

Abstract
Objectives:
Prader-Willi syndrome (PWS) is a genetic disorder (linked to chromosome 15q11-13) characterized by hypotonia and developmental delay, hyperphagia and obesity, hypersomnia and abnormal sleep, and behavioral problems. Such patients may also be at increased risk of obstructive sleep apnea (OSA), although whether this risk is explained by known risk factors has not previously been directly tested. Our aim was to compare sleep and breathing in an older group of patients with Prader-Willi syndrome with a control group—matched on the basis of age, sex, and body mass index (BMI)—in order to determine which specific features are not explained by these known confounders.
Methods:
Consecutive patients with PWS attending the PWS clinic at Royal Prince Alfred Hospital Sydney, Australia, were recruited. Age-, sex-, and BMI-matched controls were selected from the Sleep Investigation Unit at Royal Prince Alfred Hospital, and polysomnography-derived sleep and other parameters were compared across the groups.
Results:
Nineteen subjects with PWS (14 males) were included in the study. Eighteen (95 %) had a total respiratory disturbance index (TRDI) of greater than 5 events per hour, with 4 (21%) having severe obstructive sleep apnea (TRDI ≥ 30 events/hour) and 9 (47%) having evidence of obesity hypoventilation syndrome. Patients with PWS, as compared with the control group, had evidence of more nocturnal hypoxemia, with lower oxyhemoglobin saturations and percentages of sleep time at less than 80% oxyhemoglobin saturation (all p values < 0.05). There were no significant differences in sleep architecture; however, there was a reduction in rapid eye movement latency seen in the PWS group (p < 0.05). Serum leptin was higher than the reference range in the PWS group but was not measured in the control group.
Conclusion:
Patients with PWS drawn from an adult and adolescent PWS clinic have a high rate of sleep-disordered breathing. There is evidence that patients with PWS may have more nocturnal hypoventilation than a well-matched control group. These data suggest that the chromosome region 15q11-13 may be involved in some aspects of the regulation of breathing, although whether putative molecular mechanisms act directly or indirectly will require further investigation.
Citation:
Yee BJ; Buchanan PR; Mahadev S; Banerjee D; Liu PY; Phillips C; Loughnan G; Steinbeck K; Grunstein RR. Assessment of sleep and breathing in adults with Prader-Willi syndrome: a case control series. J Clin Sleep Med 2007;3(7):713–718.
Keywords: Prader-Willi syndrome, obstructive sleeps apnea, obesity hypoventilation syndrome, nocturnal hypoxemia, leptin
Prader-Willi syndrome (PWS) is a genetic disorder with anu estimated prevalence of between approximately 1 in 10,000 and 1 in 25,000 live births.1–4 PWS is characterized by hypotonia and developmental delay, short stature, small extremities and characteristic facies, hyperphagia leading to obesity, and other behavioral problems.5,6 Symptoms of obstructive sleep apnea (OSA) may be difficult to elicit in patients with PWS, although caregivers often describe snoring, nocturnal breathing difficulties, and restless sleep in patients with PWS.7 Excessive daytime sleepiness, arising from a variety of causes, is a cardinal feature of both OSA syndrome and PWS.8,9 Patients with PWS may be at increased risk of OSA10–12 due to obesity, facial dysmorphism,11 and sticky secretions.12 However, the associated hypothalamic disorders, including hypogonadism, hyperphagia, and hypotonia,5 suggest that central neurologic developmental abnormalities may also directly affect sleep and breathing through unknown neurologic mechanisms. Although there have been advances in understanding of this disorder, particularly of the genetic abnormalities, including the linkage to chromosome 15q11-13,14–19 a specific Prader-Willi gene has not yet been identified. There have been several genes and transcripts mapped within the PWS critical region and 2 identified protein products, and several of these are subject to genomic imprinting. These “controlling genes” are known to effect the expression of PWS. Clinical features are specific and some improve (e.g., hypotonia) with age (after about the first 12 months of age).20 Some features may not be as severe to the same extent in all affected people. Understanding the genetic bases and putative central mechanisms that lead to the PWS phenotype may lead to molecular targets for OSA, hyperphagia, and obesity in PWS.
Clinically, establishing the prevalence of OSA in adults and adolescents with PWS is important because OSA has now been shown to be independently associated with systemic hypertension.21–25 There is growing evidence that cardiovascular morbidity and mortality is increased in patients with untreated OSA26,27 and evidence that such unfavorable consequences may be ameliorated with effective therapy.28–31 There is also evidence of sudden and unexplained death in people with Prader-Willi, which may be secondary to respiratory pathology.32–34
Early identification and treatment of OSA in PWS thus may delay or prevent these known complications. However, the prevalence of OSA in PWS is difficult to define based on published series. Reports of prevalence of OSA vary from 0% to 100%, and this wide variability clearly reflects the inclusion criteria of the PWS study population.35 Current case series have included children, adults, or both, and there have been relatively small numbers in published series, with wide age ranges and inclusion criteria. There have also been differences in types of sleep studies used, from nocturnal oximetry to full overnight polysomnography, and sometimes with poor definition of respiratory events.35
So far there have been no case-control studies comparing a group comprising adults and adolescents with PWS and an age-, sex- and body mass index (BMI)-matched control group. The aim of this study was to compare sleep and breathing variables among subjects with PWS and controls in order to (1) establish if there is an increased risk of OSA in PWS and (2) determine if this increased risk is explained by known obesity-related OSA risk factors.
