals were devoid of mononuclear cells in the organs analyzed (1C). Thus, T cells are indispensable for the disease process to occur. To quantify the degree of infiltrate, we scored sections from aire-deficient, aire/IgH DKO, and aire/TCRα DKO animals on a scale from 0 to 4. With the exception of the lung, there was no observable difference in the histological scores of aire-deficient and aire/IgH DKO animals. In stark contrast, no infiltrate was observed in any of the aire/TCRα DKO animals that we identified, even as far out as 40 weeks of age. To assess the pathogenicity of autoantibodies present in aire-deficient mice, we also transferred sera from aire-deficient NOD animals with disease into both immunodeficient and wild-type NOD animals using a protocol that was capable of transferring arthritis in the K/BxN T cell transgenic mouse model of autoimmunity (23). Passive transfer of sera using this protocol was unable to provoke autoimmunity against the organs traditionally targeted in aire-deficient animals (figure 2) or elicit weight loss (data not shown). To determine if repeat autoantibody administration was required, we also administered aire-deficient or wild-type mouse sera to recipients on a daily basis for ten days. This multi-injection protocol also failed to yield any observable disease. In sum, these data suggest that B cells have a limited role in the disease process and that autoantibodies present in aire-deficient mice do not affect the disease outcomes measured in this study. However, additional study is needed to clarify the role of B cells in the autoimmune pneumonitis observed in these animals.
Figure 1. T cells are critical for Aire-related autoimmunity.
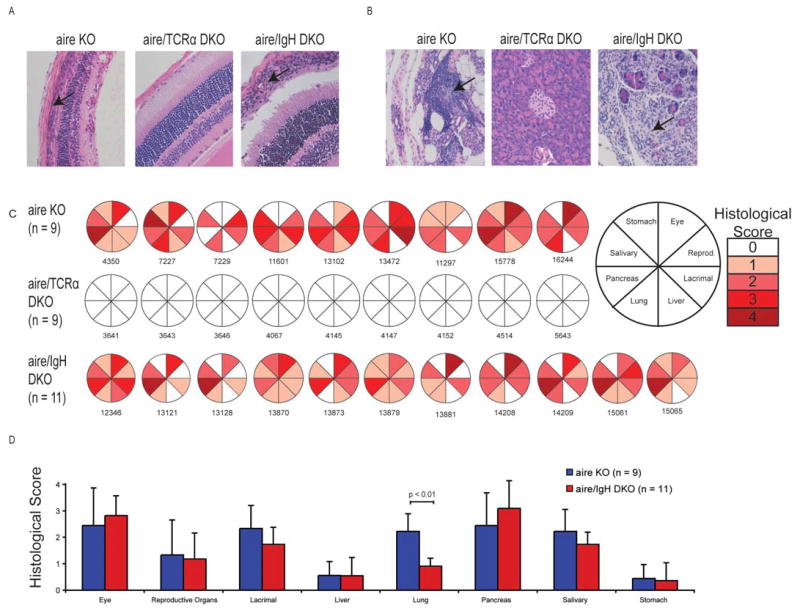
The aire mutation was bred with TCR-α or IgH-deficient mice (both backcrossed greater than 10 generations back to NOD) to determine the relative contribution of T and B cells to the disease process. aire/TCRα double knock out (DKO) mice were healthy and viable for greater than 25 weeks of age. In contrast, aire-deficient and aire/IgH DKO mice did not survive beyond 12 weeks of age. As a result, aire–deficient and aire/IgH DKO mice were aged to 6-8 weeks of age and analyzed for disease, while aire/TCRα DKO mice were aged to 25-30 weeks for analysis. aire deficient and aire/IgH DKO mice displayed a characteristic mononuclear cell infiltrate (indicated by arrow) in the retina (A) and exocrine pancreas (B) and other organs (depicted in C). In contrast, aire/TCRα DKO mice were healthy and had no infiltrates in any tissues analyzed. Shaded sections of pie graph indicate the presence of mononuclear infiltrates in the designated organ and each circle and corresponding number represent an individual mouse for that group (C). The degree of shading reflects the amount of autoimmune damage. Shown in (D) is the average histological score + / - standard deviation for each organ.
Figure 2. Transfer of sera is not capable of eliciting autoimmunity or tissue damage.
