

Abstract
1,2,3,4-Diepoxybutane (DEB), an in vivo metabolite of 1,3-butadiene (BD), is a carcinogen and a potent mutagen. Previously, DEB was shown to react with 2′-deoxyguanosine (dG) under physiological conditions to produce seven major nucleoside adducts resulting from alkylation at the N1- (P8 and P9), N7- (P5 and P5′) and both the N1- and N2-positions of dG to form six-membered (P4-1 and P4-2) and seven-membered fused ring systems (P6), respectively (Zhang and Elfarra, Chem. Res. Toxicol. 2003, 16, 1606; ibid, 2004, 17, 521). In the present study the stabilities and decomposition products of the seven adducts under in vitro physiological conditions (phosphate buffer containing KCl, pH 7.4, 37 °C) were investigated. The results showed that P4-1, P4-2 and P6 were stable, whereas P5, P5′, P8 and P9 were labile with half-lives of 2.6, 2.7, 16 and 16 h, respectively. P5 and P5′ decomposed initially by the loss of the deoxyribose moiety to yield the corresponding guanine adduct P5D, which exhibited a half-life of 33 h and decomposed through opening of the remaining oxirane ring by dihydrogen phosphate ion, water or chloride ion. Decomposition of P8 yielded P4-1, P6 and nucleoside products resulting from opening of the oxirane ring by dihydrogen phosphate ion, water or chloride ion. Similarly, decomposition of P9 led to formation of P4-2, P6, and nucleoside products resulting from opening of the oxirane ring by dihydrogen phosphate ion, water or chloride ion. These results indicate that the initial products of the reaction of DEB with dG are P5, P5′, P8 and P9, whereas P4-1, P4-2 and P6 are secondary products. The results may also facilitate development of useful biomarkers of exposure to DEB.
Introduction
BD1 is a petrochemical widely used in manufacturing of synthetic rubber and plastic. Long-term inhalation exposure of mice and rats to BD caused development of tumors at multiple sites (1-3). BD is also considered a “Human Carcinogen” mostly because occupational exposure to BD has been associated with excess mortality from lymphatic and/or hematopoietic cancers (4-8). Bioactivation of BD in animal and human tissues to yield reactive epoxides is an important determinant of the mutagenic and carcinogenic effects of BD (9).
BD is converted to 3,4-epoxy-1-butene (EB2) by both cytochrome P450- and myeloperoxidase-mediated oxidation (10-12). Further cytochrome P450-mediated oxidation of EB yields DEB2 (13). Both EB and DEB are direct-acting mutagens, which are widely believed to play important roles in the mutagenicity/carcinogenicity of BD. Because the in vitro mutagenicity of DEB was ∼100 times higher than that of EB (14-17), DEB may play a prominent role in the carcinogenic effects of BD.
Racemic DEB3 has recently been demonstrated to readily react with dG under in vitro physiological conditions (pH 7.4, 37 °C) to produce seven major nucleoside adducts (18). Six of the seven adducts were structurally characterized as three pairs of diastereomers, namely, 7-hydroxy-3-(2-deoxy-β-D-erythro-pentofuranosyl)-6-hydroxymethyl-5,6,7,8-tetrahydro-10H-pyrimido[1,2-a]purin-10-one4 (P4-1/P4-25), 2′-deoxy-7-(2-hydroxy-2-oxiranylethyl)guanosine (P5/P5′) and 2′-deoxy-1-(2-hydroxy-2-oxiranylethyl)guanosine (P8/P9) (Scheme 16). The seventh adduct was characterized as 7,8-dihydroxy-3-(2-deoxy-β-D-erythro-pentofuranosyl)-3,5,6,7,8,9-hexahydro-11H-[1,3]diazepino[1,2-a]purin-11-one (P6), and was assumed to also be a pair of diastereomers unresolved on the HPLC. As previous results have shown that these adducts exhibited different stabilities and decomposition rates under the reaction conditions, the goal of the present study was to characterize the stabilities and decomposition products of the seven adducts under in vitro physiological conditions to clarify the initial products of the DEB-dG reaction.
Scheme 1.

Structures of the Major DEB-dG Nucleoside Adducts Formed at Physiological Conditions (pH 7.4, 37 °C) and the Corresponding Acid Hydrolysis and Decomposition Products (the Structures of Phosphoric Acid Esters Resulting from Opening of the Oxirane Rings of P5D and P8/P9 by Dihydrogen Phosphate Ion Are Not Shown for Simplicity)
Experimental Procedures
Caution
DEB is a known mutagen and carcinogen and must be handled using proper safety measures.
Materials
Racemic DEB, dG (monohydrate), trifluoroacetic acid (TFA), dimethyl sulfoxide-d6 (DMSO-d6) and TMS were obtained from Sigma-Aldrich Chemical Co. (Milwaukee, WI). HPLC grade acetonitrile was purchased from EM Science (Gibbstown, NJ). Sephadex LH-20 resin was purchased from Pharmacia LKB Biotechnology (Sweden). HPLC mobile phases were prepared using acetonitrile and water, and the pH was adjusted to 2.5 with 20% (v/v) TFA if necessary. The pH 7.4 phosphate buffer (100 mM) containing 100 mM KCl was prepared with KH2PO4 and KCl using KOH to adjust the pH. P4-1, P4-2, P5, P5′, P6, P8 and P9 were prepared and purified from the reaction of DEB and dG according to the procedures described previously (18).