METHODS
Subjects
The PWS clinic at Royal Prince Alfred Hospital is a tertiary referral facility for adult or adolescent patients with PWS. Nineteen subjects with PWS (14 males, 5 females) were recruited consecutively between 2001 and 2003 regardless of symptoms of OSA. None of the subjects with PWS were being treated with growth hormone, ketogenic diet, or stimulant medications. Thirty five consecutive contemporaneous age-, sex- and BMI-matched control subjects were recruited from the Royal Prince Alfred Hospital Sleep Investigation Unit. Both the PWS and Sleep clinics are colocated tertiary referral centres, which subserve the same geographic population. Controls were taken from consecutive patients (same sex) with an age between 18 and 29 years and a BMI range between 35 and 45 kg/m2 within 6 months of their polysomnogram. We aimed to recruit 2 controls for each patient, but, due to matching difficulties, in some cases only 1 control for each patient was found. All controls snored or had other clinical features of OSA syndrome that required further investigations. None of the control subjects had previously been diagnosed with OSA or had previous therapy for snoring or OSA. Subjects were excluded if they had any known lung disease or uncontrolled metabolic or cardiovascular disease. Subjects were also excluded if they were on medications that would affect sleep and breathing (e.g., opiates, benzodiazepines) or had significant alcohol intake. For the PWS group, subject consent was acquired through the primary caregiver or guardian. The study was approved by the Ethics Committee (Royal Prince Alfred Hospital zone) of the Sydney South West Area Health Service.
Anthropometrics
All anthropometric measurements were performed in duplicate using standardized techniques.36 Height was measured (± 0.1 cm) at midinspiratory level using a stadiometer. Subjects were weighed (± 0.1 kg) on a digital scale after voiding and without shoes. BMI was calculated as weight (kg) divided by height squared (m2).
Polysomnography
A standard polysomnogram (Compumedics, Melbourne, Australia) was performed in all subjects at baseline and 24 weeks. The electroencephalogram leads were placed according to the 10–20 system with 4 electrodes attached to the scalp (C3, O2, A1 and A2) and with 2 electrooculogram electrodes and 2 submental electromyogram leads. There were also 2 electrocardiograph leads, 1 near the right shoulder and a second in the sixth intercostal space on the left side of the chest. Sensor leads were placed on each leg over the anterior tibialis for recording leg movements. A nasal pressure transducer monitored breathing. Inductive respiratory effort bands were placed around the chest and abdomen to monitor thoracic and abdominal wall movement. A position sensor was attached to monitor body position during sleep. A finger-probe oximeter recorded oxyhemoglobin saturation continuously.
Sleep stages were scored manually according to standard criteria by Board of Registered Polysomnographic Technologists certified individuals using Compumedics software.37 All scorers were blinded to group membership and undergo regular quality assurance to achieve interobserver concordance of greater than 90% for sleep staging and 85% to 90% for respiratory scoring. Arousals were scored according to the Atlas Task Force of the American Sleep Disorders Association.38 For scoring of respiratory events, thoracic and abdominal bands and nasal pressure were used as measures of breathing. An apnea was defined as cessation of airflow for at least 10 seconds (with or without a 3% oxygen desaturation or electroencephalographic arousal), and a hypopnea was defined as a clear reduction (> 20%, compared with the baseline over the preceding 2 minutes) in 1 of the measures of breathing during sleep for at least 10 seconds in association with at least a 3% oxygen desaturation, an electroencephalographic arousal, or both.38 The total respiratory disturbance index (TRDI) was defined by the number of apneas plus hypopneas per hour of sleep. The REM RDI was the number of apneas and hypopneas in rapid eye movement (REM) sleep. The NREM RDI was the number of apneas and hypopneas in non-REM (NREM) sleep. Time less than 90% and 80% was defined as total sleep times at less than 90% and 80% oxyhemoglobin saturation, respectively.
Venipuncture and Arterial Blood Gases
Blood samples obtained during fasting and arterial blood gases were collected the morning after polysomnography but only in subjects with PWS in order to further characterize this group. Blood was drawn from the antecubital vein and analyzed by the Departments of Biochemistry and Endocrinology at Royal Prince Alfred Hospital. Serum leptin was determined by microparticle enzyme immunoassay (MEIA) using an automated system (Abbott Axsymm System, Germany). Serum insulin was quantified using a specific double antibody radioimmunoassay kit (Linco Research Inc. St Charles, MO). Arterial blood was obtained from the wrist, and blood gases (PaO2, PaCO2) and oxyhemoglobin saturation were measured using an automated arterial blood gas analyzer (Radiometer ABL 520, Copenhagen, Denmark).