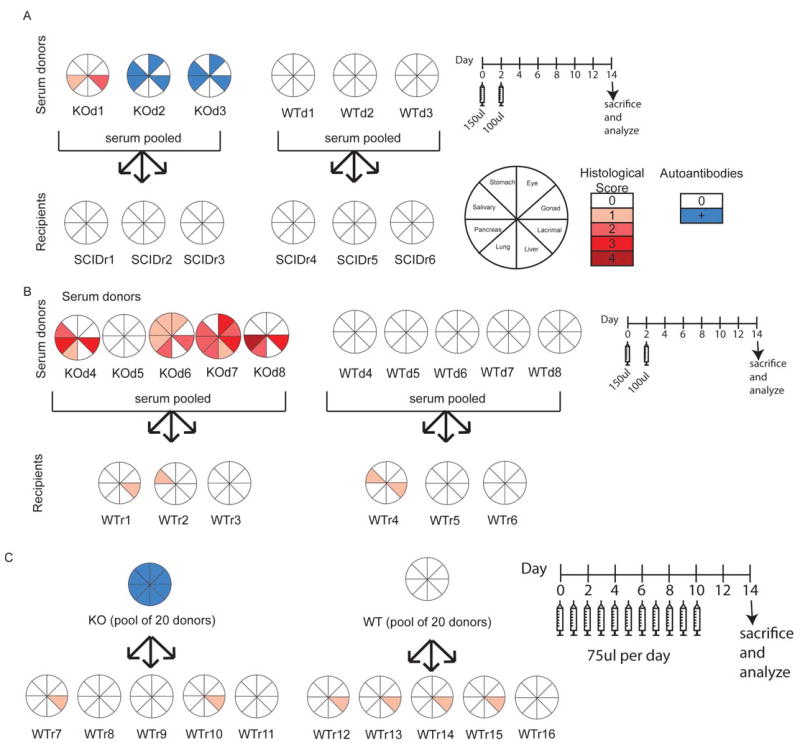
Using studies on the pathogenic role of autoantibodies in the K/BxN T cell transgenic mouse model as a guide, we performed sera transfers into either immunodeficient (A) or wild-type (B) hosts. We transferred 150ul of sera from either aire-deficient mice with active disease or aire-sufficient controls at day 0, followed by an additional 100ul of sera at day 2. The weight of recipient animals was measured on alternating days and the animals were sacrificed at day 14. Tissues were assessed for immune damage by histological analysis. Histological scores are denoted in shades of red. For animals for which histology was not available, autoantibody reactivity is denoted in blue. KOd# = aire deficient sera donor, WTd# = aire-sufficient sera donor, SCIDr# = immunodeficient sera recipient, WTr# = wild type sera recipient. For KOd2 and KOd3, histological analysis was not performed and immunoreactivity shown is based on autoantibody reactivity. To determine if repeat administration of autoantibodies was required, we administered 75ul of aire-deficient or wild-type animal sera daily for 10 days, then harvested at day 14 (C). Tissues were assessed for immune damage as described.
CD4+ and CD8+ T cells contribute to disease
Having confirmed that T cells are a critical cell type required for autoimmune pathogenesis, we sought to further delineate the role of CD4+ and CD8+ T cells in this process. To this end, we adoptively transferred aire-deficient lymphocyte populations depleted of CD4+ or CD8+ T cells into immunodeficient hosts. In this setting, aire-deficient lymphocytes devoid of CD4+ T cells had a diminished capacity to infiltrate target tissues. In fact, no evidence of immune infiltrates was observed in the lung, pancreas, reproductive organs (figure 3A) or eye (7). In addition, autoimmune infiltrates were reduced in the other organs analyzed – the lacrimal and salivary glands and the liver (figure 3A). Recipients of CD8+ T cell depleted lymphocyte populations, however, developed immune infiltrates that were no different than animals that received whole lymphocyte populations (figure 3A). These data suggested to us that CD4+ T cells are a key mediator of the autoimmune response in aire-deficient mice.
Figure 3. Immune damage in a majority of the organs involves both CD4 and CD8 T cells.
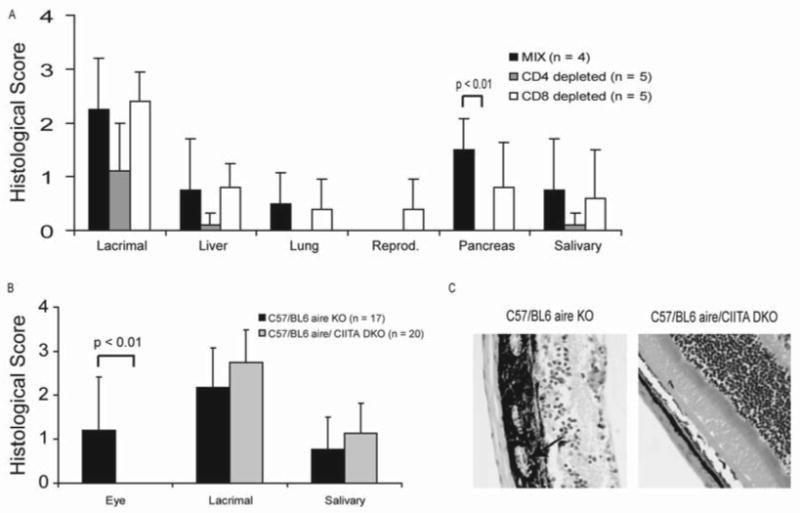
To determine if CD4 or CD8 cells were required for the disease process, aire-deficient lymphocytes depleted of either CD4+ or CD8+ T cells were adoptively transferred into immunodeficient NOD.SCID recipients. Recipient animals were also treated with neutralizing antibody against either CD4 (GK1.5) or CD8 (YTS-169.4) to further remove residual cells. Recipient animals were then aged for 4 weeks before histological analysis (A). To confirm a critical role for CD4+ T cells in the disease process, aire-deficient animals were bred to CIITA-deficient mice on the C57/BL6 background, aged to 20 weeks of age, then analyzed for disease (B). These experiments were conducted on the C57/BL6 background, where autoimmunity is predominantly found in the eye, lacrimal gland, and salivary gland (24). Absence of CD4+ T cells prevented immune infiltrates from entering the retina (C, arrow indicates immune infiltrates).
