

Abstract
3-Iodoindoles have been prepared in excellent yields by coupling terminal acetylenes with N,N-dialkyl-o-iodoanilines in the presence of a Pd/Cu catalyst and subsequent electrophilic cyclization of the resulting N,N-dialkyl-o-(1-alkynyl)anilines using I2 in CH2Cl2. Aryl-, vinylic-, alkyl- and silyl-substituted terminal acetylenes undergo this process to produce excellent yields of 3-iodoindoles. The reactivity of the carbon-nitrogen bond cleavage during cyclization follows the order: Me > n-Bu, Me > Ph, and cyclohexyl > Me. Subsequent palladium-catalyzed Sonogashira, Suzuki and Heck reactions of the resulting 3-iodoindoles proceed smoothly in good yields.
Introduction
The substituted indole nucleus is prevalent in numerous natural products and is extremely important in medicinal chemistry.1 For example, lescol has been identified as an HMG-CoA reductase inhibitor1c and indomethacin is an anti-tumor agent for endometrial cancers.1d Thus, research directed towards concise and novel syntheses of substituted indoles is highly desirable.

Electrophilic cyclization via “vinylic cations” to make heterocycles, such as benzo[b]furans and benzo[b]thiophenes, has been explored and the mechanism of this process has been studied by Taniguchi.2 However, significant limitations have been found in generating the “vinylic cations”. Only recently, the electrophilic cyclization of functionally-substituted alkynes has been reported to provide very general and efficient preparations of benzo[b]thiophenes,3 isoquinolines and naphthyridines,4 isocoumarins and α-pyrones,5 benzofurans,6 furans,7 indoles,8 furopyridines,9 cyclic carbonates,10 2,3-dihydropyrroles and pyrroles,11 pyrilium salts,12 bicyclic β-lactams,13 isochromenes,14,12a phosphaisocoumarins,15 isoindolin-1-ones,16 benzopyrans and 1,2-dihydroquinolines.17 The intramolecular transition metal-catalyzed hydroarylation of alkynes has also proven useful for the synthesis of heterocycles and carbocycles.18 This chemistry is generally viewed as proceeding through electrophilic addition to the alkyne by a cationic intermediate.
Our success employing this process for the synthesis of benzo[b]thiophenes3a,b inspired us to explore the possibility of preparing indoles by this same approach. During our investigation of the electrophilic cyclization of N,N-disubstituted o-(1-alkynyl)-anilines to substituted indoles, Barluenga8a and Knight8c reported similar electrophilic cyclizations of o-(1-alkynyl)anilines and their carbamate, sulfonamide and amide derivatives bearing nitrogen-hydrogen bonds, using IPy2BF4/HBF4 and I2/K2CO3 respectively. However, the use of either a strong acid or a base during these cyclizations might affect certain acid or base-sensitive functional groups. The use of an expensive iodonium salt, IPy2BF4, and the strong, toxic acid HBF4, the apparent need to carefully control the reaction temperature, and the highly variable yields make the former approach less appealing. 8a approach apparently only works on The latter I2 sulfonamides bearing a nitrogen-hydrogen bond. 8c From preliminary results,8b our approach, utilizing neutral reaction conditions, appeared to be more efficient and convenient, and appeared to produce N-alkylindoles in high yields.
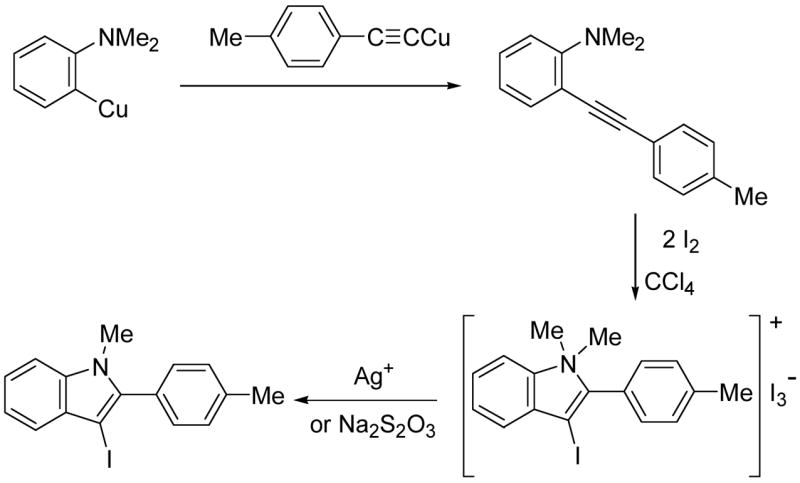
Hoedt has reported an approach to the synthesis of 1-methyl-2-p-tolylindole and its 3-iodo- derivative by a related process involving iodocyclization (Scheme 1).19 Unfortunately, the unusual synthesis of the starting acetylene, the separation of the key triiodide intermediate salt, and the stepwise addition of an expensive silver reagent or sodium thiosulfate are not particularly attractive synthetically. Furthermore, the yield of the above process was only 66% or less and the scope of the cyclization step has not been examined. Herein, we report a more convenient synthesis of the starting 2-(1-alkynyl)anilines and the successful application of this electrophilic cyclization strategy as a convenient and general synthetic method for the synthesis of 3-iodoindoles.
SCHEME 1.
