

Abstract
The costimulatory requirements required for peripheral blood T regulatory cells (Tregs) are unclear. Using cell-based artificial APCs (aAPC) we found that CD28, but not ICOS, OX40, 4-1BB, CD27, or CD40-L costimulation, maintained high levels of Foxp3 expression and in vitro suppressive function. Only CD28 costimulation in the presence of rapamycin consistently generated Tregs that consistently suppressed xeno-GVHD in immunodeficient mice. Restimulation of Tregs after 8-12 days of culture with CD28 costimulation in the presence of rapamycin resulted in >1000-fold expansion of Tregs in less than 3 weeks. Next, we determined whether other costimulatory pathways could augment the replicative potential of CD28-costimulated Tregs. We observed that while OX40 costimulation augmented the proliferative capacity of CD28-costimulated Tregs, Foxp3 expression and suppressive function were diminished. These studies indicate that the costimulatory requirements to expand Tregs differ from T effector cells, and furthermore, they extend findings from mouse Tregs to demonstratethat human post-thymic Tregs require CD28 costimulation to expand and maintain potent suppressive function in vivo.
Keywords: Autoimmunity, T cells, Human, Signal Transduction, Cell Activation
Introduction
T regulatory cells (Tregs) play a critical role in maintaining peripheral tolerance, and their loss results in severe autoimmune disease in both humans and in mice (1-3). In murine models of type 1 diabetes (T1D) (4-9), experimental autoimmune encephalomyelitis (10,11), and inflammatory bowel disease (12), adoptive transfer of Tregs has ameliorated autoimmune pathology. In humans, however, generation of therapeutic quantities of Tregs, particularly under GMP-compliant conditions, has proven problematic (13,14). Due to the relative paucity of Tregs, their lack of unambiguous cell surface markers, and their hypoproliferative nature, ex vivo expansions for adoptive transfer will likely start with enriched but not pure populations of Tregs. Since T effector cells have a replicative advantage in most culture systems, low level contamination of input cultures by effector T cells after Treg enrichment presents an obstacle to developing therapeutic adoptive transfer strategies with Treg cultures after several weeks of ex vivo expansion. Thus, determination of the optimal costimulatory requirements of Tregs is essential to develop culture systems useful for therapeutic applications.
Several groups have demonstrated that rapamycin preserves the suppressive function of ex vivo expanded Tregs (15-18). However, the underlying mechanism remains controversial. One study suggested that rapamycin selects for Tregs in culture (19) , while another study suggested that rapamycin induced transient suppressive function in T effector cells (20). Recently, our group demonstrated that Tregs are programmed to be rapamycin-resistant by the constitutive, Foxp3-dependent expression of the serine/threonine kinase pim 2 (21). Thus, Tregs are preferentially selected in rapamycin-containing cultures, due to their decreased susceptibility to rapamycin's blockade of the mTOR pathway. However, Tregs are not completely resistant to the antiproliferative effects of rapamycin, and the overall level of Treg expansion in the presence of rapamycin may be insufficient to achieve therapeutic numbers of Tregs.
Tregs require costimulatory signals to become fully functional. This first became apparent in mice with the observation that spontaneous diabetes is exacerbated in CD28-deficient NOD mice (4,22). Several other studies have addressed the role of CD28 costimulation in Treg formation and function in mice. For example, CD28 costimulation is required both for Foxp3 induction in thymocytes (23) and peripheral homeostasis (24). However, in vivo, peripheral Treg function does not require CD28, nor is CD28 required for the acquisition of inducible Treg suppression in mice (25). Nonetheless, CD28 costimulation also serves as a survival factor for induced Tregs (25,26). CD27 is another costimulatory molecule constitutively expressed on resting naïve T cells and T regulatory cells. In humans, effector T cells tend to lose CD27 during differentiation (27), while functional Tregs maintain CD27 expression (18,28). CD27's ligand, CD70, is overexpressed in lymphomas and blockade of CD70, but not CD80 or CD86, interferes with the induction of Foxp3 in CD4+ CD25− negative human T cells (29).
Tregs also express costimulatory molecules that are induced upon T cell activation (30). The role of OX40 costimulation is perhaps the most perplexing. OX40 costimulation is required to generate Tregs after nitric oxide exposure (31) and it promotes Treg survival in vivo (32). However, other studies indicate that OX40 costimulation exerts a negative effect on Tregs by inhibiting FoxP3 gene expression (33,34) and blocking the generation of IL-10 producing murine Tr1 cells (35). More recently, OX40 costimulation was shown to inactivate Tregs and make effector T cells resistant to the effects of Tregs in a murine tumor model (36). The role of 4-1BB stimulation is also unclear. Engagement of 4-1BB on human Tregs blocks both their function and their expansion (37), and systemic treatment of mice with agonist 4-1BB Abs promotes viral and tumor clearance (38-40). Other studies, however, suggest that 4-1BB costimulation may augment the Treg response. Stimulation via a soluble form of 4-1BBL promotes expansion of murine T regulatory cells (41) and 4-1BB stimulation reduces autoimmune pathology in murine models of arthritis and asthma (42-44). Similarly, 4-1BB costimulation delays the antiviral response when provided early in the immune response (42). However, in these latter studies, the direct effect of 4-1BB stimulation on Treg number and function was not examined. Several studies have addressed the role the CD40-CD40-L costimulatory axis plays in Treg function and development. Both CD40 and CD154 deficient mice have reduced numbers of Tregs in both the thymus and the periphery (45-48) and CD154 blockade results in a loss of Tregs which can be overcome by administration of IL-2. Thus, in mice CD154 costimulation appears to be important for Treg function, but this pathway also promotes both B and T cell effector responses. Loss of ICOS expression results in greatly augmented resistance to mouse Chlamydia infection, due in part to the lack of a robust Treg response (49). A recent study indicated that while CD28 costimulation plays an important role in the generation of T effector and T regulatory responses in mice, ICOS costimulation may play a similar role in sustaining both T effector and regulatory responses (50).
