

Abstract
Potassium [(trifluoroboratophenyl)methyl]triphenylphosphonium chlorides have been prepared from the corresponding benzyl chlorides and PPh3. In the presence of 1.2 equiv of K2C O3 and various aldehydes, these mixed salts are easily converted to the corresponding unsaturated organotrifluoroborates via the intermediate phosphorus ylides. A protocol for a one-pot transformation has also been developed.
The Suzuki-Miyaura cross-coupling reaction is an effective method for the formation of new carbon-carbon bonds under mild reaction conditions.1 Although they have become the standards by which all other cross-coupling reagents are measured, the boronic acids and boronate esters used in these transformations still exhibit several limitations. Because many boronic acids are waxy solids, they are difficult to purify. Furthermore, at equilibrium boronic acids form trimeric, cyclic anhydrides (boroxines), and these are not easily distinguished from the boronic acids, which themselves are not readily assayed.2 This equilibrium has no direct bearing on the cross-coupling process itself, but does influence the mass of material required for stoichiometric reactions with organic halide electrophiles. Consequently, for this reason and to overcome loss of the coupling partner as a result of protodeboronation,3 practioners often utilize excess boronic acid to insure an adequate amount of this nucleophile is available.
Most importantly, trivalent organoboron species undergo competitive, undesirable reactions with many classes of reagents commonly used in organic synthesis, including oxidants, bases and nucleophiles. Because of this reactivity, boronic acids and boronate esters are normally either purchased or prepared and then coupled immediately. These boron reagents are not typically carried through further synthetic sequences prior to coupling, making them less attractive for early incorporation into synthetic schemes in which the carbon skeleton and functional groups might be elaborated while leaving the valuable carbon-boron bond intact for subsequent processing.3 An organoboron reagent capable of withstanding extensive manipulation would address these issues and expand the range of imaginable retrosynthetic strategies in which Suzuki-Miyaura coupling could play an integral role.
For all of these reasons, there has been an increasing interest in the use of potassium organotrifluoroborates as useful coupling partners for the Suzuki-Miyaura reaction.4 The inherent air- and moisture stability of these monomeric, tetracoordinate boron species, combined with their ease of preparation as white crystalline solids, makes them an excellent alternative to boronic acids and boronate esters. Taking advantage of their inherent stability, we have generated various useful organotrifluoroborate derivatives through direct epoxidation,5 cis-dihydroxylation,6 nucleophilic substitution,7 1,3-dipolar cycloaddition,8 alcohol oxidation9 and halogen-lithium exchange reactions.10
Although Wittig and Horner-Wadsworth-Emmons reactions have been carried out on aldehyde derivatives of boronate esters,11,12 to the best of our knowledge similar reactions using functionalized boronic acids have not been reported in the literature, presumably because of the presence of acidic protons on the boron species. Recently, our group revealed the synthesis of unsaturated potassium organotrifluoroborates via the Wittig and Horner-Wadsworth-Emmons reaction of the corresponding trifluoroborato-substituted benzaldehydes.13
The complementary process, i.e., generation and reaction of boron functionalized phosphorus ylides with aldehydes, has apparently never been observed for any organoboron species. Herein, we report the preparation of potassium [(trifluoroboratophenyl)methyl]triphenylphosphonium chlorides, followed by the novel synthesis of unsaturated organotrifluoroborates via the direct Wittig reaction of these salts with various aldehydes in the presence of a base.
Initially, we prepared potassium [(trifluoroboratophenyl)methyl]triphenylphosphonium chlorides from the corresponding potassium 2-, 3-, and 4-(chloromethyl)phenyltrifluoroborates in the presence of 1.5 equiv of triphenylphosphine (PPh3) in 3-pentanone at reflux (eqs 1–3).
 |
(1) |
 |
(2) |
 |
(3) |
In the first attempts (eqs 1, 2 and 3), the potassium [(trifluoroboratophenyl)methyl]triphenylphosphonium chlorides were isolated as white solids in 93%, 94% and 86% yields, respectively. The excess PPh3 was removed from the products by trituration with ether. All of the products are stable under normal atmospheric conditions.
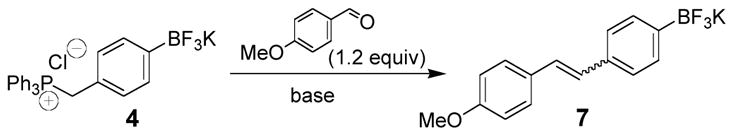
Subsequently, we optimized the Wittig reaction of the ylides derived from these salts using p-anisaldehyde and organotrifluoroborate 4. The reaction conditions explored are summarized in Table 1.
Table 1.
Optimization of Wittig Reaction Conditionsa
 | ||||
|---|---|---|---|---|
| entry | solvent | base
(1.2 equiv) |
temp
(°C) / 5 h |
conversionb (%, E/Z) |
| 1 | THF | n-BuLi | −78 to rt | 48 (1.6/1) |
| 2 | THF | NaH | 0 to rt | 46 (2.8/1) |
| 3 | DMSO-d6 | NaH | 0 to rt | 53 (2.3/1) |
| 4 | DMSO-d6 | K2CO3 | rt | 16 (2.2/1) |
| 5 | DMSO-d6 | K2CO3 | rt to 80 | 84 (2.5/1)c |
All reactions were carried out on 0.1 mmol scale in 4 mL of THF in a 10 mL round bottom flask or in 800 μL of DMSO-d6 in an NMR tube, and sonicated for 20 min.
Percent conversion was calculated by 1H NMR. The E/Z ratio was based on the integration of peaks at 6.91 (E) ppm and 6.78 (Z) ppm, respectively.
Yield is given for the isolated products.
When the reaction was performed with n-BuLi or NaH in THF, the Wittig reactions did not completely generate 7 due to the poor solubility of 4 in THF (Table 1, entries 1 and 2). However, the yield of 7 slightly increased to 53% when the reaction was repeated with DMSO-d6 in an NMR tube (Table 1, entry 3). When K2CO3 was used, although the conversion rate was lower at room temperature, a good isolated yield (84%) was obtained when the reaction temperature was increased to 80°C (Table 1, entries 4 and 5).
With the optimized conditions for the formation of 7 (Table 1, entry 5) in hand, we examined the scope of the Wittig reaction for the synthesis of olefin-functionalized organotrifluoroborates using various aryl aldehydes. The results are summarized in Table 2.
Table 2.
Wittig Reaction of Potassium [(Trifluoroboratophenyl)methyl]triphenylphosphonium Chlorides with Various o-, m- and p-Substituted Aryl Aldehydesa
 | |||||
|---|---|---|---|---|---|
| entry | A | aldehyde | reaction time (h) | product | yield
(%)b |
| 1 | 4 |
|
5 |