Results and Discussion
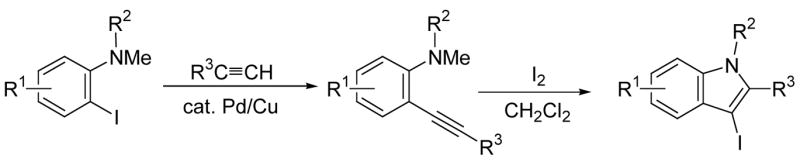
A two step approach to 3-iodoindoles has been examined involving (i) the Sonogashira coupling20 of N,N-dialkyl-o-iodoanilines with terminal alkynes, and (ii) electrophilic cyclization (Scheme 2).
SCHEME 2.
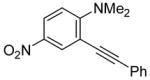
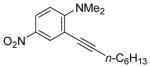
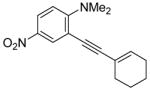
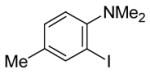
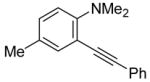
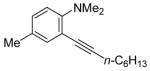
To assess the generality of this overall approach, the scope of the Sonogashira coupling of N,N-dialkyl-o-iodoanilines and terminal alkynes was first studied. Treatment of N,N-dialkyl-o-iodoanilines bearing different functionality with a variety of terminal alkynes under standard Sonogashira coupling conditions (5 mmol of N,N-dialkyl-o-iodoaniline, 1.2 equivs of terminal alkyne, 2 mol % of PdCl2(PPh3)2, 1 mol % of CuI, 12.5 ml of Et3N at room temperature for 5–24 h) affords high yields of the coupling products (eq 1, Table 1).
TABLE 1.
Sonogashira Coupling of N,N-Dialkyl-2-iodoanilines and Terminal Acetylenes.a
| entry | 2-iodoaniline | terminal acetylene | arylalkyne | reaction time (h) | yield (%) | ||
|---|---|---|---|---|---|---|---|
| 1 |
|
1 | PhC≡CH |
|
2 | 7 | 84 |
| 2 | t-BuC≡CH |
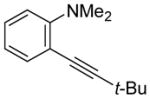
|
3 | 7 | 85 | ||
| 3 | n-C6H13C≡CH |
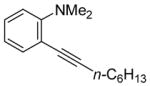
|
4 | 7 | 94 | ||
| 4 |
|
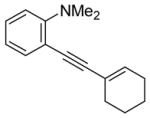
|
5 | 5 | 84 | ||
| 5 | Me3SiC≡CH |
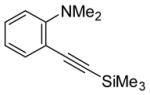
|
6 | 7 | 70 | ||
| 6 | NC(CH2)3C≡CH |
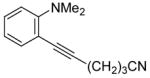
|
7 | 7 | 40 | ||
| 7 | Cl(CH2)3C≡CH |
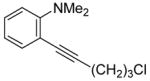
|
8 | 7 | 87 | ||
| 8 |
|
9 | PhC≡ CH |
|
10 | 5 | 89 |
| 9 | n-C6H13C≡CH |
|
11 | 5 | 83 | ||
| 10 |
|
|
12 | 5 | 100 | ||
| 11 |
 |
13 | PhC≡CH |
|
14 | 5 | 98 |
| 12 | n-C6H13C≡CH |
|
15 | 5 | 67 | ||
| 13 |
|
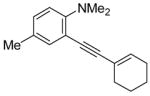
|
16 | 5 | 83 | ||
| 14 |
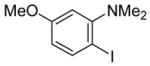
|
17 | PhC≡CH |
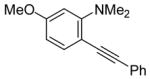
|
18 | 24 | 98 |
| 15 | n-C6H13C≡CH |
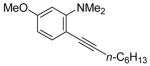
|
19 | 24 | 93 | ||
| 16 |
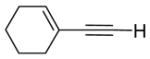
|
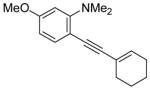
|
20 | 24 | 100 | ||
| 17 |
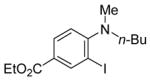
|
21 | PhC≡CH |
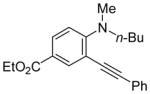
|
22 | 5 | 100 |
| 18 | n-C6H13C≡CH |
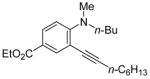
|
23 | 5 | 54 | ||
| 19 |
|
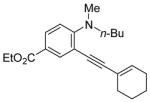
|
24 | 5 | 95 | ||
| 20 |
|
25 | PhC≡CH |
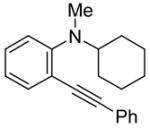
|
26 | 24 | 80 |
| 21 |

|
27 | PhC≡CH |
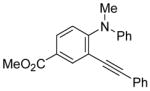
|
28 | 24 | 84 |
All reactions were run with 5 mmol of the N,N-dialkyl-2-iodoaniline, 1.2 equiv of the terminal acetylene, 2 mol % of PdCl2(PPh3)2, 1 mol % of CuI, and 12.5 mL of Et3N at 50 °C.
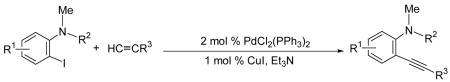 |
(1) |
N,N-Dialkyl-o-iodoanilines show high reactivity towards the Sonogashira coupling and this allows the preparation of a wide variety of functionalized N,N-dialkyl-o-(1-alkynyl)anilines. N,N-Dialkyl-o-iodoanilines bearing nitro, ester, methyl and methoxy groups undergo smooth Sonogashira coupling with alkynes bearing various substituents, including aryl, vinylic, alkyl and silyl groups. High yields were obtained in almost all cases (Table 1). Most terminal alkynes have afforded high yields in this Sonogashira reaction. However, because it is extremely reactive towards homocoupling, 5-hexynenitrile affords only a relatively low yield and a large amount of the homocoupling diyne product is formed (entry 6).