Thus, to date, no single identified costimulatory pathway promotes Treg activity without simultaneously and similarly modulating T effector activity. Rather, the effect a particular costimulatory pathway has on promoting or attenuating the immune response will likely be context-dependent. These factors make the development of agents that predictably modulate Treg activity in vivo challenging. This was unfortunately highlighted by the tragic outcome of administration of superagonistic anti-CD28 Abs to healthy volunteers (51) that was not predicted by studies in rodents. In contrast, adoptive T cell transfer approaches, in which autologous T cells are expanded ex vivo, followed by reinfusion into the patient, provide the opportunity for target cell enrichment and control over the environment in which costimulatory signals are delivered. In this manuscript, we explore the costimulatory requirements for optimal ex vivo expansion of functional human post-thymic T regulatory cells for adoptive T cell therapy. Using a T regulatory cell purification process that is GMP compatible along with cell-based artificial antigen presenting cells (aAPCs) suitable for clinical use, we found that CD28 costimulation was unique in its ability to promote T regulatory cell expansion. Enriched Tregs expanded with CD28 costimulation in the presence of rapamycin consistently maintained high Foxp3 expression in vitro and had potent in vivo suppressive function. In contrast to mouse T cells, other costimulatory pathways tested were unable to promote expansion of functional Tregs, even in the presence of CD28 costimulation and rapamycin. Importantly, by restimulating Tregs with aAPCs that deliver CD28 costimulation after 8-12 days of culture, we were able to consistently expand functional Tregs over 1000-fold. These studies demonstrate that human post-thymic Tregs are dependent upon CD28 costimulation for ex vivo expansion and provide the rationale for the design of culture systems to generate T regulatory cells with sufficient potency for adoptive T cell therapy.
Materials and Methods
Cell Isolation and Purification
Peripheral blood mononuclear cells (PBMCs) were obtained by leukapheresis of healthy volunteer donors by the University of Pennsylvania Human Immunology Core. All specimens were collected under a University Institutional Review Board-approved protocol, and informed consent was obtained from each donor. CD4+ bulk T cells were purified from freshly elutriated human peripheral blood lymphocytes using the CD4 Negative Selection Kit II (Miltenyi Biotec Inc, Auburn, CA) according to the manufacturer recommendations on the AutoMACS cell separator (Miltenyi Biotec Inc). Purified CD4+ T cells were resuspended at a concentration of 25 million per ml in running buffer (PBS containing 2 mM EDTA and 0.5% human serum albumin). 10 μl of anti-human CD127-PE (BD Biosciences, San Jose, CA) were added per 1 ml of cell suspension and the cells were incubated for 15 min at 4°C. The cells were washed once with running buffer, and CD127 positive cells were removed using anti-PE beads (Miltenyi Biotec Inc), following the manufacturer's recommendations. CD4+CD127− cells were resuspended at a concentration of 107 cells/ 80μl of running buffer and 20 μl of CD25 beads (Miltenyi Biotec Inc) per 107 cells were added. After 15 min incubation at 4°C, the cells were washed once, and CD25 positive cells were separated on the AutoMACS using the Possel s program, enriching for the final population of CD4+25+127− cells.
Mice
All animal experiments were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. NOD/scid/IL2Rγcnull (NOG) mice were purchased from the Jackson Laboratory (Bar Harbor, Maine). Animals were also bred in the Animal Services Unit of the University of Pennsylvania. The mice were housed under specific pathogen-free conditions in microisolator cages and given ad libitum access to autoclaved food and acidified water. Animals of both sexes were used for experiments at 6-9 weeks of age.
Preparation of Artificial Antigen-Presenting Cells (aAPCs)
The K562 based aAPC lines K32 (CD32 only), K32.86 (CD32 and CD86), K32.4-1BBL, K32.OX40L, K32.86.4-1BBL, K32.86.OX40L, K64 and K64.86 were previously described (52). K64.ICOS-L, K64.70 and K32.40 were created as follows: ICOS-L (NM_015259) was isolated by PCR from human DC cDNA; CD40 (X60592) was isolated by PCR from a transformed B cell line (Raji) and CD70 (NM_001252) was isolated by PCR from cDNA clone provided by Open Biosystems (Huntsville, AL). All amplified gene products were cloned into the lentiviral vector pCLPS and high titer lentiviral vectors were produced as previously described (53). K562 cells were engineered to express multiple genes by sequential transduction with the appropriate vectors. Single-cell clones of each aAPC were generated using a MoFlo sorter (Dako, Carpinteria, CA). Clones were screened for uniform high expression by flow cytometry with the appropriate Ab (BD Biosciences) as previously described (52). aAPC culture medium consisted of AIM V medium (Gibco BRL/Life Technologies, Grand Island, NY) containing 3% human AB serum (Valley Biomedical, Winchester, VA).