7E:Z = 2.5:1 |
84 |
| 2 | 5 |

|
6 |

8 E:Z = 4.1:1 |
85 |
| 3 | 6 |

|
24 |

9 E:Z = 4.7:1 |
(82)c |
| 4 | 4 |

|
4 |

10 E:Z = 1.7:1 |
99
(94)d |
| 5 | 4 |
|
3 |

11 E:Z = 1:1.2 |
97 |
| 6 | 4 |
|
1 |

12 E:Z = 1:2.1 |
87 |
| 7 | 4 |

|
1 |

13 E:Z = 2.0:1 |
97 |
All reactions were carried out on 0.1 mmol scale in 800 μL of DMSO-d6 and sonicated for 20 min.
Yield is given for the isolated products, and the E/Z ratios were determined by 1H NMR.
Product was contaminated with 2.6% of the starting potassium 2-[(trifluoroboratophenyl)methyl]triphenylphosphonium chloride (6).
Reaction was performed on 1 mmol scale in 3 mL of DMF.
In the study of the steric effect of the trifluoroborate unit using the ylides generated from the corresponding organotrifluoroborates 4–6, there was little difference in the product yields (82–85%). The E/Z ratio and the reaction times increased according to the order of the ortho>meta>para-positioned trifluoroborate unit in the aromatic ring (Table 2, entries 1–3).
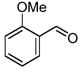
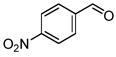
Interestingly, when 3- and 2-anisaldehydes were used (Table 2, entries 4 and 5), the reaction rates, yields and ratios of the Z-isomer were increased compared to the 4-anisaldehyde (Table 2, entry 1). Both 4-nitrobenzaldehyde and 2-cyanobenzaldehyde-containing substrates were converted to the corresponding unsaturated organotrifluoroborates in high yields (Table 2, entries 6 and 7). The Z-isomer was predominant for compound 12.
On the basis of the results in Table 2, we examined the Wittig reactions of potassium [(4 -trifluoroboratophenyl)methyl]triphenylphosphonium chloride (4) with various aliphatic and heteroaryl aldehydes (Table 3).
Table 3.
Wittig Reaction of Various Aldehydes with 4a
 | ||||
|---|---|---|---|---|
| entry | aldehyde | reaction time (h) | product | Yield
(%)b |
| 1 |
|
4 |
14 E:Z = 1.7:1 |
82 |
| 2 |