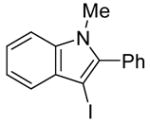
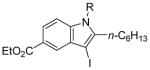
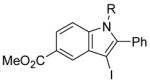
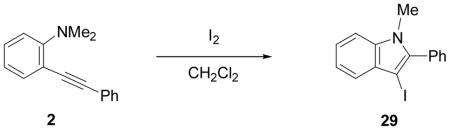
We have found that N,N-dimethyl-o-(phenylethynyl)aniline (2), when treated with I2 in CH2Cl2, undergoes smooth iodocyclization at room temperature and affords an essentially quantitative yield of the corresponding 3-iodo-1-methyl-2-phenylindole (29) (eq 2; Table 2, entry 1). Only a trace of the triiodide salt was observed upon TLC analysis. However, under Hoedt‘s reaction
TABLE 2.
Synthesis of 3-Iodoindoles.a
| entry | alkyne | time (h) | product | isolated yield (%) | ||
|---|---|---|---|---|---|---|
| 1 |
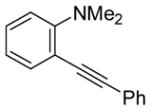
|
2 | 2 |
|
29 | 100 |
| 2 |
|
3 | 2 |
|
30 | 96 |
| 3 |
|
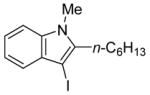
4 | 2 |
|
31 | 94 |
| 4 |
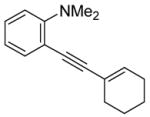
|
5 | 2 |
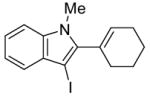
|
32 | 85 |
| 5 |
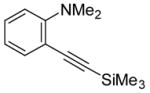
|
6 | 4 |
|
33 | ? |
| 6 |
|
7 | 4 |
|
34 | 86 |
| 7 |
|
8 | 4 |
|
35 | 73 |
| 8 |
|
10 | 0.5 |
|
36 | 100 |
| 9 |
|
11 | 0.5 |
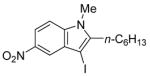
|
37 | 95 |
| 10 |
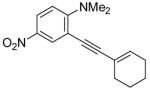
|
12 | 0.5 |
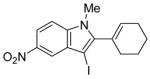
|
38 | 100 |
| 11 |
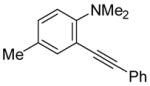
|
14 | 0.5 |
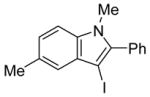
|
39 | 100 |
| 12 |
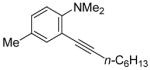
|
15 | 0.5 |
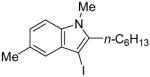
|
40 | 100 |
| 13 |
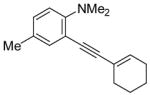
|
16 | 0.5 |
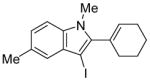
|
41 | 98 |
| 14 |
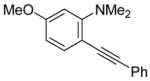
|
18 | 2 |
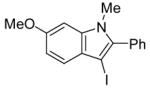
|
42 | 84 |
| 15 |
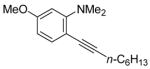
|
19 | 2 |
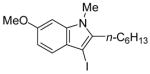
|
43 | 84 |
| 16 |
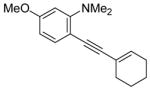
|
20 | 2 |
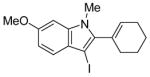
|
44 | 100 |
| 17 |
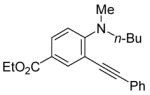
|
22 | 0.5 |
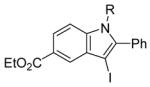
R = n-Bu (45)R = Me (46) |
98(72:28) b(45:46) | |
| 18 |
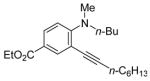
|
23 | 0.5 |
R = n-Bu (47)R = Me (48) |
84(62:38) b(47:48) | |
| 19 |
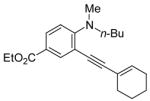
|
24 | 0.5 |
R = n-Bu (49)R = Me (50) |
92(66:34) b(49:50) | |
| 20 |
|
26 | 2 |
R = Me (27) |
50(90:10) b(27:51) | |
| 21 |
|
28 | 1 |
R = Ph (52)R = Me (27) |
80(100:0) b(52:27) |
All reactions were run with 0.25 mmol of the alkyne and 2 equiv of I2 in 5 mL of CH2Cl2 at 25 °C, followed by the addition of 5 mL of satd aq Na2S2O3 to remove the excess I2.
Ratio of the products shown immediately below.
 |
(2) |
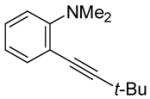
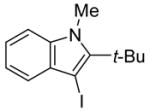
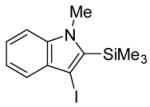
conditions,19 the triiodide salt appears to be the major product. The mild reaction conditions, as well as the high yield of this reaction, encouraged us to extend this methodology to a range of N,N-dimethyl-o-(1-alkynyl)anilines (Table 2, entries 1–7). The cyclization proceeds smoothly when the substituent on the distal end of the alkyne is aryl, vinylic, alkyl, functionally-substituted alkyl or silyl. Even hindered tert-butyl- (3) (entry 2) or trimethylsilyl- (6) (entry 5) substituted alkynes react rapidly in high yields. However, 3-iodo-1-methyl-2-(trimethylsilyl)indole formed by the iodocyclization of alkyne 6 is extremely unstable. Although the cyclization product was detected by TLC and 1H NMR spectroscopic analysis of the crude product, we were not able to separate and fully characterize this 3-iodoindole.