Stimulation and Expansion of Human CD4+ and CD4+CD25+ T Cells
K32-based aAPCs (those using CD32 as a Fc receptor) were lethally irradiated with 100Gy, washed once, and resuspended at 1 × 106 cells/ml in culture medium (X-VIVO 15 medium (LONZA, Walkersville, MD) containing 10 % heat inactivated human serum AB (Valley Biomedical, Winchester, VA) and 0.2% N-acetylcysteine (Ben venue Labs Inc, Bedford, OH). 1 μg/ml of either anti-human CD3 alone (OKT3, Orthoclone, Bridgewater, NJ), or, where indicated, 1 μg/ml of both anti-human CD3 and anti-human CD28 (9.3 MAb) were added to the aAPCs. After incubation for 10 min at 22°C the Ab loaded K32 cells were added to the cultures at a ratio 1 K32 cell: 2 CD4 cells. K64-based aAPCs (those using CD64 as an Fc receptor) were washed and resuspended in serum-free culture medium (X-VIVO 15 containing 0.2% N-acetylcysteine) 24 hours prior to antibody loading. The cells were irradiated with 100Gy and washed, followed by the addition of 1 μg/ml anti-CD3, either alone or in combination with 1 μg/ml anti-CD28. The cells were rotated at 4°C for 30 min, after which unbound antibody was removed by washing three times. Ab-loaded K64 cells were resuspended in serum-free culture medium at a density 1 × 106 cells/ml, and combined with CD4 cells (also in serum-free medium) at a final ratio of 1 K64 cell: 2 CD4 cells.
Where indicated, rapamycin (Calbiochem, San Diego, CA) was added on day 0 to a final concentration of 100 ng/ml. After 24 hrs of culture, human AB serum was added to a final concentration of 10%, while on day 2 of culture, human IL-2 (CHIRON Therapeutics, Emeryville, CA) was added to a final concentration of 300 IU/ml. Cultures were monitored for cell volume and cell density using a Coulter Multisizer 3 (Beckman Coulter, Fullerton, CA) on days 5, 8, 12, 15 and 19 of culture. Following counting, the culture was adjusted to 3 × 105 cells/ml and 300 IU/ml of IL2 was added. 100 ng/ml of rapamycin was added only to the media used to feed the cultures.
In Vitro Suppression Assay
Following harvest of expanded T cells (Tregs and control CD4 cells), varying numbers of T cells were plated in 100 μl of media in round bottom 96 well plates (Corning Incorporated, Corning, NY). Frozen autologous PBMCs were thawed and CFSE-labeled. The 5mM stock solution of CFSE (Invitrogen, Carlsbad CA) in DMSO was diluted 1:100 in PBS. PBMCs were resuspended in PBS containing 5% FCS (PBS-FCS) at a concentration of 1 × 107 cells/ml. 110 μl of CFSE solution in PBS were added per ml of cells, mixed rapidly; after 3.5 min at room temperature 10 volumes of PBS-FCS were added to the suspension, the cells were spun down for 5 min at 300g (20°C), and the supernatant was removed. Cells were washed two more times with PBS-FCS and one more time with the culture medium, then counted. CFSE-labeled PBMCs were resuspended in culture medium at 1×106 /ml, and anti-CD3 beads (Invitrogen Dynal AS, Oslo, Norway) were added at a ratio of 1 bead per cell. 100 μl of PBMC cell suspension (1× 105 cells) was placed to one well in 96 well plate. CFSE-labeled PBMCs without CD3 beads were used as negative controls; CFSE-labeled PBMCs stimulated with anti-CD3 were used as positive controls. The cultures were harvested 4 days later and stained with APC conjugated anti-CD8 antibodies (BD Pharmingen, San Diego, CA). Data were acquired on a FACS Calibur (BD Biosciences) flow cytometer using Cell Quest Pro software and analyzed using FlowJo software (Tree Star Inc., Ashland, OR). For quantitative analysis of Treg suppression capacity the gates were placed on live CD8+ T cells. The amount of cells in each generation was analyzed by using FlowJo software. The percentage of undivided (PU) cells was calculated as percentage of cells in generation 0 at the end of the assay: PU = N (0)/(N (0) + N (1) + …+ N (m))X100. Fold expansion (FE) was calculated as the number of offspring cells at the end of the assay divided by the number of cells at the beginning of the assay: FE = (N (1) + N (2) + …+ N (m))/N estimated (0), where N estimated (0) = N (0) + N (1)/2 + N (2)/4 + …+ N (m-1)/2m−1. In preliminary experiments we found that expanded CD4 bulk T cells show weak suppressive activity at 1:1 and 1:2 dilutions, and that expanded Tregs reproducibly inhibit at 1:8 and 1:16 dilutions. However, Treg activity is difficult to measure precisely at the low end of the titration curve. We have found that the 1:4 Treg:PBMC dilution is the most reproducible dilution for measuring Treg activity in vitro. A full dilution curve from 1:1 to 1:16 was done for each experiment, and for purposes of clarity, only the data for 1:4 dilutions is presented.
In Vivo Assessment of Treg activity to prevent xenogeneic GVHD
Cryopreserved, autologous PBMCs were thawed and a mixture of 1 × 107 PBMCs and 2 × 106 expanded Tregs were injected intraperitoneally into NOG mice. The cells were mixed immediately before injection. Animals were monitored regularly for symptoms of xenogeneic graft-versus-host disease (XGVHD), such as weight loss, ruffled fur, hunched posture, and diminished activity. Peripheral blood specimens were collected at regular intervals by retro-orbital bleeding. The absolute number of human cells per μl of peripheral blood was determined using TruCount tubes (BD Biosciences). Animals were euthanized by CO2 asphyxiation when determined to have advanced XGVHD.