Genetic Testing
Genetic testing was performed using fluorescence in situ hybridization or methylation.39,40
Statistical Analyses
Statistical analyses were performed using SPSS version 10.0 (SPSS, Inc., Chicago, IL). To determine whether the distributions of all continuous data were normal, a Kolmogorov-Smirnov test was performed. When such an assumption could not be met, the data were summarized in terms of medians and interquartile range rather than means and standard deviation (SD). To compare data of a parameter between the 2 groups (PWS vs control), parametric (nonpaired 2 independent group t test) or nonparametric testing (Mann-Whitney U test) was carried out as appropriate. Two-tailed tests were used, and the level of significance was taken as p < 0.05.
RESULTS
Demographics
Nineteen subjects with PWS (14 males, 5 females) were recruited from the PWS clinic. Thirty-five patients (24 men) without PWS were recruited from the adult sleep-disordered breathing clinic. Subjects with PWS and control subjects were well matched (see Table 1).
Table 1.
Demographic and Physiologic Variables of Patients with Prader-Willi Syndrome and control subjects
| Patients (n = 19) | Controls (n = 35) | p Value | |
|---|---|---|---|
| Age, y | |||
| Mean | 25.6 (10.1) | 25.8 (3.3) | NS |
| Range | 14–45 | 18 – 29 | |
| BMI, kg/m2 | 42.3 (10.1) | 41.1 (8.1) | NS |
| Men, % | 73 | 69 | NS |
| Pco2, mmHg | 46.6 (3.8) | Not tested | |
| Po2, mmHg | 80 (13.0) | Not tested | |
| hco3, mmol/L | 27.7 (1.3) | Not tested |
Data are expressed as mean (SD). BMI refers to body mass index.
Polysomnography
All but 1 of the patients in the PWS group had evidence of OSA (definition: total RDI > 5 events/h). Although the mean total RDI was the same between the PWS and control groups, there is evidence that there is more nocturnal hypoxemia in the subjects with PWS. REM RDI was higher, and lowest oxyhemoglobin saturation and oxyhemoglobin saturation time less than 80% were significantly lower in the PWS group (all p < 0.05) than in the control subjects. PWS and control subjects were also categorized depending on whether total RDI was greater than or equal to 5 per hour or 10 per hour. However, proportions did not differ significantly (2/19 vs 10/35, p = 0.17 Fisher exact test and 9/19 vs 15/35, p = 0.78 Fisher exact test). There was also a trend for subjects with PWS to have longer hypopneas and apneas; however, this did not reach statistical significance (Table 2). There was no evidence of central sleep apnea in any of the subjects.
Table 2.
Nocturnal Sleep Respiratory Variables of Patients with Prader-Willi Syndrome and Control Subjects
| Variable | Patients (n = 18) | Controls (n = 35) | p Value |
|---|---|---|---|
| RDI, events/h | |||
| NREM | 4.6 (2.6–24.6) | 8.0 (3.0–33.0) | NS |
| REM | 39.2 (23.5) | 22.6 (25.1) | 0.02 |
| Total | 11.3 (5.8–29.5) | 12.0 (4.0.–33.0) | NS |
| Lowest Sao2 %* | 77.0 (67.0-86.5) | 88.0 (81.0-90.0) | 0.005 |
| Percentage of time spent at an Sao2 less than | |||
| 90% | 1.6 (0–46.0) | 0 (0–4.0) | NS |
| 80%* | 0.17 (0-.70) | 0 (0–0) | 0.013 |
| Arousal Index | 23.6 (2.4) | 28.5 (35.9) | NS |
| Longest apnea, sec | 31.8 (24.7) | 21.6 (23.4) | NS |
| Longest hypopnea, sec | 69.9 (45.5) | 52.5 (25.2) | NS |
| Mean nocturnal Sao2, % | 94.6 (2.4) | 95.5 (2.7) | NS |
Data are presented as mean (SD) except those denoted with an asterisk, which are expressed as median (25th–75th percentile). NREM refers to non-rapid eye movement; REM, rapid eye movement; Sao2, oxygen saturation.
Using the current definition of obesity hypoventilation syndrome,41 (defined as a BMI > 30 kg/m2 and daytime Paco2 > 45 mm Hg not explained by other known causes of hypoventilation), 9 out of 18 subjects with PWS fulfilled the definition of obesity hypoventilation syndrome.
All subjects had adequate total sleep time and sleep efficiency. Although there were no significant differences in sleep architecture between the control and PWS groups, REM latency (time lapsed to enter REM sleep) was significantly reduced in the PWS group (p < 0.018) (Table 3).
Table 3.
Sleep Variables of Patients with Prader-Willi Syndrome and Control Subjects
| Variable | Patients (n = 19) | Controls (n = 35) | p Value |
|---|---|---|---|
| Sleep stage, % | |||
| 1 | 7.2 (3.9) | 9.8 (8.1) | NS |
| 2 | 53.4 (13.1) | 56.6 (11.4) | NS |
| 3 | 10.3 (7.2) | 10.7 (6.7) | NS |
| 4 | 11.0 (11.5) | 10.6 (11.0) | NS |
| REM | 16.2 (6.7) | 13.8 (12.8) | NS |
| TST, min | 303.9 (103.7) | 325.8 (94.4) | NS |
| Sleep latency, min | 26.2 (35.2) | 29.8 (42.1)) | NS |
| REM latency, min | 102.4 (65.1) | 161.1 (93.3) | 0.013 |
| Sleep efficiency, % | 74.9 (15.6) | 75.1 (17.1) | NS |
| PLMI, no./h | 0 | 0 | NS |
Data are expressed as mean (SD). REM refers to rapid eye movement sleep; TST, total sleep time; PLMI, periodic limb movement index.