To further investigate the role of CD4 T cells in the disease process, we bred aire-deficient mice to CIITA-deficient mice, which lack class II transactivator (CIITA) and peripheral class II expression, resulting in a near complete lack of mature CD4+ T cells (16). To confirm the critical role of CD4+ T cells in the pathogenesis of disease, we analyzed animals that were deficient for both aire and CIITA. In these mice, we found no evidence of autoimmunity against the eye or infiltrate into the retina (figure 3B and 3C), confirming our previous experiments demonstrating that CD4+ T cells were critical for IRBP-specific autoimmunity (7) The salivary and lacrimal glands, however exhibited the same degree of immune infiltrate in the aire-deficient and the aire/CIITA DKO animals (figure 3C). Residual CD4+ T cells have been reported in these mice, as the deficiency in class II expression is not enforced in the thymus. Using immunohistochemistry, we were able to confirm the presence of CD4+ T cells in the infiltrated salivary and lacrimal glands (data not shown), a finding confirmed by flow cytometry of lymph nodes and spleen (data not shown). Thus, we hypothesize that the residual CD4+ T cells present in aire/CIITA DKO mice are capable of eliciting salivary and lacrimal gland autoimmunity, but are not of sufficient numbers to elicit autoimmunity against an immunoprivileged organ such as the eye.
T cells present in aire-deficient mice are Th1 polarized
During maturation, CD4+ T cells polarize into effector lineages including Th1-type, Th2-type, or Th17-type helper cells. To determine if a specific path of development was being emphasized in the disease process in aire-deficient mice, we analyzed lymph node cells (LNCs) and organ-infiltrating lymphocytes for cytokine polarization. Lymphocytes from the cervical lymph node, spleen, and salivary gland were procured from aire-deficient C57BL/6 mice (n = 5) between 18 and 20 weeks of age and cervical lymph node and spleen of aire-sufficient C57BL/6 mice (n = 5) that were age and gender matched. Cells were stimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin and intracellular cytokine production was measured by flow cytometry. CD4+ and CD8+ T cells derived from wild type or aire-deficient spleens and cervical lymph nodes produced equivalent amounts of the cytokines IFN-γ, IL-4, IL-10, and IL-17 (figure 4). In contrast, T cells present in the autoimmune lesions of the salivary gland of aire-deficient mice produced dramatically increased levels of IFN-γ when stimulated with PMA and ionomycin (figure 4A). This increase in IFN-γ production was documented in both CD4+ and CD8+ subsets (figure 4B and 4C). Expression of TNF-α was also observed in these cells (data not shown). We also observed a similar Th1 bias in the cells that infiltrate the lacrimal gland and retina (data not shown). In contrast, little expression of the Th2-cytokines IL-4 and IL-10 or the Th17 cytokine IL-17 was observed in cells derived from the autoimmune infiltrate. The data shown in figure 4 is representative of four independent experiments. Additionally, we believe that this polarization skewing was not due to the genetic background of the animals, as aire-deficient mice on the BALB/c and NOD background showed the same predisposition towards Th1 polarization (data not shown)
Figure 4. CD4+ T cells present in the infiltrate are polarized to a Th1 phenotype.