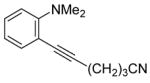
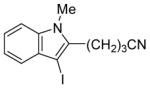
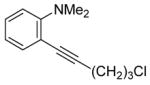
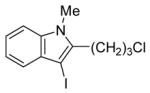
The substituents on the triple bond of the N,N-dimethyl-o-(1-alkynyl)anilines affect the yields of the cyclization reactions. Substrates with substituents, which are in conjugation with the triple bond, such as the phenyl group in alkyne 2 and the vinylic group in alkyne 5, appear to cyclize more rapidly and generally produce higher yields of products. The bulky trimethylsilyl group in 6 tends to hinder cyclization and requires a longer reaction time (entry 5). The reactions of 6-[2-(dimethylamino)phenyl]-5-hexynenitrile (7) and N,N-dimethyl-2-(5-chloro-1-pentynyl)aniline (8) with iodine (entries 6 and 7) afford somewhat lower yields than the simple alkyl-substituted alkyne 4 (entry 3) and require a significantly longer reaction time. This may be because the electron-withdrawing cyano- and chloro- groups on the alkyl chain make the corresponding vinylic cation intermediates less stable (see the later mechanistic discussion). However, no products involving simple addition of the electrophile I2 to the alkyne triple bond are observed with any of these alkynes.
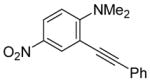
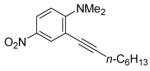
The substituents on the aniline moiety also play a significant role in the cyclization. The presence of electron-withdrawing groups, such as a nitro or an ester group, para to the aniline nitrogen enhance the electrophilic cyclization. The reaction time is shorter and higher yields are generally obtained (entries 8–10 and 17–19). Similarly, for a weak electron-donating methyl group, short reaction times and high yields are also observed (entries 11–13). However, the stronger electron-donating methoxy group meta to the aniline nitrogen requires longer reaction times and tends to lower the yield (entries 14–16).
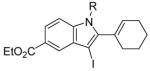
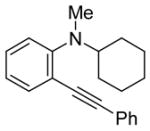
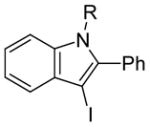
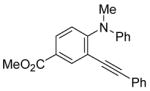
The use of two different alkyl groups on the nitrogen of the aniline affords interesting selectivity. Compounds 22–24 with a methyl and an n-butyl group on the nitrogen undergo electrophilic cyclization smoothly and the total conversions are quite high (entries 17–19). The less hindered methyl group is more easily removed than is the n-butyl group, suggesting that alkyl cleavage is proceeding by an SN2 process. The corresponding N-methyl-3-iodoindoles and N-n-butyl-3-iodoindoles were obtained in approximately a 2 to 1 ratio. Compound 26 bearing a methyl and a cyclohexyl group on the nitrogen of the aniline would thus be expected to afford exclusively the corresponding N-cyclohexylindole (entry 20). In fact, we isolated a 45% yield of N-methylindole and only about a 5% yield of the N-cyclohexylindole was formed. These results suggest that loss of the alkyl group can occur by either an SN1 or an SN2 mechanism or perhaps loss of the cyclohexyl group occurs by an E2 elimination instead. When this reaction was carried out in an NMR tube using CD2Cl2 as the solvent, MeI, cyclohexene and cyclohexyl iodide were observed in the 1H NMR spectrum. Unfortunately, the yield of these by-products could not be easily determined. Finally, compound 28 containing a methyl and a phenyl group on the aniline nitrogen affords exclusively the expected N-phenylindole 52 in an 80% yield (entry 21). Unfortunately, neither 1-[2-(phenylethynyl)phenyl)]pyrrolidine or N-methyl-N-[2-(phenylethynyl)phenyl]acetamide afforded the desired indole product under our standard reaction conditions.
In order to explore the scope of this indole synthesis, four other electrophiles, Br2, NBS, p-O2NC6H4SCl and PhSeCl, have also been examined. Only iodine exhibits high reactivity in this electrophilic cyclization. The other electrophiles provide only very poor or no yields of the desired cyclization products. The predominant reaction with these electrophiles is addition across the C-C triple bond.