Instruments and Methods
Mass spectra were obtained on an Applied Biosystems MDS Sciex API 365 LC/MS/MS triple quadrupole electrospray ionization mass spectrometer (Foster City, CA). Products were first purified by HPLC and the purified compounds were dissolved in 1:1 acetonitrile-water (v:v) for mass spectrometric analyses using electrospray ionization. MS-MS data of protonated molecular ions were also collected to obtain fragment patterns. NMR data of P4-1 and P4-2 were obtained on a Varian Unity Inova 900 MHz, 21.1 T standard bore magnet NMR spectrometer and a Bruker Instruments DMX-500 Avance console, 11.74 T standard bore magnet NMR spectrometer, HMQC spectrum of H3 was collected on a Bruker Instruments DMX-600 Avance console, 14.1 T standard bore magnet NMR spectrometer and 1H NMR spectra of P5D and H3 were recorded on a Bruker Instruments DMX-400 Avance console, 9.4 T wide-bore magnet NMR spectrometer. The compounds were dissolved in DMSO-d6 and TMS was added as the internal standard. Analytical HPLC separation was performed on a Beckman Ultrasphere 5 μm ODS reverse-phase analytical column (250×4.6 mm), using a Beckman gradient-controlled HPLC system (Irvine, CA) equipped with a Beckman diode array detector (Model 168). Acidic (pH 2.5) and pH-unadjusted mobile phases were both used for analytical separation. UV absorption spectra of the products were extracted from data collected by the diode array detector after running. A linear gradient program was used, starting at 2 min from 0% pump B to 10% pump B over 1 min [pump A, 1% (v/v) acetonitrile at either pH 2.5 or pH-unadjusted; pump B, 10% (v/v) acetonitrile at either pH 2.5 or pH-unadjusted] at a flow rate of 1.0 mL/min, then at 10 min from 10 to 20% pump B over 1 min, at 11 min from 20 to 30% pump B over 9 min, at 20 min from 30 to 65% pump B over 1 min, at 25 min from 65 to 100% pump B over 12 min, finally at 44 min from 100 to 0% pump B over 1 min and stopped at 45 min. Preparative separation and purification were carried out on a Beckman Ultrasphere 5 μm ODS reverse-phase semi-preparative column (250×10 mm). The gradient program and the mobile phases used on pump A and pump B were identical to those used with the analytical column and the flow rate was 3.0 mL/min. Samples were loaded onto both analytical and semi-preparative columns manually. For preparative separation, fractions were collected using a Gilson fraction collector (Model 202) with all collection beakers kept on ice. The collected fractions were lyophilized on a Labconco lyophilizer (Kansas City, MO).
Determination of Half-Lives of P4-1, P4-2, P5, P5′, P5D, P6, P8 and P9
Purified adducts were dissolved in the pH 7.4 phosphate buffer containing 100 mM KCl; the concentrations were adjusted so that the HPLC peak heights of the adducts would not reach maximal absorbance (approximately 2.2) at both 220 and 260 nm when samples (5 μL) were loaded onto the analytical column. The solutions of the adducts (100-200 μL) were then incubated at 37 °C with gentle shaking. Aliquots were taken periodically and injected onto the analytical HPLC column. With the unstable adducts (P5, P5′, P5D, P8 and P9), at least six aliquots were taken for HPLC analysis before the adducts decomposed completely. Natural logarithm of the peak areas of the adducts obtained from the chromatograms were plotted against incubation time. The data were regressed and half-lives of the adducts were calculated accordingly.
Isolation and Characterization of Decomposition Products of P5, P5′, P8 and P9
Solutions of P5, P5′, P8 and P9 in pH 7.4 phosphate buffer containing 100 mM KCl were incubated at 37 °C until these adducts decomposed fully as monitored by HPLC. Decomposition products were then isolated from the incubations using semi-preparative HPLC. Products were characterized by mass spectrometry, HPLC coelution experiments and/or by conducting acid hydrolysis experiments. The structure of P5D was confirmed by additional NMR data.
Acid Hydrolysis of Nucleoside Adducts and Their Decomposition Products
To characterize decomposition products of DEB-dG adducts, acid hydrolysis experiments were carried out. To a solution of an adduct or a decomposition product (100-500 μg) in water, one tenth volume of concentrated HCl was added so that the final HCl concentration was approximately 1 M. The acidic solution was heated for 1 h in a boiling-water bath, cooled to room temperature and neutralized to pH 6-7 using concentrated ammonium hydroxide. The neutralized solution was then analyzed on an analytical HPLC column.
When hydrolysis was carried out at lower HCl concentrations (0.1 and 0.01 M), the work-up procedures were the same as above except one tenth volume of 1.1 or 0.11 M HCl was used instead of concentrated HCl.
Results
Stabilities and Half-Lives of P4-1, P4-2, P5, P5′, P6, P8 and P9
At physiological conditions (pH 7.4, 37 °C), P4-1, P4-2 and P6 were stable; no decomposition was observed even after the three adducts were incubated for up to 520 h. On the contrary, P5, P5′, P8 and P9 were labile. Half-lives of these adducts were determined to be 2.6, 2.7, 16 and 16 hours, respectively (Table 1). Figure 1 shows the stability of P4-1 and the fitted exponential decay curves of P5 and P8 at pH 7.4 and 37 °C.
Table 1.
The Measured Half-Lives (h)a of P5, P5′, P5D, P8 and P9 when Incubated at pH 7.4 and 37 °C
| P5 | P5′ | P5D | P8 | P9 |
|---|---|---|---|---|
| 2.6 ± 0.1 | 2.7 ± 0.1 | 33 ± 1 | 16 ± 1 | 16 ± 1 |
Each value was an average of results obtained from two different incubations.
Figure 1.
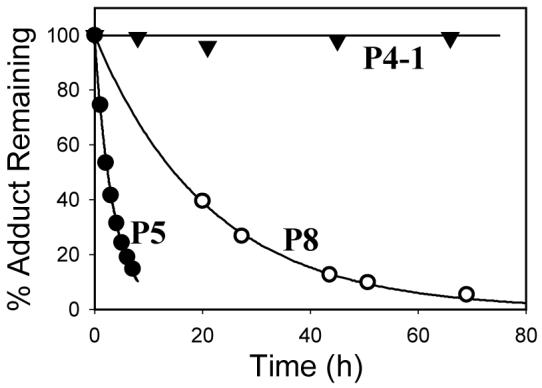
Stability of P4-1 and fitted decay curves of P5 and P8 at pH 7.4 and 37 °C (the points represent the experimental data that were fitted using exponential decay)
Characterization of Decomposition Products of P5 and P5′
Time-course experiments demonstrated that P5 and P5′, which were eluted at 19.3 and 21.5 min, respectively, first overwhelmingly decomposed to an intermediate (P5D) (Figure 2, upper panel) that exhibited a shorter retention time (13.5 min) and a longer half-life (33 h, Table 1). P5D further decomposed to several final products, among which three predominant products were eluted at 6.6, 8.8 and 18.0 min and their peaks were numbered from 1 to 3 (Figure 2, lower panel). The product with a retention time of 6.6 min had a molecular weight of 335 as determined by ESI-MS and its MS-MS spectrum exhibited peaks corresponding to loss of water and phosphoric acid, indicating that this decomposition product was a phosphoric acid ester. Obviously, this product was formed by the reaction of the oxirane ring of P5D with dihydrogen phosphate ion present in the phosphate buffer. The product eluted at 18.0 min coeluted with reference 2-amino-7-(4-chloro-2,3-dihydroxybutyl)-1,7-dihydro-6H-purin-6-one (H3, see below) (18, 19), and also possessed the same UV absorption spectrum as that of H3. Thus, this product was characterized as H3. Similarly, the decomposition product eluting at 8.8 min was characterized as 2-amino-7-(2,3,4- trihydroxybutyl)-1,7-dihydro-6H-purin-6-one (H4′), also a known compound (19, 20).