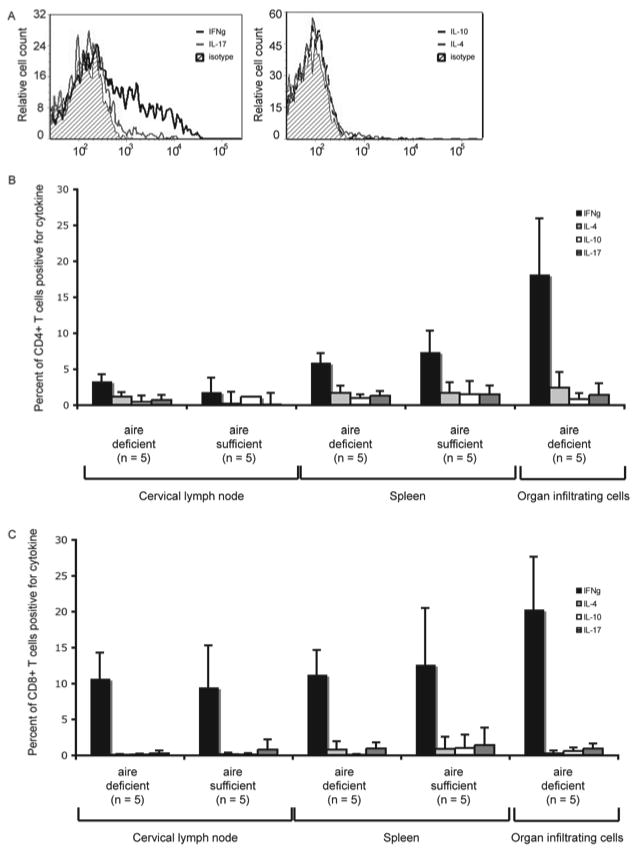
Lymphocytes from aire-deficient and aire-sufficient C57BL/6 mice were isolated from the spleen and cervical lymph node (aire-deficient and wild-type) or salivary gland immune infiltrates (aire-deficient) and stimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin to induce cytokine expression. No lymphocytes were present in the salivary glands of wild-type C57BL/6 animals. The amount of intracellular cytokine expression was measured by flow cytometry using antibodies specific for each cytokine or an isotype control. Overlay plots gated on CD4 and CD45 demonstrate that T cells in the target tissue are Th1 polarized and produce IFN-γ (A). The percentage of CD4+ T cells (B) or CD8+ T cells (C) expressing each cytokine is shown for spleen, draining lymph node, and target tissue. These results are representative of four independent experiments.
Genetic studies on the role of Th1 and Th2 polarization
To determine whether or not Th1 polarization was relevant to the disease process in aire-deficient mice, we generated mice deficient for both aire and either STAT4, a transcription factor required for potent Th1 polarization (14), or STAT6, a transcription factor required for Th2 polarization (15). To determine whether or not the disruption in STAT signaling resulted in a direct effect on the autoimmune response, tissue sections were prepared from the organs known to be infiltrated in age matched aire-deficient, aire/STAT4 DKO, and aire/STAT6 DKO mice. In the absence of STAT4 signaling, the degree of disease severity was decreased in the pancreas (figure 5A & B), whereas in the absence of STAT6 signaling, the disease severity was increased in liver and salivary glands. We also performed a histological analysis on 20 week old aire/STAT4 DKO mice. Disease as measured by histological score was ultimately indistinguishable from aire-deficient animals. Thus, even in the absence of STAT4 signaling the autoimmune disease is ultimately capable of progressing. It should be noted that CD4+ T cells from aire/STAT4 DKO mice are still capable of producing IFN-γ. Specifically, when we tested splenic CD4+ T cells from 20 week old aire/STAT4 DKO mice using PMA and ionomycin, they produced more interferon-gamma than CD4+ T cells from a 6 week old aire-deficient mouse (data not shown). Thus, we assume that the delay in the production of gamma resulting from the absence of STAT4 signaling prevents rapid onset of the disease in the double deficient animals.
Figure 5. Genetic analysis of the role of Th1 polarization in the disease process.
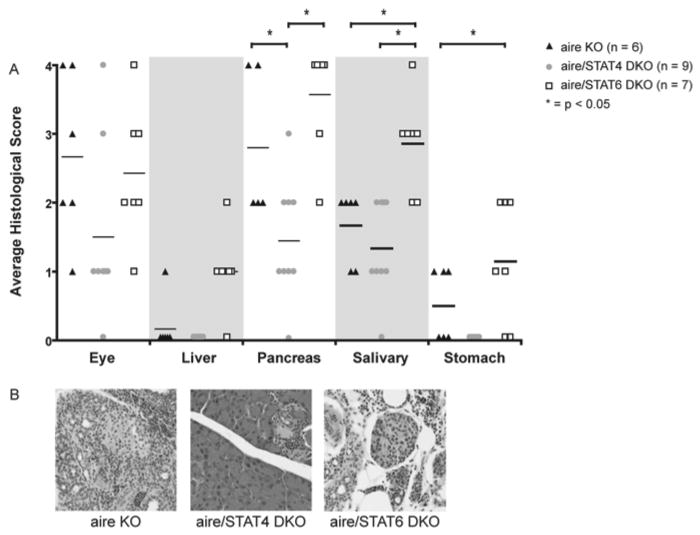
To assess the relevance of Th1 polarization in vivo, we generated aire/STAT4 DKO and aire/STAT6 DKO mice on the NOD background and analyzed the disease process in these mice. Exocrine pancreatitis was significantly decreased in aire/STAT4 DKO animals (A, B). In contrast, aire/STAT6 DKO animals had significantly worse autoimmunity targeting the liver and salivary glands (A). Representative histology for the exocrine pancreas is shown in (B).