The PWS group had normal levels of fasting glucose, 5.3 mmol/L ± 1.7 mmol/L (mean ± SD; normal range 3.6–6.6 mmol/L) but elevated insulin, 113.4 pmol/L ± 96.9 pmol/L (normal range 15–60 pmol/L), and leptin, 51.7 ng/mL ± 22.2 ng/mL (normal range 2.0-5.6 ng/mL)n compared with a published nonobese reference group.42 There was a significant correlation between OSA (as measured by total RDI) and serum leptin (p < 0.05) (Figure 1). PWS was confirmed genetically in all subjects: 68% of the subjects studied had the deletion genotype, a prevalence similar to that of other reports. We did not notice any differences among the subjects with PWS in terms of demographics, sleep, and breathing (all p > 0.05).
Figure 1.
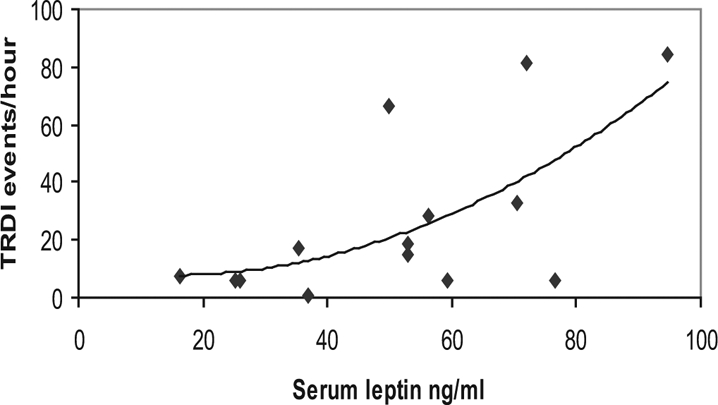
Correlation between total respiratory disturbance index (TRDI) and leptin levels in subjects with Prader-Willi syndrome (n = 14). r=0.62, p =0.018
DISCUSSION
This study shows for the first time that adolescents and adults with PWS have more severe oxygen desaturation during sleep and shorter REM latency, as compared with a well-matched obese control population. However, respiratory indices were largely comparable, meaning that most features were explained by established OSA risk factors. However, these data still do suggest that at least some of the sleep and sleep-breathing features of PWS (e.g., REM latency) are not explained by obesity or obesity-related factors, thereby implicating hitherto unknown factors in the chromosomal region 15q11-13. This is of physiologic interest in identifying potential molecular mechanisms involved in the control of sleep and may be of relevance to other conditions such as narcolepsy or idiopathic hypersomnia. It should also be noted that subjects with PWS were not selected because of OSA symptoms and were recruited from an adolescent and adult PWS clinic, whereas control subjects were recruited from the sleep investigation unit (where the prevalence of OSA would be expected to be higher than in an unselected community population). Although previous studies of PWS have demonstrated variable prevalence rates of OSA, none of these studies had compared an adolescent and adult PWS group with a carefully matched control group.35 Of importance, both groups were drawn from highly specialized tertiary referral centers subserving the same geographic population, thereby providing some assurance that cases and controls were matched for variables that we did not measure.
Any increase in nocturnal hypoventilation in PWS may be attributable to various mechanisms. It is known that PWS is associated with a restrictive impairment in pulmonary function,43,44 as may be the case for other non-PWS subjects with extreme obesity. Both basal and sleeping metabolic rates are lower in subjects with PWS than in controls,45,46 likely as a consequence of reduced nonfat body mass, i.e., muscle, and low muscle mass may contribute to respiratory muscle weakness and a restrictive lung defect. Awake ventilatory responses to both hypoxia and hypercapnia are decreased in subjects with PWS independent of the degree of obesity.47,48 There is also evidence that the ventilatory response to hypercapnia during sleep is impaired.49,50 Thus another potential mechanism for increased sleep-disordered breathing in PWS is the abnormal arousal responses or thresholds to hypoxia and hypercapnia, although we did not measure this. Abnormal arousal thresholds may lead to a prolongation of obstructive apneas and hypopneas, which will lead to more significant nocturnal hypoventilation.