Figure 2.
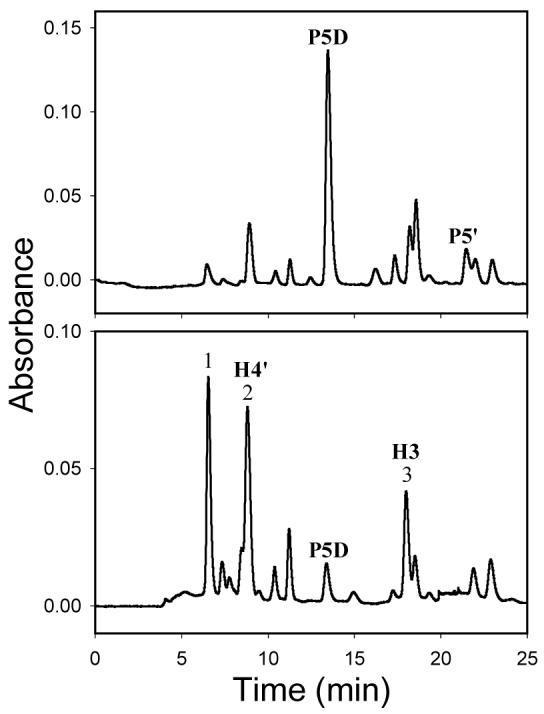
HPLC chromatograms of decomposition mixture of P5′ after incubation for 7 h (upper panel) and 121 h (lower panel) under physiological conditions (pH 7.4, 37 °C) (acidic mobile phase, monitored wavelength: 260 nm). Because of the rapid decomposition of P5′ (Table 1), the purity of the starting material in these experiments was approximately 50% with P5D being the major impurity in the sample.
Molecular weight of P5D was 237 as determined by ESI-MS, indicating that P5D was the corresponding guanine product of P5 and P5′ after cleavage of the deoxyribose moiety. 1H NMR spectrum of P5D confirmed loss of the deoxyribose moiety (Table 2). Hydrolysis of P5D by 0.1 M HCl yielded two products. One product was identified as H3 based upon the results of HPLC coelution experiments and matching the UV absorption spectrum of this product to that of the standard. The other product was similarly determined as H4′. These results confirmed that the structure of P5D was 2-amino-7-(2-hydroxy-2-oxiranylethyl)-1,7-dihydro-6H-purin-6-one (Scheme 1).
Table 2.
1H NMR Spectroscopic Data (ppm) for H3, P5D, P4-1 and P4-2 in DMSO-d6a
| H3 | P5Db | P4-1 | P4-2 | ||||
|---|---|---|---|---|---|---|---|
| peaks | assignments | peaks | assignments | peaks | assignments | peaks | assignments |
| 10.67 (s, 1H) | H-1 | 11.06 (br s, 1H) | H-1 | 7.90 (s, 1H) | H-8 | 7.91 (s, 1H) | H-8 |
| 7.79 (s, 1H) | H-8 | 8.18 (s, 1H) | H-8 | 7.64 (s, 1H) | NH | 7.66 (d, 1H) | NH |
| 6.04 (s, 2H) | NH2 | 6.47 (s, 2H) | NH2 | 6.10 (t, 1H) | H-1′ | 6.11 (dd, 1H) | H-1′ |
| 5.29 (d, 1H) | 3′-OH | 5.47 (br s, 1H) | 2′-OH | 5.31 (s, 1H) | 2”-OH | 5.32 (d, 1H) | 2”-OH |
| 5.01 (d, 1H) | 2′-OH | 4.38 (dd, 1H) | H-1′ | 5.27 (s, 1H) | 3′-OH | 5.28 (d, 1H) | 3′-OH |
| 4.30 (dd, 1H) | H-1′ | 4.20 (dd, 1H) | H-1′ | 4.95 (s, 1H) | 5′-OH or 4”-OH | 4.96 (br s, 2H) | 5′-OH, 4”-OH |
| 4.14 (dd, 1H) | H-1′ | 3.68 (m, 1H) | H-2′ | 4.94 (s, 1H) | 5′-OH or 4”-OH | ||
| 3.92 (m, 1H) | H-2′ | 2.96 (m, 1H) | H-4′ | 4.34 (s, 1H) | H-3′ | 4.34 (m, 1H) | H-3′ |
| 3.66 (dd, 1H) | H-4′ | 2.69 (m, 1H) | H-4′ | 4.26 (dd, 1H) | H-1” | 4.28 (m, 1H) | H-1” |
| 3.50 (dd, 1H) | H-4′ | 4.12 (s, 1H) | H-2” | 4.13 (s, 1H) | H-2” | ||
| 3.49 (m, 1H) | H-3′ | 3.81 (m, 1H) | H-4′ | 3.81 (m, 1H) | H-4′ | ||
| 3.55 (m, 2H) | H-5′, H-1” | 3.55 (m, 2H) | H-5′, H-1” | ||||
| 3.50 (m, 1H) | H-5′ | 3.47 (m, 2H) | H-5′, H-4” | ||||
| 3.44 (m, 1H) | H-4” | ||||||
| 3.38 (m, 1H) | H-4” | 3.34 (m, 2H) | H-4”, H-3” | ||||
| 3.30 (m, 1H) | H-3” | ||||||
| 2.54 (m, 1H) | H-2′ | 2.52 (m, 1H) | H-2′ | ||||
| 2.18 (m, 1H) | H-2′ | 2.18 (m, 1H) | H-2′ | ||||
The data of P4-1 were obtained in this study. The data of P4-2 from previous results (18) are also listed here because peak assignments were revised based upon the results from this study.