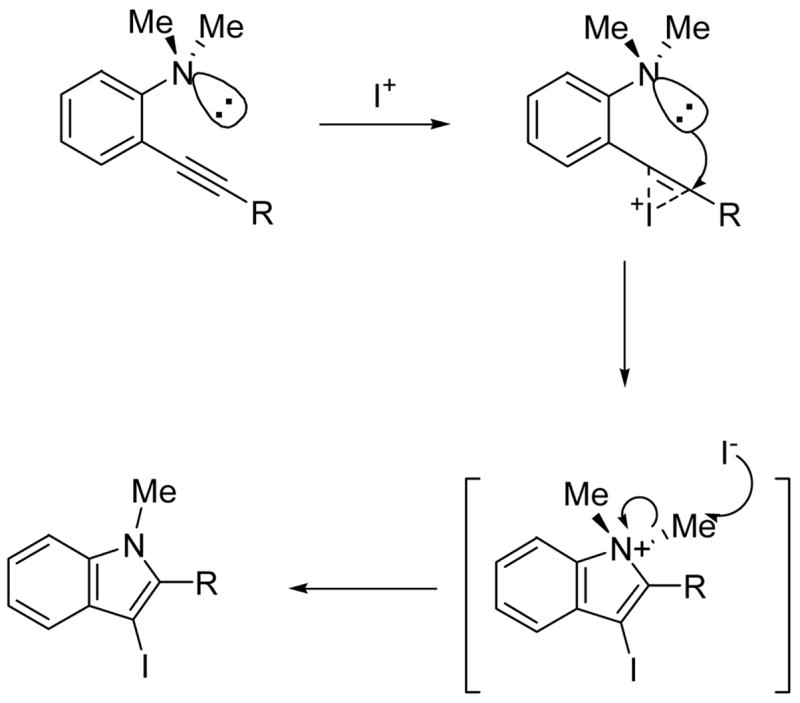
We believe that these cyclizations proceed by anti attack of the electrophile and the nitrogen of the N,N-dialkylamino group on the alkyne to produce an indolium salt, which loses an alkyl group via either SN1 or SN2 substitution or possibly E2 elimination promoted by the iodide nucleophile present in the reaction mixture (Scheme 3). The success of this reaction is presumably due to several factors. First, the two alkyl groups on the nitrogen make the nitrogen highly nucleophilic. Second, the interaction between the two alkyl groups and the internal triple bond favors an orientation of the nitrogen with its lone pair of electrons pointing towards the triple bond. Third, the highly nucleophilic iodide ion formed after the cyclization facilitates removal of the methyl or other alkyl group.
SCHEME 3.
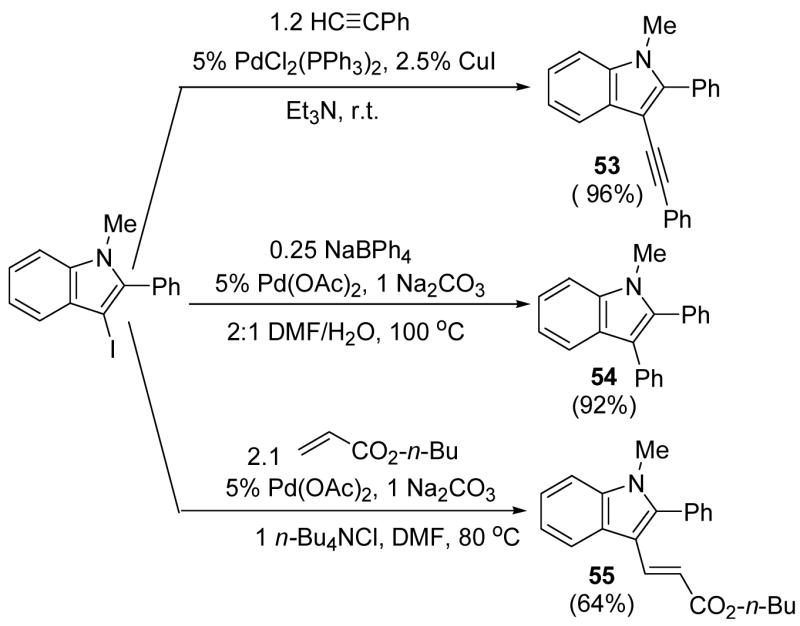
The 3-iodoindoles produced by this chemistry should be very useful for the synthesis of a wide variety of substituted indoles. For example, the 3-iodoindoles produced by this strategy can be further functionalized by applying palladium-catalyzed coupling reactions. We have found that N-methyl-2-phenyl-3-(phenylethynyl)indole (53), N-methyl-2,3-diphenylindole (54) and n-butyl E-3-[1-methyl-2-phenylindol-3-yl]propenoate (55) can be obtained in 92%, 90% and 64% overall yields respectively from N,N-dimethyl-o-iodoaniline and phenyl acetylene by our two step coupling/cyclization process, followed by palladium-catalyzed cross-couplings (Scheme 4). One should be able to prepare many other 2,3-disubstituted indoles using these iodo-substrates and other known palladium methodology.
SCHEME 4.
Conclusions
A very efficient synthesis of 2,3-disubstituted indoles has been developed by a two step approach involving the Sonogashira cross-coupling of terminal alkynes and N,N-dialkyl-o-iodoanilines, followed by electrophilic cyclization using I2 in CH2Cl2. While I2 gives 3-iodoindoles in excellent yields, Br2, NBS, p-O2NC6H4SCl and PhSeCl give mixtures of both cyclization products and products of simple addition to the triple bond with the latter predominating. A wide variety of aniline-containing acetylenes with various functional groups undergo this overall process in good to excellent yields. The steric and electronic effects of the substituents on the carbon-carbon triple bond of the N,N-dialkyl-2-(1-alkynyl)aniline intermediates have been studied.
Experimental Section
General Procedure for Preparation of the N,N-Dimethyl-o-iodoanilines
These compounds were prepared by a procedure reported by Cadogan.21 A solution of the corresponding aniline (2.0 mmol) and iodomethane (0.85 g, 6.0 mmol) in DMF (10 ml) containing K2CO3 (0.55 g, 4.0 mmol) was stirred for 48 h at room temperature. Water (10 ml) was then added, and the solution was extracted with diethyl ether (3 × 10 ml). The organic extracts were washed with water (4 × 20 ml) to remove any remaining DMF, dried (Na2SO4), and the solvent was removed under reduced pressure to yield the crude product, which was further purified by flash chromatography on silica gel using ethyl acetate/hexane as the eluent.