Cellular therapies targeting CD4+ T cells
In light of the genetic evidence suggesting that CD4+ T cells are Th1 polarized and given the availability of anti-CD4 neutralizing antibodies, we decided to investigate their potential as a therapeutic protocol in the modulation of disease. Administration of neutralizing anti-CD4 antibody has been shown to dramatically reduce the levels of circulating CD4+ T cells in the blood and lymph nodes (25). In addition, depleting CD4 has shown efficacy in other T cell mediated autoimmune models (26). We injected aire-deficient NOD mice with a regimen of 500 micrograms of anti-CD4 antibody (GK1.5) or isotype control once a week, starting at three weeks of age. Administration of anti-CD4 with our protocol resulted in a significant depletion of CD4+ T cells in treated animals (data not shown). This treatment protocol also significantly ameliorated immune infiltration in multiple organs. The histological slides were scored as described and demonstrated a significant decrease in immune infiltrate in the retina, liver, lung, and reproductive organs (figure 6A). Histology revealed dense mononuclear infiltrates in the retina and lung along with a loss of normal tissue architecture in both tissues, but no obvious immune infiltrate or defects in anti-CD4 treated mice (figure 6B).
Figure 6. Depletion of CD4+ T cells ameliorates disease in vivo.
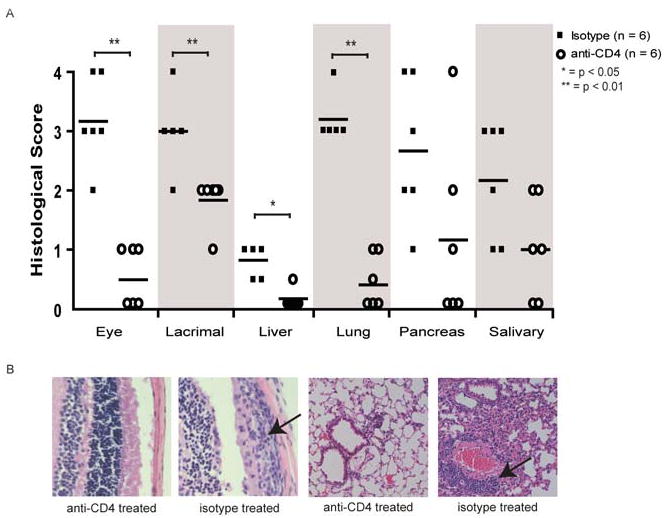
To determine if the disease process could be abrogated in vivo, neutralizing CD4 antibody was administered via intraperitoneal injection. Aire-deficient animals on the NOD background were treated with 500 micrograms of anti-CD4 (GK1.5) or isotype control once a week starting at 21 days post-birth. Animals were sacrificed at 49 days of age and disease was evaluated by histological analysis. Average histological score for all the organs (A). Representative images of the histology from the eye and lung (B), arrow indicates the immune infiltrates.
Discussion
Autoimmune Polyglandular Syndrome Type 1 is known to be a monogenic, recessive autoimmune disease that primarily affects organs of the endocrine axis. Due to the complications of studying disease in humans, little is known about the pathogenicity of B and T cells in APS1. While many previous reports have focused on the presence of autoantibodies in both aire-deficient animals and APS1 patients, no reports have demonstrated a pathogenic role for either B or T cells. Here, we show that T cells are absolutely critical to autoimmunity by generating mice deficient for both aire and TCRα. These mice, which lack αβ T cells, are completely healthy and free of immune infiltrates. To further delineate the effective contribution of CD4+ and CD8+ T cells to this process, we performed adoptive transfer of lymphocyte populations depleted of CD4+ or CD8+ T cells and analyzed CIITA deficient mice. In both cases, CD4+ T cells appeared to play a major role in disease pathogenesis. Upon further dissection, many of the CD4+ T cells resident in the infiltrated tissues produced the Th1 cytokine IFN-γ while few produced IL-17 or IL-10. The importance of Th1-like cells being effectors in aire-deficient mice was confirmed by genetic studies using STAT4 and STAT6 deficient animals. Due to these findings, we reasoned that depletion of CD4+ T cells in vivo would be an effective means of modulating disease. Indeed, administration of neutralizing antibodies significantly altered the degree of infiltration in tissues targeted by the immune system.
While the number of case reports described in the primary literature is limited, published studies of immunosuppressive regimens in APS1 patients suggest synergy with our data. In a patient of French-Canadian descent, several aspects of the disease responded to cyclosporine A treatment, including a remission of exocrine pancreatitis, keratoconjunctivitis, and alopecia (27). A 5 year old patient of Iranian descent who underwent two liver transplants responded poorly to the immunosuppressive regimen initiated during the first transplant, but well to the regimen initiated during the second transplant. In the former, prednisone, azathioprine, and cyclosporine A was administered, however, autoantibodies were detected to multiple known organ targets and disease progression was unchecked. In the latter case, the patient was given tacrolimus, prednisone, and mycophenolate mofetil and all APS1-related symptoms (including candidiasis and autoantibodies) decreased (28). Finally, in a study on children with autoimmune hepatitis and APS1, two of three patients responded well to steroids and azathioprine (29). Our own results in the mouse model would suggest that T-cell targeted therapies may hold the best promise, as specific depletion of CD4+ T cells significantly ameliorates disease.