The signal of the proton at position 3′, which was covered by that of residual water, is not listed.
Unlike H3, H4′, P5D and the phosphoric acid ester, which were detected directly from the decomposition mixture of P5 and P5′ at physiological conditions, identification of 2,6-diamino-5-(3,4-dihydroxy-1-pyrrolidinyl)- 4(3H)-pyrimidinone (H3′) required acid hydrolysis of the decomposition mixtures of P5 and P5′. The HPLC chromatograms of acid-hydrolyzed decomposition mixtures of P5 and P5′ showed a peak whose retention time (10.6 min on semi-preparative column, acidic mobile phase) and UV absorption spectrum matched those of standard (H3′) (20), in addition to H3, H4′ and several uncharacterized minor products. Interestingly, the chromatogram of the acid-hydrolyzed decomposition mixture of purified P5D did not show the H3′ peak, indicating that the precursor of H3′ was directly produced from P5 and P5′ instead of P5D.
Re-characterization of H3
H3, initially isolated from the reaction mixture of DEB with dG after hydrolysis by 1 M HCl, was previously characterized as 2-amino-7-(3-chloro-2,4-dihydroxybutyl)-1,7-dihydro-6H-purin-6-one based on limited 1H NMR data (18, 19). It has been noticed that the chlorine atom in H4 (Scheme 1), an acid hydrolysis product of P8 and P9 and also initially isolated from the reaction mixture of DEB with dG after hydrolysis by 1 M HCl, is at the end carbon of the side chain (i.e., position 4′) (20). Because H3 was a major acid-hydrolysis product of P5 and P5′, a question was raised as to why P5/P5′ and P8/P9 seemed to hydrolyze by different mechanisms (SN1 vs. SN2) (20). To conclusively determine the position of the chlorine atom of H3, 1H NMR and HMQC spectra of H3 in DMSO-d6 were obtained (Supplemental Figure 1). The HMQC spectrum indicated that the two methylene carbons had low chemical shifts (48.9 and 52.5 ppm) whereas the two methine carbons had high chemical shifts (72.6 and 74.6 ppm). Obviously, the two hydroxyls were on the two methine carbons (i.e., C2′ and C3′) and the chlorine atom was on the end carbon (i.e., C4′) rather than C3′. In addition, the HMQC spectrum of H3 also showed that the two protons with signals appearing around 3.50 ppm, which were previously assigned as the two protons at C4′ (19), were not on the same carbon. Instead, one of the two protons was at C4′ and the other one was at C3′. The modified assignments of the 1H NMR signals are listed in Table 2. Therefore, the correct structure of H3 is 2-amino-7-(4-chloro-2,3-dihydroxybutyl)-1,7-dihydro-6H-purin-6-one (Scheme 1).
Characterization of Decomposition Products of P8 and P9
Decomposition of P8 and P9 in pH 7.4 phosphate buffer containing KCl at 37 °C led to formation of many products (Figure 3). The time-course experiments of decomposition of P8 and P9 suggested that P8 and P9 directly yielded the final products rather than through an intermediate (Figures 3A and 3B). Because most of the decomposition products of P8 had different retention times from those of P9, characterization of the major decomposition products of P8 and P9 is described separately below.
Figure 3.
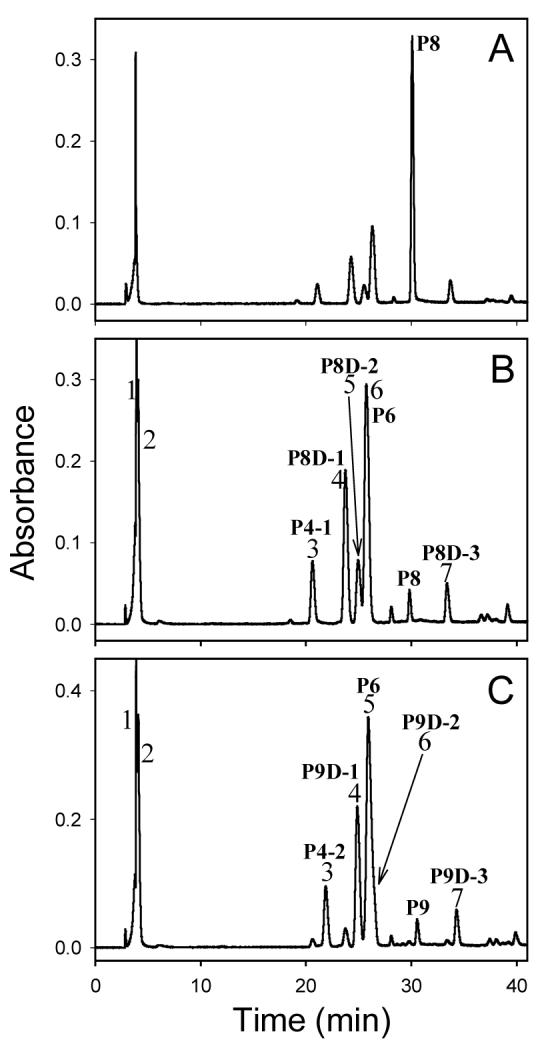
HPLC chromatograms of decomposition mixtures of P8 after incubation for 24 h (A) and 120 h (B), and P9 after incubation for 120 h (C) under physiological conditions (pH 7.4, 37 °C) (pH-unadjusted mobile phase, monitored wavelength: 260 nm)
The HPLC chromatogram of the decomposition mixture of P8 exhibited several major peaks (Figure 3B), which were numbered from 1 to 7 in the order of their retention times. The compounds that peak 4, 5 and 7 represent were designated as P8D-1, P8D-2 and P8D-3, respectively. The compound eluting at 29.8 min was undecomposed P8; Figure 4 shows that the decomposition products of P8 were formed in a time-dependent manner.