We found high rates of hypercapnic OSA in our subjects with PWS and, in the likely absence of chronic obstructive lung disease (although lung function testing was not part of the investigation protocol, the relatively young age range, absence of reported respiratory symptoms and minimal smoking history in the PWS group argues against the presence of chronic obstructive lung disease), this indicates that a large percentage of these subjects would have fulfilled the criteria for obesity hypoventilation syndrome. This finding has potential for significant long-term consequences.41,51 Although we did not measure arterial blood gases in our control group, we speculate that obesity hypoventilation syndrome would be more common in subjects with PWS than in control subjects due to the increased hypoxemia seen during the night. Limited published data on obesity hypoventilation syndrome in OSA indicates a low prevalence: 9.8% (58 of 590 patients) with a BMI from 30 to 40, and 23.6% (41 of 174 patients) with a BMI greater than 40.52
Another finding from our study was the reduced REM latency observed in men and women with PWS, compared with the control group. Although the actual mean REM latency was within normal limits for both groups, this still supports earlier findings showing reduced REM latency independent of sleep-disordered breathing in people with PWS.53,54 Reduced REM latency is consistent with a primary disorder of hypothalamic control of sleep state, perhaps as part of the overall PWS neuropathology in the hypothalamus where other syndromic features (e.g., hypogonadism, hyperphagia, temperature dysregulation) are thought to be anatomically centered. This, together with the greater sleep hypoxemia indicating altered sleep breathing drive and reduced arousal, suggests potential common abnormalities in brain centers regulating both sleep and breathing. Although a specific Prader-Willi gene has not yet been identified, candidate genes such as Necdin and Magel2 are expressed in neuronal tissue in the region in animal models, thereby making this implication plausible.55–57
This study adjusted for important obesity-related risk factors and provides novel data implicating a direct role of genes in the control of sleep, and breathing during sleep, in patients with PWS. Further preliminary data, similar to ours, implicating the importance of this region in identifying novel mechanisms that control sleep and breathing, are needed.
Insulin and leptin were elevated in these subjects with PWS, as expected for an obese population such as this. Previous studies have shown subjects with PWS to have elevated leptin levels, but these are generally similar to age-, weight-, and sex-matched controls.58 There was a positive correlation between serum leptin and more severe disordered breathing (total RDI), which has not been previously shown in older patients with PWS. The biologic relevance of this association is unclear. Nevertheless, in other settings, leptin is associated with ventilatory control.59 A significant correlation between leptin and Pco2 was detected (r2 = 0.38, p = 0.032). However, after accounting for the effect of total RDI, this association was no longer significant (r2 = 0.15, p = 0.17).
Although we did not measure ghrelin levels in these patients with PWS, others have shown abnormalities of ghrelin responses to food intake and gastric ghrelin density in PWS60,61 consistent with central neuronal dysregulation of this orexigenic hormonal regulator, and we could speculate such central dysregulation may also involve leptin.
A limitation of this study is that incidence rates of OSA in patients with PWS were not compared with an age-, sex-, and BMI-matched community sample. However the rates are still much higher than projected community rates,62 suggesting that the increased prevalence is PWS related. The addition of an age-, sex-, and BMI-matched control group drawn from an OSA clinic population merely served as a comparator for assessing whether OSA severity was worse in PWS. The results indicated that REM-related OSA and hypoxemia were worse in the PWS group. A second limitation is that, without transcutaneous Pco2, it is difficult to determine whether the above-mentioned differences in REM OSA and hypoxemia explain the elevated daytime Pco2 seen in the PWS group. Apart from blood gases, future studies would need to include nocturnal Tcco2 measurements together with lung function tests to more clearly resolve this question. Nevertheless, we believe that these findings provide novel and direct data implicating non-obesity–related factors in the pathogenesis of some aspects of OSA in men and women with PWS.
In summary, we have reported sleep-breathing data in an older group of subjects with PWS showing a high incidence of OSA and accompanied by daytime hypercapnia, suggesting the possible presence of sleep hypoventilation. Subjects with PWS also demonstrated early REM-onset and increased REM-related OSA with worse hypoxemia, when compared with a matched obese control group with OSA. We postulate genetic central hypothalamic dysfunction as a likely basis of these findings.
Footnotes
Disclosure Statement
This was not an industry supported study. The authors have indicated no financial conflicts of interest.