Antibodies, Surface and Intracellular Staining
Anti-CD4-APC (555349), anti-CD8-APC (555369), anti-CD25-PE (555432), anti-CD27-PE (555441), anti-CD32-APC (559769), anti-CD40-PE (555589), anti-CD62L-APC (559772), anti-CD64-FITC (555527), anti-CD70-FITC (555834), anti-CD127-PE (557938), anti-CD137L-PE (559446), anti-OX40L-PE (558164) and anti-ICOSL-PE (552502) were obtained from BD Biosciences, anti-CD25-PE (120-001-311) was purchased from Miltenyi Biotec Inc, and anti-FoxP3-AlexaFluor 488 was purchased from Biolegend (San Diego, CA). Intracellular FoxP3 staining was performed using the FOXP3 Fix/Perm Buffer set (Biolegend) according to the manufacturers' recommendations.
Statistical Analysis
Survival data were analyzed by lifetable methods using log rank analysis performed using SysStat (Systat Software, Inc. San Jose, CA). Other data were analyzed by ANOVA or student's t-test using the same software package. Probability (P) values ≤ .05 were considered statistically significant.
Results
CD28 costimulation is required to consistently obtain expanded cultures enriched for Foxp3 expression
We first developed an efficient process to enrich input populations of polyclonal Tregs for culture that could be used to identify conditions for optimal costimulation. Starting with PBMCs, our yield of >95% pure CD4+ CD25+ cells was ∼0.1%. 60-85% of the cells in this enriched population expressed the Treg master transcription factor Foxp3 (Fig. 1A). We also developed genetically defined cell-based aAPCs that rapidly expand primary human effector CD4 (54) and CD8 T cells (55). More recently, we used lentiviral vectors to engineer aAPCs (52) to express costimulatory molecules in combinatorial sets. Here, K562 cells were transduced with lentiviral vector(s) to express CD86 and/or an Fc binding receptor (CD32 or CD64). The Fc receptor permits the loading of anti-CD3 Abs onto the aAPC. Single-cell clones were isolated by sorting, expanded, and characterized for CD64 and CD86 expression (K64.86) (Fig. 1B). We have previously shown that lentiviral vector-engineered K562-based aAPCs stably express introduced molecules without the need for selection agents, and equivalent T cell proliferation is observed whether CD32 or CD64 was used to load anti-CD3 (52). The advantage of using genetically defined cell-based aAPCs as opposed to natural APCs is that we can study the influence of particular costimulatory pathways in isolation.
Figure 1. CD28 costimulation is required to consistently obtain expanded cultures enriched for Foxp3 expression.

A. Analysis of CD25 expression in purified CD4 T cells (Left panel), CD25 (Middle Panel) and Foxp3 (Right Panel) expression after CD127 depletion and CD25 selection of CD4 T cells. This data is representative of the enrichment of all input Treg populations used in this study. B. Expression of CD64 and CD86 was analyzed on K64 (Left panel) and K64.86 (Right panel) aAPCs by flow cytometry. C. 200,000 enriched Tregs were stimulated with anti-CD3 Ab loaded K32 aAPCs, and anti-CD3 loaded K32.86 aAPCs cultured in the presence and absence of rapamycin (RAPA) for two weeks and population doubling rate was measured by cell counting. Each data point represents the average of four independent experiments (error bars represent standard deviation). D and E Analysis of Foxp3 (D) or CD27 and CD62-L (double positive) (E) expression on Tregs expanded with anti-CD3 Ab loaded K32 or K32.86 aAPCs in the presence of rapamycin. The panels on the left show a representative experiment and the box plot on the right is data compiled from 4 independent experiments.
Previous studies have shown that CD28 is required for the development of Tregs in the thymus (23). First we evaluated whether costimulation was required for human post-thymic Treg expansion. We have previously shown that anti-CD3-loaded cell-based aAPCs deliver a more potent signal to T cells than plate- or bead-bound anti-CD3 (54), so we asked whether cell-based anti-CD3 stimulation, in the presence of exogenous IL-2, was sufficient to promote Treg expansion. While rapamycin preserves human T regulatory activity (15-18), it also limits the extent of Treg expansion. Therefore, we tested whether Treg expansion, either in the presence or absence of rapamycin, required costimulatory molecules. In cultures stimulated by cell-bound anti-CD3 Ab alone, a considerable lag time preceded Treg expansion, and addition of rapamycin markedly reduced this already modest level of expansion (Fig. 1C). In contrast, Treg cultures that received CD28 costimulation via either anti-CD28 Ab (data not shown) or CD86 (K32.86) entered exponential growth phase upon stimulation. However, inclusion of rapamycin in these cultures resulted in a 5-10 fold reduction in Treg expansion (Fig 1C). Rapamycin is reported to select for Foxp3+ cells (15-17). Therefore, it was surprising to observe (Figs 1D and 1E) that rapamycin in the absence of CD28 costimulation was insufficient to select for cells expressing Foxp3 and naïve markers that have been correlated to potent Treg activity (6,18,28,56). Collectively, these studies indicate that both CD28 costimulation and rapamycin are required to consistently maintain the Treg phenotype during ex vivo expansion. However, in ∼25% of the cultures expanded using CD28 costimulation, rapamycin was not required to maintain high levels of Foxp3 expression and in vitro suppressive function (data not shown), indicating that in some donors, Tregs can be expanded without rapamycin, providing sufficient CD28 costimulation is present. The experimental basis for the donor to donor heterogeneity vis a vis the requirement for rapamycin remain unclear, however, for consistent generation of suppressive T regs by ex vivo expansion, the routine incorporation of rapamycin seems justified.