|
12 |

15 E:Z = 4.6:1 |
87 |
| 3 |
|
10 |

16 E:Z = 7.6:1 |
63 |
| 4 |

|
7 |

17 E:Z = 2.9:1 |
72
(65)c |
| 5 |
|
5 |
18 E:Z = 4.0:1 |
76 |
| 6 |
|
11 |

19 E:Z = 1.5:1 |
98 |
| 7 |
|
1 |

20 E:Z = 2.8:1 |
92
(87)c |
| 8 |

|
2 |

21 E:Z = 1:1.7 |
83
(94)c |
All reactions were carried out on 0.1 mmol scale in 800 μL of DMSO-d6 and sonicated for 20 min.
Yield is given for the isolated products and the E/Z ratios were determined by 1H NMR.
Reaction was performed on 1 mmol scale in 3 mL of DMF.
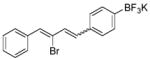
In the reactivity test of various saturated alkyl aldehydes, the reaction with the linear alkyl aldehyde (Table 3, entry 1), proceeded faster than the reactions with cyclic or branched alkanals. Moreover, the stereoselectivity of the E-isomer of products 15 and 16 were significantly influenced by the sterically more hindered saturated alkyl aldehydes such as cyclohexanecarboxaldehyde and 2,6-dimethyl-5-heptenal (Table 3, entries 2 and 3). Furthermore, conjugated diene-functionalized organotrifluoroborates 17 and 18 were obtained successfully from trans-cinnamaldehyde and α-bromocinnamaldehyde in 72% and 76% yields, respectively (Table 3, entries 4 and 5).
For the Wittig reaction of aryl aldehydes containing a heteroatom such as oxygen, sulfur and nitrogen (Table 3, entries 6–8), the desired products were obtained in good yields. The reactivity of 2-pyridinecarboxaldehyde was higher when compared to that of other heterocyclic aldehydes (Table 3, entry 8).
The Wittig reactions could be scaled up to a 1 mmol scale (Table 3 entries 4, 7 and 8) yielding similar results. Moreover, a one-pot Wittig reaction was developed. In this case, potassium [(4-trifluoroboratophenyl)methyl]-triphenylphosphonium chloride ( 4 ), the intermediate generated by reaction of 1 with triphenylphosphine, was reacted directly with 2-thiophenecarboxaldehyde in the presence of a base, and the desired product was isolated in 87% overall yield (eq 4).
 |
(4) |
To demonstrate the viability and utility of this method further, we used a Pd/C-catalyzed hydrogenation to reduce the diastereomeric mixture of alkene isomers to a single compound. In the event, 10 was reduced to 22 in 86% yield using 20 wt % Pd/C in acetonitrile (eq 5).
 |
(5) |
In summary, we have successfully prepared potassium [(trifluoroboratophenyl)methyl]triphenylphosphonium chlorides in 86–94% yield using PPh3 and the corresponding potassium (chloromethyl)phenyltrifluoroborates in 3-pentanone under refluxing conditions. We have developed a novel synthetic method for creating an alkene within potassium organotrifluoroborates via the Wittig reaction with various aldehydes in 63–99% yield. Additionally, these Wittig reactions can be performed via a one-pot process.
Supplementary Material
Experimental procedures, spectral characterization, and copies of 1H, 13C, 19F and 11B NMR spectra for all the new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported in part by the Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2005-214-C00170). We thank the National Institutes of Health (GM35249), Merck Research Laboratories, Amgen and Johnson Matthey for their generous support.
References
