

The clean, efficient utilization of carbon monoxide as a C1 source continues to represent a significant chemical challenge. Many homogeneous late metal-mediated industrial processes have been developed which center on the utility of well-studied transition metal acyl intermediates.1 Stoichiometric reactions of carbon monoxide with simple alkyl and dialkylmetal complexes usually lead cleanly to carbonylation products. This is true irrespective of the precise conditions employed, although some substrates may require more forcing conditions (e.g., high temperature or pressure) than others. Through this stoichiometric chemistry, many of the classic primary steps in homogeneous late metal-mediated catalytic organometallic reactions, such as migratory CO insertion, have been identified. Herein we describe the carbonylation reaction of the dimethylniobium complex (BDI)Me2Nb(NtBu) (1, BDI = β-diketiminate),2 in which the relative amounts of an unusually wide range of products change dramatically in response to very modest changes in reaction conditions. Four different stable products, in which CO insertion takes place coupled with metal reduction, enediolate formation and C—H activation, are formed in these transformations. By careful adjustment of reaction conditions, the majority of these materials have been isolated and fully characterized.
On adding 3.0 equiv of CO to a solution of 1 in THF at —72 °C, stirring rapidly for 5 min, then warming directly to room temperature, a brown solution was obtained from which a green product was isolated by crystallization. The products were identified as acetone and the Nb(III) dicarbonyl adduct (BDI)(CO)2Nb(NtBu)(2, 42% isolated yield, Scheme 1), which can be considered to result from transfer of both methyl groups to one CO, followed by the displacement of acetone by two molecules of CO (νCO (2) = 1988, 1893 cm−1). The formation of acetone was confirmed by both GC—MS (20% yield)3 and isolation of the 2,4-dinitrophenylhydrazone derivative (43% yield). The release of a ketone and formation of a dicarbonyl adduct suggest analogies with previous work on dialkyl metallocenes of the group 4 triad4 and on η2-acetone complexes in group 5 systems.5,6
The outcome of the reaction of 1 with CO may be altered dramatically by using pentane as the solvent in place of THF and by allowing the reaction mixture to remain at —72 °C for 2 h prior to warming to room temperature. In so doing, a second product may be isolated by crystallization, whose analytical data are consistent with its formulation as the enediolate species (BDIiPr)(Me2C2O2)Nb(NtBu) (3, 54% yield, Scheme 1), presumably formed via the intramolecular coupling of two Nb-acyl fragments,7-10 though other mechanisms cannot be ruled out. NMR spectral analysis of the crude reaction mixture indicates that the product distribution has inverted from 4:1 mixtures of 2:3 obtained when THF is used as the solvent to 1:4 mixtures of 2:3 obtained when pentane is used and longer reaction times are employed.
A third product from the reaction of CO with 1 is obtained by slowly introducing 1.0 equiv of CO into the headspace above a solution of 1 in THF in a flask cooled to —72 °C, followed by immediately allowing the system to warm to room temperature. After stirring the resulting mixture for 36 h and removing the volatile materials, a new product may be crystallized from pentane, yielding yellow crystals of {ArNC(Me)CHC(Me)N[2-(CHMeCH2)-6-iPr-C6H3]}(Me2HCO)Nb(NtBu) in 37% yield (4, Scheme 1). The structure of 4, determined crystallographically, indicates that the product is formed in a net process of transfer of both methyl groups to the CO carbon and the formal addition of a ligand C—H bond across the Nb—O—C linkage.11
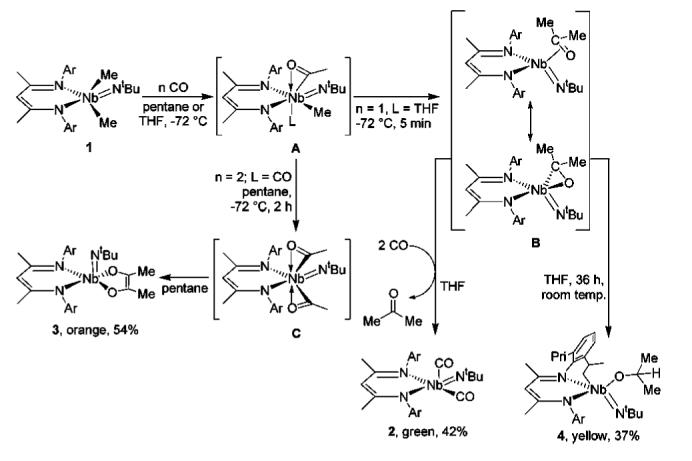
The utility of the CO reaction scheme presented here lies in the ability to perturb the ultimate fate of common intermediates that appear along the course of the reaction. Compounds 2—4 presumably originate from an initial monoacyl complex (A, Scheme 1). Indeed, NMR studies on a mixture of 13CO and 1 at —80 °C allowed us to observe a Nb monoacyl species (BDI)Me(Me13CO)Nb(NtBu)(5, Scheme 2), displaying a strong 13C NMR signal at 299 ppm. Still, while the signal lies within a region characteristic of early metal acyl complexes,12 no 1H coupling information could be discerned, presumably owing to a complicated coupling pattern.