REFERENCES
- 1.Smith A, Egan J, Ridley G, et al. Birth prevalence of Prader-Willi syndrome in Australia. Arch Dis Child. 2003;88:263–4. doi: 10.1136/adc.88.3.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burd L, Vesely B, Martsolf J, Kerbeshian J. Prevalence study of Prader-Willi syndrome in North Dakota. Am J Med Genet. 1990;37:97–9. doi: 10.1002/ajmg.1320370122. [DOI] [PubMed] [Google Scholar]
- 3.Vogels A, Van Den Ende J, Keymolen K, et al. Minimum prevalence, birth incidence and cause of death for Prader-Willi syndrome in Flanders. Eur J Hum Genet. 2004;12:238–40. doi: 10.1038/sj.ejhg.5201135. [DOI] [PubMed] [Google Scholar]
- 4.Akefeldt A, Gillberg C, Larsson C. Prader-Willi syndrome in a Swedish rural county: epidemiological aspects. Dev Med Child Neurol. 1991;33:715–21. doi: 10.1111/j.1469-8749.1991.tb14950.x. [DOI] [PubMed] [Google Scholar]
- 5.Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91:398–402. [PMC free article] [PubMed] [Google Scholar]
- 6.Couper R. Prader-Willi syndrome. J Paediatr Child Health. 1999;35:331–4. doi: 10.1046/j.1440-1754.1999.00397.x. [DOI] [PubMed] [Google Scholar]
- 7.Richdale AL, Cotton S, Hibbit K. Sleep and behaviour disturbance in Prader-Willi syndrome: a questionnaire study. J Intellect Disabil Res. 1999;43:380–92. doi: 10.1046/j.1365-2788.1999.043005380.x. [DOI] [PubMed] [Google Scholar]
- 8.Hiroe Y, Inoue Y, Higami S, Suto Y, Kawahara R. Relationship between hypersomnia and respiratory disorder during sleep in Prader-Willi syndrome. Psychiatry Clin Neurosci. 2000;54:323–5. doi: 10.1046/j.1440-1819.2000.00697.x. [DOI] [PubMed] [Google Scholar]
- 9.Manni R, Politini L, Nobili L, et al. Hypersomnia in the Prader Willi syndrome: clinical-electrophysiological features and underlying factors. Clin Neurophysiol. 2001;112:800–5. doi: 10.1016/s1388-2457(01)00483-7. [DOI] [PubMed] [Google Scholar]
- 10.Clift S, Dahlitz M, Parkes JD. Sleep apnoea in the Prader-Willi syndrome. J Sleep Res. 1994;3:121–6. doi: 10.1111/j.1365-2869.1994.tb00115.x. [DOI] [PubMed] [Google Scholar]
- 11.Richards A, Quaghebeur G, Clift S, Holland A, Dahlitz M, Parkes D. The upper airway and sleep apnoea in the Prader-Willi syndrome. Clin Otolaryngol Allied Sci. 1994;19:193–7. doi: 10.1111/j.1365-2273.1994.tb01213.x. [DOI] [PubMed] [Google Scholar]
- 12.Hertz G, Cataletto M, Feinsilver SH, Angulo M. Developmental trends of sleep-disordered breathing in Prader-Willi syndrome: the role of obesity. Am J Med Genet. 1995;56:188–90. doi: 10.1002/ajmg.1320560215. [DOI] [PubMed] [Google Scholar]
- 13.Hart PS. Salivary abnormalities in Prader-Willi syndrome. Ann N Y Acad Sci. 1998;842:125–31. doi: 10.1111/j.1749-6632.1998.tb09640.x. [DOI] [PubMed] [Google Scholar]
- 14.Wharton RH, Loechner KJ. Genetic and clinical advances in Prader-Willi syndrome. Curr Opin Pediatr. 1996;8:618–24. doi: 10.1097/00008480-199612000-00013. [DOI] [PubMed] [Google Scholar]
- 15.State MW, Dykens EM. Genetics of childhood disorders: XV. Prader-Willi syndrome: genes, brain, and behavior. J Am Acad Child Adolesc Psychiatry. 2000;39:797–800. doi: 10.1097/00004583-200006000-00021. [DOI] [PubMed] [Google Scholar]
- 16.Muscatelli F, Abrous DN, Massacrier A, et al. Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum Mol Genet. 2000;9:3101–10. doi: 10.1093/hmg/9.20.3101. [DOI] [PubMed] [Google Scholar]
- 17.Glenn CC, Nicholls RD, Robinson WP, et al. Modification of 15q11-q13 DNA methylation imprints in unique Angelman and Prader-Willi patients. Hum Mol Genet. 1993;2:1377–82. doi: 10.1093/hmg/2.9.1377. [DOI] [PubMed] [Google Scholar]
- 18.Reed ML, Leff SE. Maternal imprinting of human SNRPN, a gene deleted in Prader-Willi syndrome. Nat Genet. 1994;6:163–7. doi: 10.1038/ng0294-163. [DOI] [PubMed] [Google Scholar]
- 19.MacDonald HR, Wevrick R. The necdin gene is deleted in Prader-Willi syndrome and is imprinted in human and mouse. Hum Mol Genet. 1997 Oct;6(11):1873–8. doi: 10.1093/hmg/6.11.1873. [DOI] [PubMed] [Google Scholar]
- 20.Burman P, Ritzen EM, Lindgren AC. Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH. Endocrine reviews. 2001;22:787–99. doi: 10.1210/edrv.22.6.0447. [DOI] [PubMed] [Google Scholar]
- 21.Lavie P, Herer P, Hoffstein V. Obstructive sleep apnoea syndrome as a risk factor for hypertension: population study. BMJ. 2000;19:479–82. doi: 10.1136/bmj.320.7233.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nieto F, Young T, Lind B, et al. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA. 2000;283:1829–36. doi: 10.1001/jama.283.14.1829. [DOI] [PubMed] [Google Scholar]