One question unanswered by the human data is the relative role of B cells in disease pathogenesis, as the immunosuppressive regimens administered equally affect B and T lymphocytes. Despite the identification of many autoantibody specificities in both the aire-deficient mouse and APS1 patients, our data suggests that B cells play a limited role in disease pathogenesis in the mouse model. Sera transfers of autoantibodies were unable to elicit any observable autoimmunity and a genetic deficiency in the B cell compartment had a limited effect on the autoimmune infiltrates. Why then is the generation of multiple autoantigen specificities observed so clearly? The loss of central tolerance in the absence of Aire permits the escape of high-affinity autoreactive T cells (20, 21). It is likely that these T cells, upon their escape into the periphery, provide B cell help and result in the generation of high affinity autoantibodies. It remains possible, however, that B cells influence additional aspects of the disease not analyzed in this work. For example, APS1 patients have been reported whose clinical history include autoimmune diseases such as Graves and idiopathic thrombocytopenic purpura (1), in which a clear role for a pathogenic autoantibody has been established. To date, no one has established such a disease mechanism in the mouse model.
Recently, there has been a vigorous discussion in the literature about the potential parallels between this animal model and human patients (30-32). Our findings in the animal model, when compared with the extensive clinical experience of clinicians (1, 33), indicate that a majority of the organs targeted in the absence of Aire expression are shared between the two species (1, 34). Thus, aire-deficient mice and APS1 patients have been reported to develop adrenal failure, hypoparathyroidism, premature ovarian failure, thyroiditis, autoimmune hepatitis, exocrine pancreatitis, gastritis, pneumonitis, dacryoadenitis and sialitis. Further strengthening the connection between the mouse model and APS1 is preliminary data in our lab where we have found an APS1 patient with photoreceptor-specific autoantibodies and retinitis that is highly similar to our published work (7) detailing the uveitis in aire-deficient mice (unpublished data).
The larger view of what drives autoimmunity in the absence of Aire is becoming increasingly clear. Previous results from our lab identified IRBP as the primary target of the immune response in the retina. Confirming an important role of aire in autoantigen expression within mTECs, transplantation of a thymus derived from an IRBP-deficient mouse into nude recipients was sufficient to result in autoimmunity (7). Thus, absence of a single antigen within the thymic compartment, despite the presence of functional aire protein, allowed for the generation of an aberrant immune response. This immune response likely results from a failure in negative selection, rather than a deficit in positive selection of regulatory T cells. In the murine model, no defect in the number of or selection of FoxP3+ regulatory T cells has been reported (20), although data in humans remains controversial (35). Recent data also suggests that the innate immune response, in particular signals generated through Toll like receptors, plays a limited role in driving the autoimmunity in the mouse model (36)
In what direction, then, does the future of APS1 treatment lie? Our results suggest that specific targeting of T cells may be an effective way to modulate disease. While depletion of CD4+ T cells in APS1 patients may not be a practical clinical approach given the severe immunosuppression associated with it, other antibody-based therapies may prove useful. For example, immunomodulation using monoclonal antibodies against CD3 has been proven to delay onset of the T-cell driven disease, type 1 diabetes and also is associated with more acceptable levels of immunosuppression (37). Likewise, targeting of Th1-related cytokines such as IFN-γ or TNF-α may prove to be efficacious given our data supporting a role for Th1 T these cells as key effectors in the mouse model. Finally, identifying the autoantigens that are targeted in APS1 patients could result in antigen-specific therapies involving the suppression of activated autoreactive cells, via regulatory T cells, or other forms of antigen specific tolerance such as coupled cell tolerance (38, 39). In support of this notion is recent data from our group that demonstrated that the uveitis in the mouse model is driven by a single self-antigen (7). The studies presented here should help provide the framework for targeted immunotherapy that can be used in the treatment of this rare, but severe clinical syndrome.
Acknowledgments
We thank Jeffrey Bluestone, Abul Abbas, M. Cheng, J. Gardner, and M. Su for critical review of the manuscript; members of the Anderson Laboratory for helpful discussions; and the UCSF Biomolecular Resource Center Mass Spectrometry Facility for mass spectrometry.
Grant support: This work was supported by grants from the NIH (MSA and ECS), the Pew Scholars Program in Biomedical Sciences (MSA), the Sandler Foundation (MSA) the Burroughs Wellcome Fund (MSA), the RPB James S. Adams Scholar Award (ECS), the Ocular Immunology Fund (ECS) and the Giannini Foundation (JDV).
Footnotes
This is an author-produced version of a manuscript accepted for publication in The Journal of Immunology (The JI). The American Association of Immunologists, Inc. (AAI), publisher of The JI, holds the copyright to this manuscript. This version of the manuscript has not yet been copyedited or subjected to editorial proofreading by The JI; hence, it may differ from the final version published in The JI (online and in print). AAI (The JI) is not liable for errors or omissions in this author-produced version of the manuscript or in any version derived from it by the U.S. National Institutes of Health or any other third party. The final, citable version of record can be found at www.jimmunol.org.