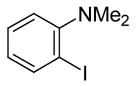
N,N-Dimethyl-2-iodoaniline (1)
The product was obtained as a yellow oil: 1H NMR (300 MHz, CDCl3) δ 2.76 (s, 6H), 6.76 (t, J = 8.2 Hz, 1H), 7.08 (d, J = 9.3 Hz, 1H), 7.30 (t, J = 7.7 Hz, 1H), 7.83 (d, J = 7.8 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 45.2, 97.3, 120.6, 125.2, 129.2, 140.4, 155.1; IR (neat, cm−1) 3002, 1944, 2830, 2783; HRMS calcd for C8H10IN 246.98580. Found 246.98605.
General Procedure for Synthesis of the N,N-Dialkyl-2-(1-alkynyl)anilines
To a solution of Et3N (12.5 ml), PdCl2(PPh3)2 (0.070 g, 2 mol %), 5 mmol of N,N-dialkyl-o-iodoaniline and 6.0 mmol of terminal acetylene (stirring for 5 min beforehand), CuI (0.010 g, 1 mol %) was added and stirring was continued for another 2 min before flushing with Ar. The flask was then sealed. The mixture was allowed to stir at room temperature for the desired time and the resulting solution was filtered, washed with a satd aq NaCl solution and extracted with diethyl ether (2 × 10 ml). The combined ether fractions were dried over Na2SO4 and concentrated under vacuum to yield the crude product. The crude product was purified by flash chromatography on silica gel using ethyl acetate/hexane as the eluent.
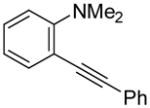
N,N-Dimethyl-2-(phenylethynyl)aniline (2)
The product was obtained as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 3.01 (s, 6H), 6.88–6.94 (m, 2H), 7.25 (t, J = 6.3 Hz, 1H), 7.31–7.37 (m, 3H), 7.49 (d, J = 5.7 Hz, 1H), 7.54 (d, J = 7.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 43.8, 89.1, 95.0, 115.3, 117.2, 120.7, 124.1, 128.2, 128.5, 129.5, 131.5, 134.6, 155.0; IR (neat, cm−1) 3059, 2943, 2833, 2785, 2209; HRMS calcd for C16H15N 221.12045. Found 221.12076.
General Procedure for Iodocyclization
To a solution of 0.25 mmol of the N,N-dialkyl-2-(1-alkynyl)aniline and 3 ml of CH2Cl2, 2 equivs of I2 dissolved in 2 ml of CH2Cl2 was added gradually. The reaction mixture was flushed with Ar and allowed to stir at room temperature for the desired time. The excess I2 was removed by washing with a satd aq soln of Na2S2O3. The aqueous solution was then extracted by diethyl ether (2 × 10 ml). The combined ether layers were dried over anhydrous Na2SO4 and concentrated under vacuum to yield the crude product, which was purified by flash chromatography on silica gel using ethyl acetate/hexanes as the eluent.
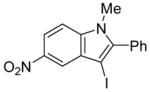
3-Iodo-1-methyl-2-phenylindole (29)
The product was obtained as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 3.63 (s, 3H), 7.21–7.28 (m, 3H), 7.43–7.51 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 32.2, 59.0, 110.0, 120.9, 121.6, 123.1, 128.6, 129.0, 130.5, 131.1, 131.8, 137.9, 141.9; IR (neat, cm−1) 3054, 2937; HRMS calcd for C15H12IN 333.00145. Found 333.00194.
1-Methyl-2-phenyl-3-(phenylethynyl)indole (53)
This indole was prepared by the following procedure. Into a well mixed Et3N solution (5 ml) containing 5.0 mmol of 3-iodo-1-methyl-2-phenylindole, 6.0 mmol of phenylacetylene and PdCl2(PPh3)2 (5 mol %), CuI (2.5 mol %) was added and the flask was flushed with Ar, sealed and allowed to stir at room temperature for 2 h. The resulting precipitate was filtered off and washed with diethyl ether (10 ml). The combined ether layers were dried over anhydrous Na2SO4 and concentrated under vacuum to afford a yellow oil: 1H NMR (300 MHz, CDCl3) δ 3.69 (s, 3H), 7.21–7.32 (m, 6H), 7.41–7.44 (m, 3H), 7.49 (t, J = 7.2 Hz, 2H), 7.65 (dd, J = 10.8, 2.0 Hz, 2H), 7.85 (dd, J = 10.4, 2.0 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 31.8, 84.5, 91.9, 110.0, 120.2, 121.0, 123.1, 124.7, 127.4, 128.1, 128.4, 128.5, 128.7, 129.0, 130.4, 131.0, 131.3, 137.4, 144.0; IR (neat, cm−1) 3004, 2962, 2923, 2204; HRMS calcd for C23H17N 307.13610. Found 307.13698.