Cell-based aAPCs expand Tregs more efficiently than bead-based aAPCs
Several groups, including ours, have expanded Tregs using anti-CD3/28 coated beads as aAPCs (57-61). However, antibody-coated beads are not an ideal platform for determining whether additional costimulatory signals can augment Treg expansion and function. Natural ligands are difficult to attach to the beads, and standardizing the amount of each Ab added to the bead is difficult. Moreover, clinical applications would require production of GMP antibody lots, a time-consuming and expensive process. Therefore, we compared the ability of anti-CD3/28-coated beads (CD3/28 BD) and anti-CD3 Ab loaded K562-based aAPCs to expand Tregs. In the absence of rapamycin, there was no statistically significant difference in the ability of the bead-and cell-based aAPCs to expand input populations of Tregs (Fig. 2A). It should be noted, however, that the majority of these cultures lost their suppressive phenotype and function (data not shown). However, in the presence of rapamycin, cell-based aAPCs expanded Tregs to a much greater degree than bead-based aAPCs (Figs 2A and B). In contrast, our previous studies of bulk human CD4 cells cultured in the absence of rapamycin, bead and K562 based aAPCs were equivalent in the ability to promote the expansion of effector CD4 T cells (54). We observed no statistically significant difference (p=0.2) in Foxp3 expression in cells expanded with anti-CD3/28 coated beads (59% ± 18%) and anti-CD3-loaded K64.86 aAPCs (66± 23%) (Fig. 2C). However, it should be noted that in every experiment (n >5), we observed more Foxp3+ cells in cultures expanded with the cell-based aAPC vs. beads (data not shown). Thus in the presence of rapamycin, cell based aAPCs promote more efficient expansion of human Tregs than anti-CD3/28 Ab coated beads.
Figure 2. CD28 ligands on cell-based aAPCs expand Tregs more efficiently than bead-based aAPCs.

A. 2×105 enriched Tregs were stimulated with either CD3/28 Ab coated beads or anti-CD3 loaded K64.86 aAPCs and cultured in the presence or absence of rapamycin for two weeks. Expansion was measured as described in the Materials and Methods. B. Box plot showing fold expansion for each culture condition using data collected from four donors. Fold expansion was determined by dividing the total number of cells at the end of culture by the initial starting number stimulated by the indicated aAPC in the presence or absence of rapamycin. C. Analysis of Foxp3 and CD25 expression in Tregs expanded with either CD3/28 Ab coated beads (BD) or anti-CD3 loaded K64.86 aAPCs. Data is representative of four independent experiments.
CD27, 4-1BB, CD40L, OX40 or ICOS costimulation cannot substitute for CD28 costimulation to promote Treg expansion
Little is known about how costimulatory molecules other than CD28 affect the expansion and function of adult human post-thymic Tregs. In the mouse, OX40 contributes to efficient Treg-mediated suppression. However, T effectors became insensitive to Treg-mediated suppression when they were exposed to OX40L-expressing cells (62). Furthermore, ICOS signaling is required to maintain tolerance in the NOD mouse (63). To determine whether other costimulatory molecules could substitute for CD28 or augment the ability of CD28 to expand Tregs, we created aAPCs expressing the ligands for CD154, CD27, 4-1BB, OX40, and ICOS by transducing CD32 or CD64 expressing K562 cells with lentiviral vectors expressing each of the costimulatory ligands. Each aAPC was loaded with anti-CD3 antibody, and used to expand Tregs in the presence of rapamycin. Only CD28-costimulated Tregs did not undergo a lag phase before expansion (Fig 3A). Importantly, only those Tregs expanded in the presence of CD28 costimulation maintained high levels of Foxp3 expression and naïve T cell markers (CD27 and CD62L) (Fig 3B). These data suggest that CD28 costimulation is required to augment the expansion of Tregs and this signal for Treg expansion cannot be delivered by the other costimulatory pathways that were tested.
Figure 3. CD27, 4-1BB, CD40L, OX40 or ICOS costimulation cannot substitute for CD28 costimulation to promote Treg expansion.

A.2×105 enriched Tregs were stimulated with a cell-based aAPC that delivered the indicated costimulatory signal and cultured the presence of rapamycin. The population doubling rates were measured by cell counting. Each data point represents the average of three independent experiments (error bars represent standard deviation). B and C Analysis of the cell populations expanded in A. after 14 days culture for Foxp3 (B) and CD62-L and CD27 (C) expression.
CD28 costimulation and rapamycin are required to consistently obtain cultures that suppress in vitro
Tregs are defined functionally by their ability to suppress immune responses. In the preceding experiments we used the level of expression of Foxp3 and Treg-related cell surface markers to demonstrate that CD28 costimulation and rapamycin maintain a Treg-like phenotype. Next, we determined whether both CD28 and rapamycin are required for Treg function using an in vitro suppression assay. Autologous PBMCs were labeled with CFSE and stimulated with anti-CD3 coated beads. Ex vivo-expanded Tregs were added at various dilutions and their ability to suppress CD8+ T cell expansion was measured. To quantify the ability of expanded Tregs to suppress, we determined the extent of CD8+ T cell suppression by counting the number of daughter cells and dividing by the number of input cells. Thus, in the absence of added expanded Tregs, we observed a 5.5 fold expansion of CD8+ T cells during 4 days of culture (Fig 4A). Tregs expanded by anti-CD3 stimulation alone, either with or without rapamycin, only had modest (20%) inhibitory activity on the CD8+ T cell proliferation. In contrast, Tregs expanded with CD28 costimulation (either by Ab engagement or CD86 ligation) exhibited significant suppressive activity, reducing CD8 T cell expansion by an order of magnitude. The inhibitory properties of these Tregs were further augmented by rapamycin addition. We compared the ability of rapamycin to augment the functional activity of CD28-costimulated Tregs in 11 donors. As shown in Fig 4B, CD28 costimulated Tregs expanded in the absence of rapamycin maintained a high level of functional activity in only a subset of the cultures. However, in all cases rapamycin augmented the functional activity of CD28-costimulated Tregs. Importantly, Tregs from all of the donors expanded by CD28 costimulation in the presence of rapamycin displayed robust functional activity (Fig 4B). Next, we determined whether CD28 costimulation was required for functionally active Tregs, or whether expanding the cells in rapamycin would be sufficient. Here we compared 5 donors (Fig. 4C). In addition to proliferating to ∼ 10 fold higher levels (Fig 2A), Treg cultures expanded using CD28 costimulation in the presence of rapamycin displayed more functional activity than cultures expanded in the presence of rapamycin but in the absence of CD28 costimulation. Thus, the culture conditions that yielded Tregs expressing high levels of Foxp3 and naïve T cell markers also yielded Tregs with the highest functional activity (compare Fig 1D and 1E to Fig 4C). This correlation between Treg marker expression and Treg functional activity was also observed in Treg cultures expanded by costimulatory molecules other than CD28. None of these cultures shown in Fig 3 functioned as well as CD28 costimulated Tregs (data not shown, see Fig 6B). Thus, both CD28 costimulation and rapamycin are required for optimal and consistent expansion of functional Tregs.