However, support for the formulation of both 5 and A as monoacyls is provided by treating the analogous monoalkylated complex (BDI)MeBrNb(NtBu) (6) with 13CO. In this case a single product is observed with a 13C NMR resonance at 292 ppm, attributable to the species (BDI)Br(Me13CO)Nb(NtBu) (7, Scheme 2). The 1H-coupled 13C NMR spectrum reveals the signal to be a pseudoquartet (93Nb, 100%, I = 9/2) with 2JC—H = 6.5 Hz, corresponding to a doublet with the same coupling constant in the 1H NMR spectrum at 2.56 ppm. This complex is stable in solution at room temperature for ca. 30 min before transforming into an unidentified species.
Assuming that products 2 and 3 arise from a common acyl intermediate A and that sufficient amounts of CO are present in both systems to allow for either reaction pathway to occur, then the factor determining the product distribution appears to be the coordinating ability of the solvent. In pentane, free CO (a weak ligand for d0 metal systems) coordinates to the metal center and undergoes insertion into the remaining Nb—Me bond to form a diacyl complex. The diacyl then undergoes intramolecular C—C coupling to form 3. In THF, however, the solvent occupies a coordination site preferentially over CO, allowing methyl transfer to the acyl group to occur on warming. This second methyl group transfer amounts to a formal reduction of Nb(V) to Nb(III). Displacement of the coordinated acetone with 2 equiv of CO then leads to the formation of 2. These hypotheses, which exclude control by differences in solvent polarity, are supported by two key observations: (i) that higher concentrations of 1 in either solvent lead to greater 3:2 ratios and (ii) that performing the synthesis of 2 with 2,5-Me2THF as the solvent leads to a 1.1:1 ratio of 3:2.
The formation of 4 could conceivably proceed through a number of pathways. The formation of 4-d6 from (BDI)(CD3)2Nb(NtBu)(1-d6) revealed that the two diastereotopic deuterium-labeled methyl groups remained intact. This excludes the possibility of pathways involving an initial C—H(D) activation at one of the methyl groups and suggests that an intermediate acetone adduct B may be involved in this mechanism.
To gain further insight into the possible intermediacy of complex B, the reaction of 1 with isocyanides, an isoelectronic variant of CO, was investigated. The reaction of 1 with 1.0 equiv of XylNC(Xyl = 2,6-Me2C6H3) in Et2O at room temperature gave a deep red solution that, after workup, yielded (BDI)(η2-XylNCMe2)-Nb(NtBu) (8, Scheme 3) as a dark red crystalline solid in 48% yield. While an X-ray diffraction study indicates the product of the reaction to be a Nb(V) azaniobacyclopropane,13 spectroscopic and chemical observations imply assignment of the oxidation state to be more complicated than the crystallographic data suggest.
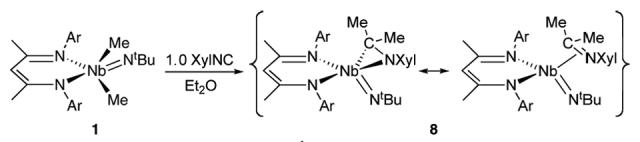
The room temperature 1H NMR spectrum of 8 displays broad resonances, indicating either general intramolecular rearrangement processes or rotation about a ketimine—Nb(III) vector. A more concrete demonstration of the ability of 8 to serve as a source of Nb(III) was provided by treating 8 with 2.0 equiv of CO in C6D6 at room temperature. Over the course of three hours, 2 grows in cleanly and quantitatively with concomitant formation of free ketimine XylN=CMe2 (Scheme 4).