1-Methyl-2,3-diphenylindole (54)
This indole was prepared according to a literature procedure.22 To 10 ml of a 2:1 DMF/H2O solution containing 5.0 mmol of 3-iodo-1-methyl-2-phenylindole, 5.0 mmol of Na2CO3 and 1.25 mmol of NaBPh4 were added and the reaction mixture was stirred for 2 min. Pd(OAc)2 (5 mol %) was then added and the flask was flushed with Ar, sealed and allowed to stir at 100 °C for 2 h. The resulting reaction mixture was extracted with diethyl ether (2 × 10 ml). The combined ether layers were dried over anhydrous Na2SO4 and concentrated under vacuum to afford a white solid. Recrystallization afforded a 92% yield of a crystalline solid. The product was obtained as white needles: mp 113–114 °C; 1H NMR (300 MHz, CDCl3) δ 3.67 (s, 3H), 7.16–7.39 (m, 13H), 7.80 (d, J = 10.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 31.1, 109.8, 115.3, 119.8, 120.4, 122.4, 125.7, 127.2, 128.2, 128.4, 128.6, 130.1, 131.4, 132.1, 135.4, 137.5, 137.9; IR (neat, cm−1) 3058, 2923, 2849; HRMS calcd for C21H17N 283.13610. Found 283.13690.
n-Butyl (E)-3-[1-methyl-2-phenylindol-3-yl]propenoate (55)
This indole was prepared by the following procedure. Into 1 ml of DMF containing 0.25 mmol of 3-iodo-1-methyl-2-phenylindole and 0.25 mmol of Na2CO3, 0.25 mmol of n-Bu4NCl and 0.525 mmol of n-butyl acrylate were added and the reaction mixture was stirred for 2 min. Pd(OAc)2 (5 mol %) was then added and the flask was flushed with Ar, sealed and allowed to stir at 80 °C for 24 h.23 The resulting reaction mixture was extracted with diethyl ether (2 × 10 ml). The combined ether layers were dried over anhydrous Na2SO4 and concentrated under vacuum to afford a pale yellow oil: 1H NMR (300 MHz, CDCl3) δ 0.94 (t, J = 7.5 Hz, 3H), 1.37–1.44 (m, 2H), 1.62–1.67 (m, 2H), 3.61 (s, 3H), 4.15 (t, J = 6.6 Hz, 2H), 6.48 (d, J = 15.9 Hz, 1H), 7.32–7.40 (m, 5H), 7.50–7.53 (m, 3H), 7.72 (d, J = 16.2 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.0, 19.4, 31.1, 31.3, 64.0, 110.2, 110.6, 113.1, 120.9, 121.9, 123.2, 125.8, 128.8, 129.4, 130.2, 131.1, 138.1, 139.0, 145.6, 168.8; IR (neat, cm−1) 3004, 2963, 2925, 1711; HRMS calcd for C22H23NO2 333.17288. Found 333.17370.
Supplementary Material
General experimental procedures and spectral data for all previously unreported starting materials and products. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We gratefully acknowledge partial financial support from the National Institute of General Medical Sciences (GM070620) and National Institutes of Health Kansas University Chemical Methodologies and Library Development Center of Excellence (P50 GM069663). We thank Johnson Matthey, Inc., and Kawaken Fine Chemicals Co., Ltd. for providing the palladium salts.
References
- 1.For reviews see: Sundberg RJ. Prog Heterocyclic Chem. 1989;1:111.Saxton JE. Nat Prod Rep. 1986;3:357. doi: 10.1039/np9860300353.1987;4:591.1989;6:1.Sundberg RJ. Indoles. Academic Press; New York: 1996. Mori H, Yoshimi N. Mutation Research. 2001;480:201. doi: 10.1016/s0027-5107(01)00200-7.Gribble GW. J Chem Soc Perkin Trans 1. 2000:1045.Toyota M, Ihara M. Nat Prod Rep. 1998;15:327.Lobo AM, Prabhakar S. J Heterocycl Chem. 2002;39:429.Tokuyama H, Fukuyama T. Kagaku Kogyo. 2001;52:416.Xiong WN, Yang CG, Jiang B. Bioorg Med Chem. 2001;9:1773. doi: 10.1016/s0968-0896(01)00070-0.Ezquerra J, Pedregal C, Lamas C. J Org Chem. 1996;61:5804.