- 23.Young T, Peppard P, Palta M, et al. Population-based study of sleep-disordered breathing as a risk factor for hypertension. Arch Intern Med. 1997;157:1746–52. [PubMed] [Google Scholar]
- 24.Grote L, Ploch T, Heitmann J, Knaack L, Penzel T, Peter J. Sleep-related breathing disorder is an independent risk factor for systemic hypertension. Am J Respir Crit Care Med. 1999;160:1875–82. doi: 10.1164/ajrccm.160.6.9811054. [DOI] [PubMed] [Google Scholar]
- 25.Bixler E, Vgontzas A, Lin H, et al. Association of hypertension and sleep-disordered breathing. Arch Intern Med. 2000;160:2289–95. doi: 10.1001/archinte.160.15.2289. [DOI] [PubMed] [Google Scholar]
- 26.Young T, Peppard PE, Gottlieb DJ. Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med. 2002;2002(165):1217–39. doi: 10.1164/rccm.2109080. [DOI] [PubMed] [Google Scholar]
- 27.Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;2005(353):2034–41. doi: 10.1056/NEJMoa043104. [DOI] [PubMed] [Google Scholar]
- 28.Dhillon S, Chung SA, Fargher T, Huterer N, Shapiro CM. Sleep apnea, hypertension, and the effects of continuous positive airway pressure. Am J Hypertens. 2005;18:594–600. doi: 10.1016/j.amjhyper.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 29.Doherty LS, Kiely JL, Swan V, McNicholas WT. Long-term effects of nasal continuous positive airway pressure therapy on cardiovascular outcomes in sleep apnea syndrome. Chest. 2005;127:2076–84. doi: 10.1378/chest.127.6.2076. [DOI] [PubMed] [Google Scholar]
- 30.Marin JM, Carrizo SJ, Vicente E, Agusti AG. Long-term cardiovascular outcomes in men with obstructive sleep apnoea-hypopnoea with or without treatment with continuous positive airway pressure: an observational study. Lancet. 2005;365:1046–53. doi: 10.1016/S0140-6736(05)71141-7. [DOI] [PubMed] [Google Scholar]
- 31.Becker HF, Jerrentrup A, Ploch T, et al. Effect of nasal continuous positive airway pressure treatment on blood pressure in patients with obstructive sleep apnea. Circulation. 2003;107:68–73. doi: 10.1161/01.cir.0000042706.47107.7a. [DOI] [PubMed] [Google Scholar]
- 32.Smith A, Loughnan G, Steinbeck K. Death in adults with Prader-Willi syndrome may be correlated with maternal uniparental disomy. J Med Genet. 2003;40:e63. doi: 10.1136/jmg.40.5.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schrander-Stumpel CT, Curfs LM, Sastrowijoto P, Cassidy SB, Schrander JJ, Fryns JP. Prader-Willi syndrome: causes of death in an international series of 27 cases. Am J Med Genet A. 2004 Jan;124:333–8. doi: 10.1002/ajmg.a.20371. [DOI] [PubMed] [Google Scholar]
- 34.Stevenson DA, Anaya TM, Clayton-Smith J, et al. Unexpected death and critical illness in Prader-Willi syndrome: report of ten individuals. Am J Med Genet A. 2004;124:158–64. doi: 10.1002/ajmg.a.20370. [DOI] [PubMed] [Google Scholar]
- 35.Nixon GM, Brouillette RT. Sleep and breathing in Prader-Willi syndrome. Pediatr Pulmonol. 2002;34:209–17. doi: 10.1002/ppul.10152. [DOI] [PubMed] [Google Scholar]
- 36.Lohmann TG, Roche A, Martello R. Anthrometrics standardization reference manual. Human Kinetics; 1988. [Google Scholar]
- 37.Rechtschaffen A, Kales A, editors. A Manual of Standardized Terminology, Techniques, and Scoring System for Sleep Stages of Human Subjects. Los Angeles: Brain Information Service/Brain Research Institute, UCLA; 1968. [Google Scholar]
- 38.Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. The Report of an American Academy of Sleep Medicine Task Force. Sleep. 1999;22:667–89. [PubMed] [Google Scholar]
- 39.Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. 1981;304:325–9. doi: 10.1056/NEJM198102053040604. [DOI] [PubMed] [Google Scholar]
- 40.Butler MG. Prader-Willi syndrome: current understanding of cause and diagnosis. Am J Med Genet. 1990;35:319–32. doi: 10.1002/ajmg.1320350306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olson AL, Zwillich C. The obesity hypoventilation syndrome. Am J Med. 2005;118:948–56. doi: 10.1016/j.amjmed.2005.03.042. [DOI] [PubMed] [Google Scholar]
- 42.Conus F, Allison DB, Rabasa-Lhoret R, et al. Metabolic and behavioral characteristics of metabolically obese but normal-weight women. J Clin Endocrinol Metab. 2004;89:5013–20. doi: 10.1210/jc.2004-0265. [DOI] [PubMed] [Google Scholar]
- 43.Mallory GB, Jr., Fiser DH, Jackson R. Sleep-associated breathing disorders in morbidly obese children and adolescents. J Pediatr. 1989;115:892–7. doi: 10.1016/s0022-3476(89)80738-3. [DOI] [PubMed] [Google Scholar]