References
- 1.Perheentupa J. APS-I/APECED: the clinical disease and therapy. Endocrinol Metab Clin North Am. 2002;31:295–320. vi. doi: 10.1016/s0889-8529(01)00013-5. [DOI] [PubMed] [Google Scholar]
- 2.Perheentupa J. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. J Clin Endocrinol Metab. 2006;91:2843–50. doi: 10.1210/jc.2005-2611. [DOI] [PubMed] [Google Scholar]
- 3.An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. The Finnish-German APECED Consortium. Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy. Nat Genet. 1997;17:399–403. doi: 10.1038/ng1297-399. [DOI] [PubMed] [Google Scholar]
- 4.Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJ, Lalioti MD, Mullis PE, Antonarakis SE, Kawasaki K, Asakawa S, Ito F, Shimizu N. Positional cloning of the APECED gene. Nat Genet. 1997;17:393–398. doi: 10.1038/ng1297-393. [DOI] [PubMed] [Google Scholar]
- 5.Bjorses P, Halonen M, Palvimo JJ, Kolmer M, Aaltonen J, Ellonen P, Perheentupa J, Ulmanen I, Peltonen L. Mutations in the AIRE gene: effects on subcellular location and transactivation function of the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy protein. Am J Hum Genet. 2000;66:378–392. doi: 10.1086/302765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 7.Devoss J, Hou Y, Johannes K, Lu W, Liou GI, Rinn J, Chang H, Caspi R, Fong L, Anderson MS. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J Exp Med. 2006;203:2727–2735. doi: 10.1084/jem.20061864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perniola R, Falorni A, Clemente MG, Forini F, Accogli E, Lobreglio G. Organ-specific and non-organ-specific autoantibodies in children and young adults with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) Eur J Endocrinol. 2000;143:497–503. doi: 10.1530/eje.0.1430497. [DOI] [PubMed] [Google Scholar]
- 9.Bensing S, Fetissov SO, Mulder J, Perheentupa J, Gustafsson J, Husebye ES, Oscarson M, Ekwall O, Crock PA, Hokfelt T, Hulting AL, Kampe O. Pituitary autoantibodies in autoimmune polyendocrine syndrome type 1. Proc Natl Acad Sci U S A. 2007;104:949–954. doi: 10.1073/pnas.0610070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuroda N, Mitani T, Takeda N, Ishimaru N, Arakaki R, Hayashi Y, Bando Y, Izumi K, Takahashi T, Nomura T, Sakaguchi S, Ueno T, Takahama Y, Uchida D, Sun S, Kajiura F, Mouri Y, Han H, Matsushima A, Yamada G, Matsumoto M. Development of autoimmunity against transcriptionally unrepressed target antigen in the thymus of Aire-deficient mice. J Immunol. 2005;174:1862–1870. doi: 10.4049/jimmunol.174.4.1862. [DOI] [PubMed] [Google Scholar]
- 11.Niki S, Oshikawa K, Mouri Y, Hirota F, Matsushima A, Yano M, Han H, Bando Y, Izumi K, Matsumoto M, Nakayama KI, Kuroda N. Alteration of intra-pancreatic target-organ specificity by abrogation of Aire in NOD mice. J Clin Invest. 2006;116:1292–1301. doi: 10.1172/JCI26971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavanescu I, Kessler B, Ploegh H, Benoist C, Mathis D. Loss of Aire-dependent thymic expression of a peripheral tissue antigen renders it a target of autoimmunity. Proc Natl Acad Sci U S A. 2007;104:4583–4587. doi: 10.1073/pnas.0700259104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 14.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 16.Fikrig E, Barthold SW, Chen M, Chang CH, Flavell RA. Protective antibodies develop, and murine Lyme arthritis regresses, in the absence of MHC class II and CD4+ T cells. J Immunol. 1997;159:5682–5686. [PubMed] [Google Scholar]
- 17.Yin DP, Ma LL, Sankary HN, Shen J, Zeng H, Varghese A, Chong AS. Role of CD4+ and CD8+ T cells in the rejection of concordant pancreas xenografts. Transplantation. 2002;74:1236–1241. doi: 10.1097/00007890-200211150-00007. [DOI] [PubMed] [Google Scholar]
- 18.Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH, Shultz LD. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med. 1996;184:2049–2053. doi: 10.1084/jem.184.5.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slattery RM, Kjer-Nielsen L, Allison J, Charlton B, Mandel TE, Miller JF. Prevention of diabetes in non-obese diabetic I-Ak transgenic mice. Nature. 1990;345:724–726. doi: 10.1038/345724a0. [DOI] [PubMed] [Google Scholar]
- 20.Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity. 2005;23:227–239. doi: 10.1016/j.immuni.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC. Aire regulates negative selection of organ-specific T cells. Nat Immunol. 2003;4:350–354. doi: 10.1038/ni906. [DOI] [PubMed] [Google Scholar]