Figure 4.
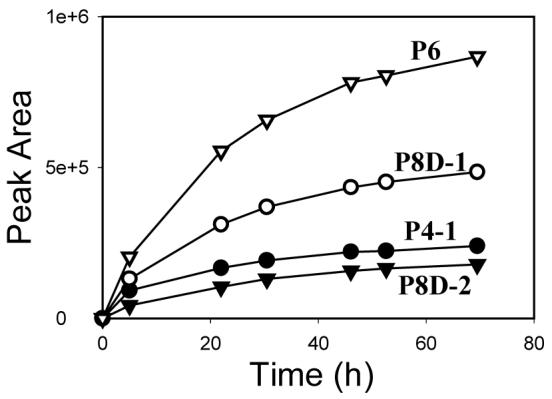
Formation of decomposition products (P4-1, P8D-1, P8D-2 and P6) of P8 over time at physiological conditions (pH 7.4, 37 °C)
Peak 1 was very sharp and overlapped with peak 2 (Figure 3B). However, when the decomposition mixture was separated on semi-preparative column, only one peak with shoulders was observed. ESI-MS of the compound that this peak represents showed a predominant ion at m/z 452, consistent with this product being a phosphoric acid ester resulting from opening of the oxirane ring of P8 by a dihydrogen phosphate ion. This product was not further characterized by NMR spectrometry.
Molecular weights of the products that peak 3 and 6 (Figure 3B) represent were determined by ESI-MS to be the same as that of P8 (i.e., 353), suggesting that the two compounds were formed through intramolecular reactions. The product that peak 3 represents was identified as 7-hydroxy-3-(2-deoxy-β-D-erythro-pentofuranosyl)-6-hydroxymethyl-5,6,7,8-tetrahydro-10H-pyrimido[1,2-a]purin-10-one (P4-1, see below), an adduct characterized before (18), based upon the results of HPLC coelution experiments and matching the UV absorption spectrum of this product to that of the standard. Furthermore, the only hydrolysis product of this product by 1M HCl was similarly identified as 7-hydroxy-6-hydroxymethyl-5,6,7,8-tetrahydro-pyrimido[1,2-a]purin-10(1H)-one (H2), the known hydrolysis product of P4-1 (20). Therefore, the product that peak 3 represents was characterized as P4-1. Similarly, the product that peak 6 represents was characterized as 7,8-dihydroxy-3-(2-deoxy-β-D-erythro-pentofuranosyl)-3,5,6,7,8,9-hexahydro-11H-[1,3]diazepino[1,2-a]purin-11-one (P6) (18).
ESI-MS spectra showed that the molecular weights of P8D-1 and P8D-2 were 371, consistent with opening of the oxirane ring of P8 by a molecule of water. Hydrolysis of P8D-1 in 1M HCl yielded 2-amino-1-(2,3,4-trihydroxybutyl)-1,7-dihydro-6H-purin-6-one (H1′), a known acid hydrolysis product of P8 and P9 (20), based upon the results of HPLC coelution experiments and matching the UV absorption spectrum of the P8D-1 acid hydrolysis product to that of the standard. Similarly, hydrolysis of P8D-2 also gave only one product, which was identified as H5′, a diastereomer of H1′ (20). Therefore, P8D-1 and P8D-2 were characterized as a pair of diastereomers of 2′-deoxy-1-(2,3,4-trihydroxybutyl)guanosine.
Molecular weight of P8D-3 was determined by ESI-MS to be 390. The isotope peak at m/z 393 with intensity of ∼1/3 of the peak at m/z 391 (protonated molecular ion) indicated that the molecule contained a chlorine atom. These results are consistent with P8D-3 being formed by opening of the oxirane ring of P8 by a chloride ion. Hydrolysis of P8D-3 in 1M HCl yielded 2-amino-1-(4-chloro-2,3-dihydroxybutyl)-1,7-dihydro-6H-purin-6-one (H4), a known acid hydrolysis product of P8 and P9 (20), based upon the results of HPLC coelution experiments and matching the UV absorption spectrum of the P8D-3 acid hydrolysis product to that of the standard. Therefore, P8D-3 was characterized as 2′-deoxy-1-(4-chloro-2,3-dihydroxybutyl)guanosine.
Like P8, seven major product peaks, numbered from 1 to 7 in the order of their retention times, could be identified in the HPLC chromatogram of P9 decomposition mixture (Figure 3C). Among these peaks, peak 6 appeared as a shoulder on peak 5. The products that peak 4, 6 and 7 represent were designated P9D-1, P9D-2 and P9D-3, respectively. The compound eluting at 30.6 min was undecomposed P9.
The products that peak 1 and 2 represent were not characterized, but at least one of these products was assumed to be phosphoric acid ester due to similar retention time and UV absorption spectrum to those of phosphoric acid ester of P8. The products that peak 3 and 5 represent were characterized as P4-2 (a diastereomer of P4-1) and P6 (18), P9D-1 and P9D-2 were characterized as a pair of diastereomers of 2′-deoxy-1-(2,3,4-trihydroxybutyl)guanosine, and P9D-3 was characterized as 2′-deoxy-1-(4-chloro-2,3-dihydroxybutyl)guanosine. Similar to P8D-1, P8D-2 and P8D-3, acid hydrolysis products of P9D-1, P9D-2 and P9D-3 were H1′, H5′ and H4, respectively. P8D-1 had a different HPLC retention time from that of P9D-1, but they both yielded the same hydrolysis product (H1′)7. Similar results were obtained with P8D-2/P9D-2 and P8D-3/P9D-3. Thus, P8D-1/P9D-1, P8D-2/P9D-2 and P8D-3/P9D-3 were characterized as three pairs of diastereomers (Scheme 1).
Re-characterization of P4-1 and P4-2
Previously, we characterized P4-1 and P4-2 as 2′-deoxy-N-(2-hydroxy-1-oxiranylethyl)guanosine based on limited NMR data (18). Obviously, this structural assignment needed to be re-examined after it was discovered that P4-1 and P4-2 were derived from P8 and P9, respectively. Hydrolysis of P4-1 and P4-2 by different concentrations of HCl (1, 0.1 and 0.01 M) only yielded one product, i.e., H2 (data not shown). If an intact oxirane ring exists in the molecules of P4-1 and P4-2 as suggested by previous structures, acid hydrolysis would likely yield multiple products.