- 44.Hakonarson H, Moskovitz J, Daigle KL, Cassidy SB, Cloutier MM. Pulmonary function abnormalities in Prader-Willi syndrome. J Pediatr. 1995;126:565–70. doi: 10.1016/s0022-3476(95)70350-0. [DOI] [PubMed] [Google Scholar]
- 45.van Mil EA, Westerterp KR, Gerver WJ, et al. Energy expenditure at rest and during sleep in children with Prader-Willi syndrome is explained by body composition. Am J Clin Nutr. 2000;71:752–6. doi: 10.1093/ajcn/71.3.752. [DOI] [PubMed] [Google Scholar]
- 46.van Mil EG, Westerterp KR, Kester AD, et al. Activity related energy expenditure in children and adolescents with Prader-Willi syndrome. Int J Obes Relat Metab Disord. 2000;24:429–34. doi: 10.1038/sj.ijo.0801175. [DOI] [PubMed] [Google Scholar]
- 47.Arens R, Gozal D, Omlin KJ, Livingston FR, Liu J, Keens TG, et al. Hypoxic and hypercapnic ventilatory responses in Prader-Willi syndrome. J Appl Physiol. 1994;77:2224–30. doi: 10.1152/jappl.1994.77.5.2224. [DOI] [PubMed] [Google Scholar]
- 48.Gozal D, Arens R, Omlin KJ, Ward SL, Keens TG. Absent peripheral chemosensitivity in Prader-Willi syndrome. J Appl Physiol. 1994;77:2231–6. doi: 10.1152/jappl.1994.77.5.2231. [DOI] [PubMed] [Google Scholar]
- 49.Arens R, Gozal D, Burrell BC, et al. Arousal and cardiorespiratory responses to hypoxia in Prader-Willi syndrome. Am J Respir Crit Care Med. 1996;153:283–7. doi: 10.1164/ajrccm.153.1.8542130. [DOI] [PubMed] [Google Scholar]
- 50.Livingston FR, Arens R, Bailey SL, Keens TG, Ward SL. Hypercapnic arousal responses in Prader-Willi syndrome. Chest. 1995;108:1627–31. doi: 10.1378/chest.108.6.1627. [DOI] [PubMed] [Google Scholar]
- 51.Nowbar S, Burkart KM, Gonzales R, et al. Obesity-associated hypoventilation in hospitalized patients: prevalence, effects, and outcome. Am J Med. 2004;116:1–7. doi: 10.1016/j.amjmed.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 52.Laaban JP, Chailleux E. Daytime hypercapnia in adult patients with obstructive sleep apnea syndrome in France, before initiating nocturnal nasal continuous positive airway pressure therapy. Chest. 2005;127:710–5. doi: 10.1378/chest.127.3.710. [DOI] [PubMed] [Google Scholar]
- 53.Vgontzas AN, Kales A, Seip J, et al. Relationship of sleep abnormalities to patient genotypes in Prader-Willi syndrome. Am J Med Genet. 1996;67:478–82. doi: 10.1002/(SICI)1096-8628(19960920)67:5<478::AID-AJMG7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 54.Hertz G, Cataletto M, Feinsilver SH, Angulo M. Sleep and breathing patterns in patients with Prader Willi syndrome (PWS): effects of age and gender. Sleep. 1993;16:366–71. doi: 10.1093/sleep/16.4.366. [DOI] [PubMed] [Google Scholar]
- 55.Kuwajima T, Nishimura I, Yoshikawa K. Necdin promotes GABAergic neuron differentiation in cooperation with Dlx homeodomain proteins. J Neurosci. 2006;26:5383–92. doi: 10.1523/JNEUROSCI.1262-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee S, Walker CL, Karten B, et al. Essential role for the Prader-Willi syndrome protein necdin in axonal outgrowth. Hum Mol Genet. 2005;14:627–37. doi: 10.1093/hmg/ddi059. [DOI] [PubMed] [Google Scholar]
- 57.Kuwako K, Taniura H, Yoshikawa K. Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J Biol Chem. 2004;279:1703–12. doi: 10.1074/jbc.M308454200. [DOI] [PubMed] [Google Scholar]
- 58.Talebizadeh Z, Butler MG. Insulin resistance and obesity-related factors in Prader-Willi syndrome: comparison with obese subjects. Clin Genet. 2005;67:230–9. doi: 10.1111/j.1399-0004.2004.00392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O'Donnell C P, Schaub CD, Haines AS, et al. Leptin prevents respiratory depression in obesity. Am J Respir Crit Care Med. 1999;159:1477–84. doi: 10.1164/ajrccm.159.5.9809025. [DOI] [PubMed] [Google Scholar]
- 60.Goldstone AP, Patterson M, Kalingag N, et al. Fasting and postprandial hyperghrelinemia in Prader-Willi syndrome is partially explained by hypoinsulinemia, and is not due to peptide YY3-36 deficiency or seen in hypothalamic obesity due to craniopharyngioma. J Clin Endocrinol Metab. 2005;90:2681–90. doi: 10.1210/jc.2003-032209. [DOI] [PubMed] [Google Scholar]
- 61.Choe YH, Song SY, Paik KH, et al. Increased density of ghrelin-expressing cells in the gastric fundus and body in Prader-Willi syndrome. J Clin Endocrinol Metab. 2005;90:5441–5. doi: 10.1210/jc.2004-1935. [DOI] [PubMed] [Google Scholar]
- 62.Young T, Peppard PE, Taheri S. Excess weight and sleep disordered breathing. J Appl Physiol. 2005;99:1592–99. doi: 10.1152/japplphysiol.00587.2005. [DOI] [PubMed] [Google Scholar]