Figure 4. Consistent suppression in vitro by Tregs expanded with CD28 costimulation in the presence of rapamycin.

A. Tregs were expanded using the indicated cell-based aAPC. In a suppression assay, autologous PBMCs were labeled with CFSE and stimulated using anti-CD3 Ab coated beads and mixed with either no Tregs (top panel) or expanded Tregs at a ratio 1:4 (Treg:PBMC). Histograms show the expansion of CD8+ cells. Fold expansion and percent suppression (parentheses) are indicated in the upper left hand corner. B and C. Scatter plots showing the degree by which CD28 costimulated Tregs expanded with and without rapamycin (B) or by which anti-CD3 Ab or anti-CD3 and CD28 costimulated Tregs expanded in rapamycin (C) suppress CD8+ T cell proliferation. Each point represents a separate culture; the average (+/− s.d ) suppressive activity is indicated. Paired T test was performed to determine the significance of differences between each group.
Figure 6. OX40 costimulation promotes CD28-mediated T regulatory expansion in the presence of rapamycin but often results in loss of suppressive activity.

A. 2×105 enriched Tregs were stimulated with cell-based aAPCs that delivered the indicated costimulatory signals in the presence of rapamycin and the population doubling rate was measured by cell counting. Each data point represents the average of three independent experiments (error bars represent standard deviation). B and C. Scatter plots of percent of Foxp3 expressing cells (B) and in vitro suppressive activity (C) measured after two weeks of culture after stimulation with the indicated cell-based aAPC. Each dot represents a separate experiment and cell donor. Mean values and standard deviation are also indicated.
T regulatory cultures expanded with CD28 and rapamycin are able to prevent xenogeneic GVHD
Tregs have been posited to control T effector responses by many mechanisms, including cytokine starvation, production of IDO, TGF-β, IL-10, IL-35 or by directly killing effectors (64). Presently, it is not clear which mechanism(s) predominates in vivo. Furthermore, it is unclear if in vitro suppression assays predict in vivo Treg function. To test whether CD28 costimulated Tregs could function in vivo we employed a xenogeneic acute GVHD model in which we could test whether expanded, autologous Tregs could block GVHD induced by human PBMC transferred into NOD/scid/IL2Rγcnull (NOG) mice. Enriched Treg cells and bulk CD4 T control cells were expanded by anti-CD3 loaded K64.86 aAPCs in the presence or absence of rapamycin. Interestingly, rapamycin-treated bulk CD4 T cells and non-rapamycin treated enriched Treg cultures had equivalent levels of Foxp3 expression at the end of culture. Enriched Tregs expanded with rapamycin had substantially more Foxp3-expressing cells whereas bulk CD4 T cells expanded without rapamycin had the least number of Foxp3-expressing cells (Fig 5A). We tested these cells for their ability to suppress in vitro CD8 T cell expansion (Fig 5B). Surprisingly, CD4 T cells expanded in the presence of rapamycin had significantly more in vitro suppression than CD4 T cells expanded in the absence of rapamycin. As previously shown in Fig 4, the functional activity of enriched Tregs expanded in the absence of rapamycin was variable whereas the functional activity of enriched Tregs expanded with rapamycin was consistent and robust (Fig 5B). Two million expanded T cells from each culture shown in Fig 5A were mixed with 10 million autologous PBMCs and injected into NOG mice. 3 weeks later the mice were bled and the number of human CD8+T cells present in the peripheral blood was determined. Importantly, only enriched Tregs expanded in the presence of rapamycin were able to prevent the expansion of human CD8 T cells (Fig 5C, note log scale of Y-axis). We also monitored GVHD by clinical examination and survival analysis (Fig 5D). There was no survival benefit conferred by bulk CD4 T cells grown in rapamycin or by enriched Tregs expanded in the absence of rapamycin. However, enriched Tregs expanded in the presence of rapamycin protected NOG mice from lethal xeno GVHD, indicating that these ex vivo expanded Treg cells can function in vivo. It should be emphasized that rapamycin was not given to the mice, indicating that Tregs expanded with rapamycin retain in vivo function in the absence of rapamycin. In addition, these results show that the results from in vitro suppression assays do not necessary correlate with in vivo GVHD assays, particularly in the case of bulk CD4 T cells which had low level but consistent suppressive activity after culture in rapamycin, while failing to suppress in vivo. Finally, the level of CD8+ T cells measured in the peripheral blood of the mice correlated well with survival (compare Fig. 5C and 5D).