Because P4-1 and P4-2 were derived from P8 and P9, respectively, they must be 1-alkylation adducts. On the other hand, no single peak containing two protons could be observed in their 1H NMR spectra, thus P4-1 and P4-2 were also N-alkylated. Taken together, the results indicated that P4-1 and P4-2 were both 1- and N-alkylated; i.e., the carbon chain of DEB formed a fused ring with the pyrimidine ring. The fused ring was determined to be six-membered, because 13C DEPT 135 experiment (Supplemental Figure 2) showed that one of the two methylene carbons on the DEB moiety had a high chemical shift (62.59 ppm, see Ref. 18, Table 2; Supplemental Figure 2), indicating that the methylene carbon was attached to a hydroxyl group. Therefore the structures of P4-1 and P4-2 were corrected as 7-hydroxy-3-(2-deoxy-β-D-erythro-pentofuranosyl)-6-hydroxymethyl-5,6,7,8-tetrahydro-10H-pyrimido[1,2-a]purin-10-one (Scheme 1). The assignments of their 1H NMR signals are thus revised accordingly (Table 2).
Discussion
Seven major nucleoside adducts have been identified from the reaction of DEB with dG under in vitro physiological conditions (18). Among these adducts, P5 and P5′ were the products formed first in the DEB-dG reaction, but they also were the first to decompose (see Ref. 18, Figure 2). The latter observation is consistent with the relatively short half-lives of purified P5 and P5′ (Table 1). The primary decomposition product P5D maintained the oxirane ring of P5 and P5′ but lost the deoxyribose moiety. Decomposition of P5D was slower than that of P5 and P5′ (Table 1) and was caused by opening of the oxirane ring by dihydrogen phosphate ion, water molecule or chloride ion to yield phosphoric acid ester, H4′ or H3, respectively.
The above-mentioned results suggest that lability of P5 and P5′ is primarily caused by the positive charge on the imidazole ring. Besides leading to the loss of the deoxyribose moiety, the positive charge also made the C8 of the purine ring a target for nucleophilic attack, which could lead to formation of many decomposition products (20); a comparison of HPLC chromatograms of decomposition mixtures of P5 and P5′ with that of P5D revealed that decomposition of P5 and P5′ led to formation of several more products than P5D (data not shown). Interestingly, H3′ was not detected when the incubation mixture of P5D was subjected to acid hydrolysis, providing further evidence that the positive charge in P5 and P5′ played a critical role in the formation of H3′.
P8 and P9 exhibited longer half-lives than P5 and P5′ (Table 1). Unlike P5 and P5′, lability of P8 and P9 stemmed from the intact oxirane ring of the DEB side chain, and decomposition of P8 and P9 yielded stable nucleoside adducts. P4-1, P4-2 and P6 are formed by intramolecular reactions, whereas the other products resulted from opening of the oxirane ring by dihydrogen phosphate ion, water or chloride ion. It is interesting to note that with both P8 and P9 the intramolecular reactions favor formation of the seven-membered products (i.e., P6 diastereomers) rather than the six-membered products (i.e., P4-1 and P4-2) possibly because of steric factors. The results also confirmed that P6 was indeed a mixture of two diastereomers that were not resolved at the used HPLC conditions.
The DEB used in these experiments is a racemic mixture of two enantiomers (R/R and S/S). As a result, the dG adducts formed are pairs of diastereomers. In fact, the configurations of the nucleoside adducts and their acid hydrolysis/decomposition products can be related to those of DEB through reaction mechanisms. Because the two chiral centers of DEB are at the two nonend carbons (i.e., C2 and C3), reactions happening at the end carbons (i.e., C1 and C4) will proceed with retention of configurations of the two chiral centers of DEB. Thus, the configurations of the DEB side chains of P5/P5′ and P8/P9 are the same as those of DEB, namely, they must be (2”R, 3”R) and (2”S, 3”S) even though our results do not allow assignment of the configurations of the diastereomers. Apparently, the P6 diastereomers derived from P8 and P9 must also have (2”R, 3”R) and (2”S, 3”S) configuration although the used HPLC conditions did not allow resolution of these diastereomers. On the other hand, P4-1 and P4-2 will likely have (2”R, 3”S) and (2”S, 3”R) configurations because of the inversion of configuration expected from the SN2 nucleophilic attack of the exocyclic amino group on the C3” of the DEB moiety.
The results in this study indicated that the primary products of the reaction of DEB with dG were P5, P5′, P8 and P9 (Scheme 1). Although P4-1, P4-2 and P6 were previously isolated from DEB-dG reaction mixtures, the results from this study indicate that they are not produced directly from DEB and dG; instead, they are secondary products, produced from primary products P8 and P9. Identification of the primary and secondary products of the DEB-dG reaction may allow a better understanding of the roles of these adducts in the mutagenicity and carcinogenicity of DEB. It may also facilitate the study of DEB biomarkers of exposure. At present H4′ has been used as a biomarker of BD exposure in vivo (21-24). However, H4′ is not an ideal biomarker because it could arise by the reaction of dG with either DEB, 3,4-epoxybutan-1,2-diol (EBD), or both. Because of significant difference in the mutagenicity of these epoxides (16, 17), it is important to develop specific biomarkers for each metabolite. In this regard, H3 may represent a useful specific biomarker for DEB because it can be formed only from DEB [the possibility that H4′ could be converted to H3 by HCl was excluded because heating H4′ in 1 M HCl did not yield H3 (data not shown)]. H3 can be produced by decomposition of P5 and P5′ in solutions containing chloride ion, or by hydrolysis of P5 and P5′ in HCl. Interestingly, H4′, rather than H3, is the predominant product of P5 and P5′ under most of the used decomposition and hydrolysis conditions; for decomposition in pH 7.4 phosphate buffer containing 0.1 M KCl, the ratio H4′:H3 was 3.7:1 for P5 and 3.1:1 for P5′ (based on the peak areas at 260 nm). The ratio H4′:H3 in acid hydrolysis of P5 and P5′ depends on the concentration of HCl: approximately 1:3 for 1 M HCl, 1:1 for 0.1 M HCl, and 5:2 for 0.01 M HCl.