- 1.(a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]; (b) Suzuki A. J Organomet Chem. 1999;576:147. [Google Scholar]; (c) Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633. [Google Scholar]; (d) Suzuki A, Brown HC. Organic Syntheses via Boranes. Vol. 3 Aldrich Chemical Company, Inc; Milwaukee: 2003. [Google Scholar]; (e) Suzuki A. Proc Jpn Acad, Ser B. 2004;80:359. [Google Scholar]; (f) Suzuki A. Chem Commun. 2005:4759. doi: 10.1039/b507375h. [DOI] [PubMed] [Google Scholar]; (g) Netherton MR, Dai C, Neuschütz K, Fu GC. J Am Chem Soc. 2001;123:10099. doi: 10.1021/ja011306o. [DOI] [PubMed] [Google Scholar]; (h) Kirchhoff JH, Dai C, Fu GC. Angew Chem, Int Ed. 2002;41:1945. [PubMed] [Google Scholar]; (i) Chemler SR, Trauner D, Danishefsky SJ. Angew Chem, Int Ed. 2001;40:4544. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n. and references therein. [DOI] [PubMed] [Google Scholar]; (j) Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4442. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]
- 2.(a) Barder TE, Buchwald SL. Org Lett. 2006;ASAP [Google Scholar]; (b) Hall DG, editor. Boronic Acids. Wiley-VCH; Weinheim: 2005. [Google Scholar]; (b) Onak T. Organoborane Chemistry. Academic Press; New York: 1975. [Google Scholar]
- 3.Jin B, Liu Q, Sulikowski GA. Tetrahedron. 2005;61:401. [Google Scholar]
- 4.(a) Molander GA, Figueroa R. Aldrichimica Acta. 2005;38:49. [Google Scholar]; (b) Genêt J-P. Eur J Org Chem. 2002:3552. [Google Scholar]; (c) Molander GA, Ellis NM. Acc Chem Res. doi: 10.1021/ar050199q. In press. [DOI] [PubMed] [Google Scholar]
- 5.Molander GA, Ribagorda M. J Am Chem Soc. 2003;125:11148. doi: 10.1021/ja0351140. [DOI] [PubMed] [Google Scholar]
- 6.Molander GA, Figueroa R. Org Lett. 2006;8:75. doi: 10.1021/ol052549e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molander GA, Ham J. Org Lett. 2006;8:2031. doi: 10.1021/ol060375a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Molander GA, Ham J. Org Lett. 2006;8:2767. doi: 10.1021/ol060826r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molander GA, Petrillo DE. J Am Chem Soc. 2006;128:9634. doi: 10.1021/ja062974i. [DOI] [PubMed] [Google Scholar]
- 10.Molander GA, Ellis NM. J Org Chem. 2006;71:7491. doi: 10.1021/jo061324u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wittig: Lautens M, Mancuso J. J Org Chem. 2004;69:3478. doi: 10.1021/jo049874k.Lautens M, Marquardt T. Org Chem. 2004;69:4607. doi: 10.1021/jo049722p.Kobayashi Y, Tokoro Y, Watatani K. Tetrahedron Lett. 1998;39:7537.Kobayashi Y, Tokoro Y, Watatani K. Eur J Org Chem. 2000:3825.
- 12.Horner-Wittig-Emmons: Schmidt U, Leitenberger V, Griesser H, Schmidt J, Meyer R. Synthesis. 1992:1248.Park KC, Yoshino K, Tomiyasu H. Synthesis. 1999:2041.Busnel O, Carreaux F, Carboni B, Pethe S, Goff SVL, Mansuy D, Boucher J-L. Bioorg Med Chem. 2005;13:2373. doi: 10.1016/j.bmc.2005.01.053.Gopalarathnam A, Nelson SG. Org Lett. 2006;8:7. doi: 10.1021/ol051861l.
- 13.Molander GA, Figueroa R. J Org Chem. 2006;71:6135. doi: 10.1021/jo060863w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, spectral characterization, and copies of 1H, 13C, 19F and 11B NMR spectra for all the new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.