- 22.Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, Wang L, Ichikawa Y, Jaenisch R, Hooper ML, et al. Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature. 1992;360:225–231. doi: 10.1038/360225a0. [DOI] [PubMed] [Google Scholar]
- 23.Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, Benoist C, Mathis D. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 24.Jiang W, Anderson MS, Bronson R, Mathis D, Benoist C. Modifier loci condition autoimmunity provoked by Aire deficiency. J Exp Med. 2005;202:805–815. doi: 10.1084/jem.20050693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buller RM, Holmes KL, Hugin A, Frederickson TN, Morse HC., 3rd Induction of cytotoxic T-cell responses in vivo in the absence of CD4 helper cells. Nature. 1987;328:77–79. doi: 10.1038/328077a0. [DOI] [PubMed] [Google Scholar]
- 26.Makhlouf L, Grey ST, Dong V, Csizmadia E, Arvelo MB, Auchincloss H, Jr, Ferran C, Sayegh MH. Depleting anti-CD4 monoclonal antibody cures new-onset diabetes, prevents recurrent autoimmune diabetes, and delays allograft rejection in nonobese diabetic mice. Transplantation. 2004;77:990–997. doi: 10.1097/01.tp.0000118410.61419.59. [DOI] [PubMed] [Google Scholar]
- 27.Ward L, Paquette J, Seidman E, Huot C, Alvarez F, Crock P, Delvin E, Kampe O, Deal C. Severe autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy in an adolescent girl with a novel AIRE mutation: response to immunosuppressive therapy. J Clin Endocrinol Metab. 1999;84:844–852. doi: 10.1210/jcem.84.3.5580. [DOI] [PubMed] [Google Scholar]
- 28.Ulinski T, Perrin L, Morris M, Houang M, Cabrol S, Grapin C, Chabbert-Buffet N, Bensman A, Deschenes G, Giurgea I. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome with renal failure: impact of posttransplant immunosuppression on disease activity. J Clin Endocrinol Metab. 2006;91:192–195. doi: 10.1210/jc.2005-1538. [DOI] [PubMed] [Google Scholar]
- 29.Lankisch TO, Strassburg CP, Debray D, Manns MP, Jacquemin E. Detection of autoimmune regulator gene mutations in children with type 2 autoimmune hepatitis and extrahepatic immune-mediated diseases. J Pediatr. 2005;146:839–842. doi: 10.1016/j.jpeds.2005.01.050. [DOI] [PubMed] [Google Scholar]
- 30.Kekalainen E, Miettinen A, Arstila TP. Does the deficiency of Aire in mice really resemble human APECED? Nat Rev Immunol. 2007;7:1. doi: 10.1038/nri2136-c1. [DOI] [PubMed] [Google Scholar]
- 31.Mathis D, Benoist C. Yes, it does. Nat Rev Immunol. 2007;7:1. doi: 10.1038/nri2136. [DOI] [PubMed] [Google Scholar]
- 32.Pontynen N, Miettinen A, Petteri Arstila T, Kampe O, Alimohammadi M, Vaarala O, Peltonen L, Ulmanen I. Aire deficient mice do not develop the same profile of tissue-specific autoantibodies as APECED patients. J Autoimmun. 2006;27:96–104. doi: 10.1016/j.jaut.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 33.Peterson P, Peltonen L. Autoimmune polyendocrinopathy syndrome type 1 (APS1) and AIRE gene: new views on molecular basis of autoimmunity. J Autoimmun. 2005;25(Suppl):49–55. doi: 10.1016/j.jaut.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 34.DeVoss JJ, Anderson MS. Lessons on immune tolerance from the monogenic disease APS1. Curr Opin Genet Dev. 2007;17:193–200. doi: 10.1016/j.gde.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 35.Kekalainen E, Tuovinen H, Joensuu J, Gylling M, Franssila R, Pontynen N, Talvensaari K, Perheentupa J, Miettinen A, Arstila TP. A defect of regulatory T cells in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Immunol. 2007;178:1208–1215. doi: 10.4049/jimmunol.178.2.1208. [DOI] [PubMed] [Google Scholar]
- 36.Gray DH, Gavanescu I, Benoist C, Mathis D. Danger-free autoimmune disease in Aire-deficient mice. Proc Natl Acad Sci U S A. 2007;104:18193–18198. doi: 10.1073/pnas.0709160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 38.Masteller EL, Tang Q, Bluestone JA. Antigen-specific regulatory T cells--ex vivo expansion and therapeutic potential. Semin Immunol. 2006;18:103–110. doi: 10.1016/j.smim.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Fife BT, Guleria I, Gubbels Bupp M, Eagar TN, Tang Q, Bour-Jordan H, Yagita H, Azuma M, Sayegh MH, Bluestone JA. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med. 2006;203:2737–2747. doi: 10.1084/jem.20061577. [DOI] [PMC free article] [PubMed] [Google Scholar]