The decomposition products of P8 and P9, especially, P4-1, P4-2, P6, and their hydrolysis products H2 and H2′, also have potential to be useful biomarkers of DEB exposure. Advantages of these compounds as potential specific biomarkers for DEB include: a) their formation is not dependent on the presence of chloride ion; b) P6 is the predominant decomposition product of both P8 and P9.
Supplementary Material
Acknowledgments
This research was supported by NIH grant ES06841 from the National Institute of Environmental Health Sciences. We thank Dr. Mark Anderson for his assistance in measuring NMR spectra. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by National Institutes of Health grants P41RR02301 (Biomedical Research Technology Program, National Center for Research Resources) and P41GM66326 (National Institute of General Medical Sciences). Equipment in the facility was purchased with funds from the University of Wisconsin, the National Institutes of Health (P41GM66326, P41RR02301, RR02781, RR08438), the National Science Foundation (DMB-8415048, OIA-9977486, BIR-9214394), and the U.S. Department of Agriculture.
Footnotes
- BD
- 1,3-butadiene
- DEB
- 1,2,3,4-diepoxybutane
- DEPT
- distortionless enhancement by polarization transfer
- dG
- 2′-deoxyguanosine
- DMSO
- dimethyl sulfoxide
- EB
- 3,4-epoxy-1-butene
- EBD
- 3,4-epoxybutan-1,2-diol
- ESI
- electrospray ionization
- HMQC
- heteronuclear multiple quantum coherence
- MS
- mass spectrometry
- MS-MS
- tandem mass spectrometry
- TFA
- trifluoroacetic acid
According to the Chemical Abstracts nomenclature rules, 3,4-epoxy-1-butene, 1,2,3,4-diepoxybutane and 3,4-epoxybutan-1,2-diol should be named ethenyloxirane, 2,2′-bioxirane and 1-oxiranyl-1,2-ethanediol, respectively. However, to be consistent with the literature, the names 3,4-epoxy-1-butene, 1,2,3,4-diepoxybutane and 3,4-epoxybutan-1,2-diol are still used in this article.
DEB has three isomers, R/S or S/R (meso), R/R and S/S. In this study, racemic DEB (dl, a 1:1 mixture of R/R and S/S isomers) was used.
The structures of P4-1 and P4-2 were prematurely characterized previously as 2′-deoxy-N-(2-hydroxy-1-oxiranylethyl)guanosine based on limited NMR data (18). Their structures were corrected based upon the results of the present study.
The adducts were designated according to the following rules: P refers to the reaction products of DEB and dG; H refers to the acid hydrolysis products of the reaction products; D following P and a number refers to the corresponding decomposition products of the reaction products. For consistency with our previous publications (18, 20), designations were made on the basis of peak resolution by HPLC, not on the basis of the stereochemistry of the reactions. For clarity, Scheme 1 shows the chemical structures of all compounds and their designations.
All compounds shown in Scheme 1 were named according to the Chemical Abstracts nomenclature rules. The purine ring, the deoxyribose moiety and the side chain were numbered as shown on the structure of P8/P9. The fused-rings of P4-1, P4-2, P6, H2 and H2′ were numbered differently, shown outside the rings of P4-1/P4-2 and P6, as required by the nomenclature rules. However, to facilitate discussion and comparison of these compounds with other adducts, the DEB moieties in these molecules were also numbered (from 1” to 4”) as shown inside the rings of P4-1/P4-2 and P6.
The acid hydrolysis product of P8D-1 is indeed an enantiomer of the acid hydrolysis product of P9D-1. However, enantiomers are not resolved at our HPLC conditions (non-chiral column and mobile phase).
References
- (1).Huff JE, Melnick RL, Solleveld HA, Haseman JK, Powers M, Miller RA. Multiple organ carcinogenicity of 1,3-butadiene in B6C3F1 mice after 60 weeks of inhalation exposure. Science. 1985;227:548–549. doi: 10.1126/science.3966163. [DOI] [PubMed] [Google Scholar]
- (2).Owen PE, Glaister JR, Gaunt IF, Pullinger DH. Inhalation toxicity studies with 1,3-butadiene: 3. Two year toxicity/carcinogenicity study in rats. Am. Ind. Hyg. Assoc. J. 1987;48:407–413. doi: 10.1080/15298668791384959. [DOI] [PubMed] [Google Scholar]
- (3).Melnick RL, Huff J. 1,3-Butadiene: toxicity and carcinogenicity in laboratory animals and in humans. Rev. Environm. Contamination Toxicol. 1992;124:111–144. doi: 10.1007/978-1-4612-2864-6_5. [DOI] [PubMed] [Google Scholar]
- (4).Santos-Burgoa C, Matanoski GM, Zeger S, Schwartz L. Lymphohematopoietic cancer in styrene-butadiene polymerization workers. Am. J. Epidemiol. 1992;136:843–854. doi: 10.1093/aje/136.7.843. [DOI] [PubMed] [Google Scholar]
- (5).Divine BJ, Wendt JK, Hartman CM. Cancer mortality among workers at a butadiene production facility. In: Sorsa M, Peltonen K, Vainio H, Hemminki K, editors. Butadiene and Styrene: Assessment of Health Hazards. IARC; Lyon: 1993. pp. 345–362. IARC Scientific Publications No. 127. [PubMed] [Google Scholar]
- (6).Delzell E, Sathiakumar N, Hovinga M, Macaluso M, Julian J, Larson R, Cole P, Muir DCF. A follow-up study of synthetic rubber workers. Toxicology. 1996;113:182–189. doi: 10.1016/0300-483x(96)03443-9. [DOI] [PubMed] [Google Scholar]