Figure 5. In vivo prevention of xeno GVHD of T regs expanded with CD28 and rapamycin.

A. A Starting cell population (SCP) of 2×105 CD4 bulk T cells (CD4) or enriched Tregs (CD25+) were stimulated with K64.86 aAPCs in the presence or absence of rapamycin. After two weeks of culture, Foxp3 (filled) expression and isotype control Ab (open) staining was measured by flow cytometry. B. In vitro suppression of each population described in A was measured as described in Fig4. Data represents four independent experiments. C. 2 million cells from each culture shown in A. were mixed with 10 million autologous PBMCs and injected into NOG mice (6 mice per group). After 8 weeks, the mice were bled and the number of human CD8+ T cells/μl of blood was determined. D. Kaplan-Meier Survival Analysis (Log-Rank) was performed with the indicated cohorts of mice. The Holm-Sidak method for multiple comparisons (significance level = 0.05) was performed in all groups and significant differences were found between in the mice that were treated with enriched Tregs expanded in the presence of rapamycin and all other groups.
OX40 costimulation promotes CD28-mediated T regulatory expansion in the presence of rapamycin but often results in loss of suppressive activity
As shown above in Fig 1, Tregs underwent 6-7 population doublings (100-300 fold) after a single round of CD28 costimulation in the presence of rapamycin. This amount of expansion may be insufficient for therapeutic applications, as studies with effector CD4 T cells have found that infusions of 1×109 to 1×1010 cells are required to affect CD4 T cell homeostasis (13). To determine if additional costimulatory pathways could augment CD28-mediated Treg expansion in the presence of rapamycin, we created aAPCs expressing CD86 and ICOS-L, CD40, OX40-L, 4-1BBL or CD70. Of these, only K64.86.OX40-L aAPCs consistently expanded Treg cultures in the presence of rapamycin 5-10 fold greater than CD28 costimulation alone (Fig 6A and data not shown). However, OX40 costimulation appeared to have a dominant effect over CD28 costimulation in that it reduced the number of Foxp3+ cells (Fig 6B) and the in vitro suppressive activity of these cells (p = 0.08) (Fig 6C). Thus, consistent with the results in Fig. 3, engagement of additional costimulatory pathways in Tregs provides no benefit to CD28 costimulation and hinders the ability of CD28 costimulatory to promote the expansion of Tregs.
Distinct restimulation requirements of effector and T regulatory cells
Once recruited into the cell cycle, T cells require repeated cycles of exposure to antigen and rest in order to maintain exponential expansion. For human CD8+ T cells, the cultures require restimulation at weekly intervals (65), while effector CD4 T cells require less frequent restimulation at 2-3 week intervals (66). Moreover, if effector cells are restimulated prematurely, they undergo activation induced cell death (AICD). Thus, the timing of restimulation is crucial to optimize T cell expansion. It is unclear whether Tregs follow the same rules of ex vivo expansion as T effector cells. We stimulated enriched Tregs in the presence of rapamycin using anti-CD3 loaded K64.86 aAPCs and monitored cell volume and and expansion using a Coulter Counter. When the Tregs stopped expanding exponentially (Day 8), the culture was split, with a portion being restimulated by anti-CD3 loaded K64.86 aAPCs while the remaining cells were maintained undisturbed with continued culture in IL-2. We observed that the restimulated Tregs re-entered exponential growth phase and continued to expand until Day 20. Superficially at least, the culture requirements for Tregs appear more to resemble CD8+ T cells rather than CD4 effector T cells with respect to the timing of restimulation. Using this modified approach, Tregs were consistently expanded to greater than 10 population doublings (1000-fold) (Fig 7A). Next, we examined Foxp3 expression in Treg cultures receiving one and two stimulations. Restimulated Tregs reproducibly had more Foxp3+ cells (Fig 7B). The mechanism behind this observation is unclear presently, but it may indicate that residual contaminating effector cells from the input population were killed by being restimulated too early. Importantly, twice -stimulated Tregs were able to function as well as singly stimulated Tregs in vitro and in vivo by protecting from GVHD lethality (Fig 7C and D).
Figure 7. Restimulation of T regulatory cells enables greater than 1000 fold expansion without loss of in vitro or in vivo suppressive activity.