- 2.(a) Sonada T, Kawakami M, Ikeda T, Kobayashi S, Taniguchi H. J Chem Soc, Chem Commun. 1976:612. [Google Scholar]; b Kitamura T, Kobayashi S, Taniguchi H, Hori K. J Am Chem Soc. 1991;113:6240. [Google Scholar]; c Kitamura T, Takachi T, Kawasato H, Taniguchi H. J Chem Soc, Perkin Trans 1. 1992:1969. [Google Scholar]
- 3.(a) Larock RC, Yue D. Tetrahedron Lett. 2001;42:6011. [Google Scholar]; b Yue D, Larock RC. J Org Chem. 2002;67:1905. doi: 10.1021/jo011016q. [DOI] [PubMed] [Google Scholar]; c Flynn BL, Verdier-Pinard P, Hamel E. Org Lett. 2001;3:651. doi: 10.1021/ol0067179. [DOI] [PubMed] [Google Scholar]
- 4.(a) Huang Q, Hunter JA, Larock RC. Org Lett. 2001;3:2973. doi: 10.1021/ol010136h. [DOI] [PubMed] [Google Scholar]; b Huang Q, Hunter JA, Larock RC. J Org Chem. 2002;67:3437. doi: 10.1021/jo020020e. [DOI] [PubMed] [Google Scholar]
- 5.(a) Yao T, Larock RC. Tetrahedron Lett. 2002;43:7401. [Google Scholar]; b Yao T, Larock RC. J Org Chem. 2003;68:5936. doi: 10.1021/jo034308v. [DOI] [PubMed] [Google Scholar]; c Oliver MA, Gandour RD. J Org Chem. 1984;49:558. [Google Scholar]; d Biagetti M, Bellina F, Carpita A, Stabile P, Rossi R. Tetrahedron. 2002;58:5023. [Google Scholar]; e Rossi R, Carpita A, Bellina F, Stabile P, Mannina L. Tetrahedron. 2003;59:2067. [Google Scholar]
- 6.Arcadi A, Cacchi S, Fabrizi G, Marinelli F, Moro L. Synlett. 1999:1432. [Google Scholar]
- 7.(a) Bew SP, Knight DW. J Chem Soc, Chem Commun. 1996:1007. [Google Scholar]; b Djuardi E, McNelis E. Tetrahedron Lett. 1999;40:7193. [Google Scholar]; c El-Taeb GMM, Evans AB, Jones S, Knight DW. Tetrahedron Lett. 2001;42:5945. [Google Scholar]; d Rao MS, Esho N, Sergeant C, Dembinski R. J Org Chem. 2003;68:6788. doi: 10.1021/jo0345648. [DOI] [PubMed] [Google Scholar]; e Sniady A, Wheeler KA, Dembinski R. Org Lett. 2005;7:1769. doi: 10.1021/ol050372i. [DOI] [PubMed] [Google Scholar]; f Yao T, Zhang X, Larock RC. J Am Chem Soc. 2004;126:11164. doi: 10.1021/ja0466964. [DOI] [PubMed] [Google Scholar]
- 8.(a) Barluenga J, Trincado M, Rubio E, Gonzalez JM. Angew Chem, Int Ed. 2003;42:2406. doi: 10.1002/anie.200351303. [DOI] [PubMed] [Google Scholar]; b) Yue D, Larock RC. Org Lett. 2004;6:1037. doi: 10.1021/ol0498996. [DOI] [PubMed] [Google Scholar]; c Amjad M, Knight DW. Tetrahedron Lett. 2004;45:539. [Google Scholar]
- 9.Arcadi A, Cacchi S, Di Giuseppe S, Fabrizi G, Marinelli F. Org Lett. 2002;4:2409. doi: 10.1021/ol0261581. [DOI] [PubMed] [Google Scholar]
- 10.Marshall JA, Yanik MM. J Org Chem. 1999;64:3798. [Google Scholar]
- 11.Knight DW, Redfern AL, Gilmore J. J Chem Soc, Chem Commun. 1998:2207. [Google Scholar]
- 12.(a) Barluenga J, Vazque-Villa H, Ballestems A, Gonzaléz JM. J Am Chem Soc. 2003;125:9028. doi: 10.1021/ja0355372. [DOI] [PubMed] [Google Scholar]; b Barluenga J, Vazque-Villa H, Ballestems A, Gonzaléz JM. Org Lett. 2003;5:4121. doi: 10.1021/ol035691t. [DOI] [PubMed] [Google Scholar]; c Yue D, Della Cá N, Larock RC. Org Lett. 2004;6:1581. doi: 10.1021/ol049690s. [DOI] [PubMed] [Google Scholar]
- 13.Ren XF, Konaklieva MI, Shi H, Dickey S, Lim DV, Gonzaléz J, Turos E. J Org Chem. 1998;63:8898. [Google Scholar]
- 14.(a) Asao N, Nogami T, Takahashi K, Yamamoto Y. J Am Chem Soc. 2002;124:764. doi: 10.1021/ja017366b. [DOI] [PubMed] [Google Scholar]; b Nakamura H, Ohtaka M, Yamamoto Y. Tetrahedron Lett. 2002;43:7631. [Google Scholar]; c Yue D, Dellá Ca N, Larock RC. Org Lett. 2004;6:1581. doi: 10.1021/ol049690s. [DOI] [PubMed] [Google Scholar]
- 15.Peng AY, Ding YX. Org Lett. 2004;6:1119. doi: 10.1021/ol0499506. [DOI] [PubMed] [Google Scholar]
- 16.Yao T, Larock RC. J Org Chem. 2005;70:1432. doi: 10.1021/jo048007c. [DOI] [PubMed] [Google Scholar]
- 17.Barluenga J, Trincado M, Marco-Arias M, Ballesteros A, Rubio E, Gonzalez JM. Chem Commun. 2005:2008. doi: 10.1039/b500303b. [DOI] [PubMed] [Google Scholar]
- 18.For a review, see: Nevado C, Echavarren AM. Synthesis. 2005:167.
- 19.Ten Hoedt RWM, Van Koten G, Noltes JG. Synth Commun. 1977;7:61. [Google Scholar]
- 20.(a) Sonogashira K. In: Metal-Catalyzed Cross-Coupling Reactions. Diederich F, Stang PJ, editors. Chapter 5. Wiley-VCH; Weinheim: 1998. pp. 203–229. [Google Scholar]; (b) Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975:4467. [Google Scholar]
- 21.Cadogan JTG, Hickson CL, Husband JB, McNab H. J Chem Soc, Perkin Trans 1. 1985:1891. [Google Scholar]
- 22.Bumagin N, Bykov VV. Tetrahedron. 1997;53:14437. [Google Scholar]
- 23.Jeffery T. J Chem Soc, Chem Commun. 1984:1287. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
General experimental procedures and spectral data for all previously unreported starting materials and products. This material is available free of charge via the Internet at http://pubs.acs.org.