- (7).Macaluso M, Larson R, Delzell E, Sathiakumar N, Hovinga M, Julian J, Muir D, Cole P. Leukemia and cumulative exposure to butadiene, styrene and benzene among workers in the synthetic rubber industry. Toxicology. 1996;113:190–202. doi: 10.1016/0300-483x(96)03444-0. [DOI] [PubMed] [Google Scholar]
- (8).Department of Health and Human Services. U. S. Public Health Services. National Toxicology Program . The Ninth Report on Carcinogens. Research Triangle Park; NC: 2000. [Google Scholar]
- (9).Elfarra A, Moll T, Krause R, Kemper R, Selzer R. Reactive metabolites of 1,3-butadiene: DNA and hemoglobin adduct formation and potential roles in carcinogenicity. In: Dansette PM, Snyder R, Delaforge M, Gibson GG, Greim H, Jollow DJ, Monks TJ, Sipes IG, editors. Biological Reactive Intermediates VI. Kluwer Academic/Plenum Publishers; New York: 2001. pp. 93–103. [DOI] [PubMed] [Google Scholar]
- (10).Csanády GA, Guengerich FP, Bond JA. Comparison of the biotransformation of 1,3-butadiene and its metabolite, butadiene monoepoxide, by hepatic and pulmonary tissues from humans, rats and mice. Carcinogenesis. 1992;13:1143–1153. doi: 10.1093/carcin/13.7.1143. [DOI] [PubMed] [Google Scholar]
- (11).Duescher RJ, Elfarra AA. 1,3-Butadiene oxidation by human myeloperoxidase. Role of chloride ion in catalysis of divergent pathways. J. Biol. Chem. 1992;267:19859–19865. [PubMed] [Google Scholar]
- (12).Duescher RJ, Elfarra AA. Human liver-microsomes are efficient catalysts of 1,3-butadiene oxidation. Evidence for major roles by cytochromes P450 2A6 and 2E1. Arch. Biochem. Biophys. 1994;311:342–349. doi: 10.1006/abbi.1994.1246. [DOI] [PubMed] [Google Scholar]
- (13).Krause RJ, Elfarra AA. Oxidation of butadiene monoxide to meso-and (±)-diepoxybutane by cDNA-expressed human cytochrome P450s and by mouse, rat, and human liver microsomes: evidence for preferential hydration of meso-diepoxybutane in rat and human microsomes. Arch. Biochem. Biophys. 1997;337:176–184. doi: 10.1006/abbi.1996.9781. [DOI] [PubMed] [Google Scholar]
- (14).Gervasi PG, Citti L, Del Monte M, Longo V, Benetti D. Mutagenicity and chemical reactivity of epoxidic intermediates of the isoprene metabolism and other structurally related compounds. Mutat. Res. 1985;156:77–82. doi: 10.1016/0165-1218(85)90009-6. [DOI] [PubMed] [Google Scholar]
- (15).Tice RR, Boucher R, Luke CA, Shelby MD. Comparative cytogenetic analysis of bone marrow damage induced in male B6C3F1 mice by multiple exposures to gaseous 1,3-butadiene. Environ. Mutagen. 1987;9:235–250. doi: 10.1002/em.2860090303. [DOI] [PubMed] [Google Scholar]
- (16).Cochrane JE, Skopek TR. Mutagenicity of butadiene and its epoxide metabolites: I. Mutagenic potential of 1,2-epoxybutene, 1,2,3,4-diepoxybutane and 3,4-epoxy-1,2-butanediol in cultured human lymphoblasts. Carcinogenesis. 1994;15:713–717. doi: 10.1093/carcin/15.4.713. [DOI] [PubMed] [Google Scholar]
- (17).Cochrane JE, Skopek TR. Mutagenicity of butadiene and its epoxide metabolites: II. Mutational spectra of butadiene, 1,2-epoxybutene and diepoxybutane at the hprt locus in splenic T cells from exposed B6C3F1 mice. Carcinogenesis. 1994;15:719–723. doi: 10.1093/carcin/15.4.719. [DOI] [PubMed] [Google Scholar]
- (18).Zhang X-Y, Elfarra AA. Identification and characterization of a series of nucleoside adducts formed by the reaction of 2′-deoxyguanosine and 1,2,3,4-diepoxybutane under physiological conditions. Chem. Res. Toxicol. 2003;16:1606–1615. doi: 10.1021/tx0341355. [DOI] [PubMed] [Google Scholar]
- (19).Tretyakova NY, Sangaiah R, Yen T-Y, Swenberg JA. Synthesis, characterization, and in vitro quantitation of N-7-guanine adducts of diepoxybutane. Chem. Res. Toxicol. 1997;10:779–785. doi: 10.1021/tx970004q. [DOI] [PubMed] [Google Scholar]
- (20).Zhang X-Y, Elfarra AA. Characterization of the reaction products of 2′-deoxyguanosine and 1,2,3,4-diepoxybutane after acid hydrolysis: formation of novel guanine and pyrimidine adducts. Chem. Res. Toxicol. 2004;17:521–528. doi: 10.1021/tx034243r. [DOI] [PubMed] [Google Scholar]
- (21).Oe T, Kambouris SJ, Walker VE, Meng QX, Recio L, Wherli S, Chaudhary AK, Blair IA. Persistence of N7-(2,3,4-trihydroxybutyl)guanine adducts in the livers of mice and rats exposed to 1,3-butadiene. Chem. Res. Toxicol. 1999;12:247–257. doi: 10.1021/tx980193s. [DOI] [PubMed] [Google Scholar]
- (22).Koivisto P, Kilpeläinen I, Rasanen I, Adler I-D, Pacchierotti F, Peltonen K. Butadiene diolepoxide-and diepoxybutane-derived DNA adducts at N7-guanine: a high occurrence of diolepoxide-derived adducts in mouse lung after 1,3-butadiene exposure. Carcinogenesis. 1999;20:1253–1259. doi: 10.1093/carcin/20.7.1253. [DOI] [PubMed] [Google Scholar]
- (23).Tretyakova NY, Chiang S-Y, walker VE, Swenberg JA. Quantitative analysis of 1,3-butadiene-induced DNA adducts in vivo and in vitro using liquid chromatography electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 1998;33:363–376. doi: 10.1002/(SICI)1096-9888(199804)33:4<363::AID-JMS643>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- (24).Koc H, Tretyakova NY, Walker VE, Henderson RF, Swenberg JA. Molecular dosimetry of N-7 guanine adduct formation in mice and rats exposed to 1,3-butadiene. Chem. Res. Toxicol. 1999;12:566–574. doi: 10.1021/tx980265f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.