The contrast between the oxidation states assigned to reactions involving 8 and the ground-state of 8 (XRD) allow for informed speculation as to the mechanism leading to 4. While intermediate B would yield 2 by displacement of acetone with 2.0 equiv of CO, B may also be described as a Nb(V) oxaniobacyclopropane, which, in the absence of excess CO, would ring-open (due to strain within in the three-membered metallacycle) by way of an intramolecular C—H bond activation to yield 4. This behavior would reflect the dual Nb(III)/Nb(V) character exhibited by the electronically and structurally analogous ketimine complex 8.
These results indicate an ability to dial in a specific product mixture based on very subtle modifications to the reaction conditions—a technique particularly useful in transforming CO into various organic species. The close energetic proximity of diverse reaction pathways for second-row transition metals14 often impedes progress in these areas. However, these initial results demonstrate an ability to make use of these small energy differences for accessing separate product distributions. Further research will be directed toward uncovering factors that would allow us to control the product distribution with higher precision.
Scheme 1.
Scheme 2.
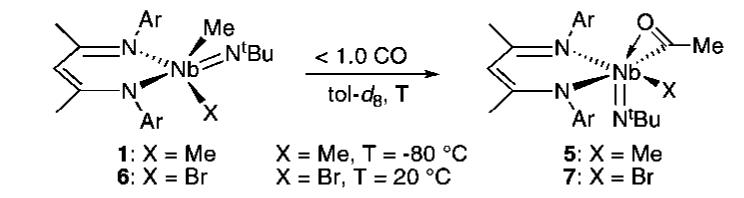
Scheme 3.
Scheme 4.
Acknowledgment
This work was supported by the American Chemical Society Petroleum Research Fund (ACS-47249AC3), the National Science Foundation (CHE-0416309), and the National Institutes of Health (R01-GM025459-29).
Supporting Information Available: Experimental procedures and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Kuhlmann EJ, Alexander JJ. Coord. Chem. Rev. 1980;33:195–225. [Google Scholar]
- (2).Mindiola DJ. Acc. Chem. Res. 2006;39:813–821. doi: 10.1021/ar0500113. For related early metal chemistry see: (a) and references cited therein. [DOI] [PubMed] [Google Scholar]
- (3). The low yield obtained by GC—MS is attributed to low reaction concentrations and the collection of acetone on glassware.
- (4).Erker G, Dorf U, Czisch P, Petersen JL. Organometallics. 1986;5:668–676. and references cited therein. [Google Scholar]
- (5).Wood CD, Schrock RR. J. Am. Chem. Soc. 1979;101:5421–5422. [Google Scholar]
- (6).Gomez M, Gomez-Sal P, Jimenez G, Martin A, Royo P, Sanchez-Nieves J. Organometallics. 1996;15:3579–3587. [Google Scholar]
- (7). Previous work has shown that early metal acyl fragments possess significant oxy-carbene character due to the oxophilicity of the d0 systems (see refs 8-10)
- (8).Castro A, Galakhov MV, Gomez M, Gomez-Sal P, Martin A, Sanchez F. J. Organomet. Chem. 2000;595:36–53. [Google Scholar]
- (9).Moloy KG, Fagan PJ, Manriquez JM, Marks TJ. J. Am. Chem. Soc. 1986;108:56–67. [Google Scholar]
- (10).Dawson DY, Arnold J. Organometallics. 1997;16:1111–1113. [Google Scholar]
- (11).Thorn MG, Fanwick PE, Rothwell IP. Organometallics. 1999;18:4442–4447. In related chemistry, an ortho-metallated Ti center bearing a secondary amido ligand was obtained following reduction of an isocyanide by (ArO)2TiMe2 (Ar = 2,6-Ph2-C6H3). The latter reaction is believed to proceed through an intermediate ketimine complex, although no direct evidence for this assumption was given, nor was a comparison made with CO reactivity. (a) [Google Scholar]
- (12).Gomez M. Eur. J. Inorg. Chem. 2003;368:1–3697. [Google Scholar]
- (13). Bond lengths about the niobacyclopropane unit are consistent with Nb—C, Nb—N, and N—C single bonds: Nb(1)—C(1) = 2.197(4) Å, Nb(1)—N(1) = 1.964(3) Å, N(1)—C(1) = 1.477(4) Å.
- (14).Hirsekorn KF, Hulley EB, Wolczanski PT, Cundari TR. J. Am. Chem. Soc. 2008;130:1183–1196. doi: 10.1021/ja074972j. [DOI] [PubMed] [Google Scholar]