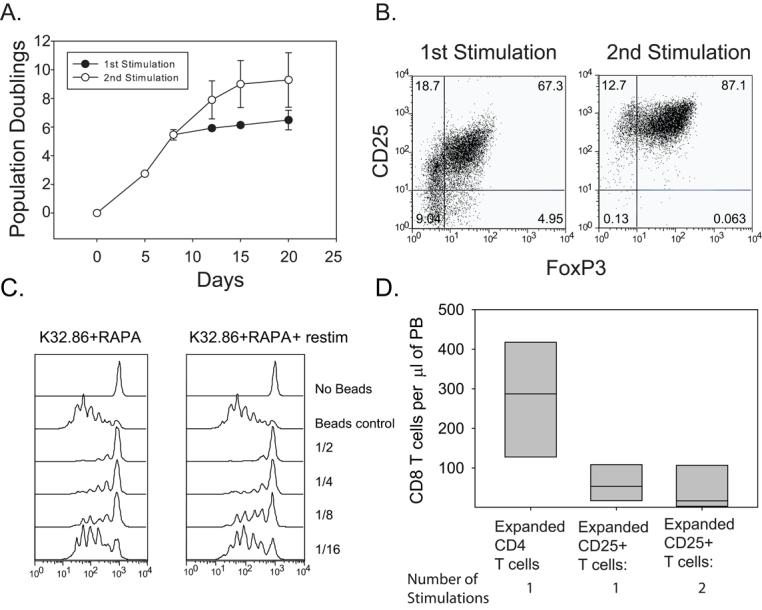
A. 2×105 enriched Tregs were stimulated by K64.86 aAPCs and expanded in the presence of rapamycin. Population doubling rate was measured by cell counting. Each data point represents the average of three independent experiments (error bars represent standard deviation). When cell expansion reached the plateau phase, the culture was restimulated with K64.86 cell-based aAPCs (open circles) or left alone (filled in circles). B. Analysis of Foxp3 and CD25 expression in enriched Tregs stimulated with K64.86 aAPCs once or twice in the presence of rapamycin after 14 days of culture. Data is representative of four independent experiments. C. The in vitro suppressive activity of cell populations shown in A and B was measured by mixing CFSE-labeled autologous PBMCs, anti-CD3 Ab coated beads, and the indicated ratio of expanded Tregs and PBMCs. After 4 days of culture, CFSE dilution was measured by flow cytometry. D. 2×106 cells from each culture shown in A. were mixed with 1×107 autologous PBMCs and injected into NOG mice (6 mice per group). After 8 weeks, the mice were bled and the number of human CD8 T cells/μl of blood was determined.
Discussion
Based on results from numerous studies in mice (1), human Tregs have considerable therapeutic potential in adoptive transfer settings (14), provided that they can be generated in therapeutic quantities. Here we compared the ability of K562 cell-based and bead-based aAPCs expressing a panel of costimulatory ligands to expand functional Tregs from enriched but still impure starting populations. In the absence of rapamycin, we observed an overall decline in the frequency of Foxp3-expressing cells in both cell- and bead-based culture systems, suggesting that both systems preferentially expanded T effectors rather than Tregs. In the presence of rapamycin, however, cell-based aAPCs expanded functional Treg populations more efficiently than bead-based aAPCs. Several mechanisms could account for the superiority of the cell based APC culture approach. Our previous data showed that Foxp3 expression promotes pim2 kinase expression, which mediates rapamycin resistance (21). Thus, if cell-based aAPCs induced higher levels of Foxp3 than bead-based aAPCs, greater pim2-mediated rapamycin resistance would lead to increased Treg proliferation. However, we were unable to observe a significant difference in the level of Foxp3 expression or in the number of Foxp3-expressing cells between bead-based and cell-based aAPC-stimulated cultures (Fig 2). However, it is possible that the expression level or stability of other molecules associated with Foxp3 and pim2 may be promoted by cell-based aAPC. For example, the expression of TIP60 and HDAC7 may differ, as Foxp3 must form higher order complexes with these molecules in order to inhibit IL-2 production (67). If stimulation with cell-based aAPCs promoted or stabilized the formation of these complexes within Tregs more efficiently than stimulation with bead-based aAPCs, then this could explain why more Foxp3 activity i.e. rapamycin resistance via pim 2 expression, is not revealed simply by absolute measurements of Foxp3 levels.
Regardless of the precise mechanism, our observation that cell-based aAPCs promote the expansion of Tregs in the presence of rapamycin has important therapeutic implications. If rapamycin is included in the culture system to expand Tregs, it will be advantageous to use cell-based aAPCs delivering CD28 costimulation either by anti-CD28 Ab or CD86. While cell-based aAPCs are superior to bead-based aAPCs or soluble anti-CD28 antibody (data not shown) in expanding rapamycin-resistant Tregs, they possess other advantages for expansion of Tregs. K562 cell-based aAPCs express multiple adhesion molecules, facilitating aAPC-T cell interactions. Moreover, these aAPCs participate in T cell/APC crosstalk by upregulating multiple modulators of T cell activation (54). Since they are lethally irradiated, K562-based aAPCs disappear from culture after ∼5 days, whereas bead-based aAPCs must be removed from culture at the time of cell harvest by passage through magnetic fields or other procedures. Furthermore, the self-replicating nature of K562-based aAPCs likely makes them more cost-effective than bead based aAPCs. Lastly, irradiated GM-CSF-expressing K562 cells were incorporated in a cancer vaccine protocol (68), suggesting that K562-based aAPCs can be used safely in humans
The use of cell-based aAPCs facilitated the identification of culture conditions for optimal expansion of functional Tregs. T cell activation requires two signals: one signal is delivered through the TCR/CD3 complex and the second is delivered by one or more members of the costimulatory receptor families. To deliver signal one, we have used Fc receptors to bind anti-CD3 Ab. Both CD32 and CD64 bind anti-CD3 Ab to promote Treg expansion, however, CD64's higher affinity for Fc allows excess Ab to be washed away prior to use, reducing the amount of anti-CD3 Ab infused into patients and the likelihood of inducing human anti-mouse Ab (HAMA) responses after multiple T cell infusions. Thus, CD64 is superior for clinical applications. Starting from enriched but not pure Treg populations, we found that CD27, CD40-L, 4-1BB, OX40 and ICOS costimulation all failed to support Treg expansion. Multiple mechanisms could account for these results, including inactivation of Tregs or conversion of Tregs to T effectors. Alternatively, these costimulatory pathways could favor expansion of T effectors at the expense of Tregs. We favor the latter interpretation because our studies using Tregs purified from cord blood (CB Tregs) revealed that in addition to CD28 costimulation, 4-1BB and OX40 costimulation significantly augmented CB Treg expansion and function (unpublished data). Because CB CD4 T cells are antigen-inexperienced and resting, highly enriched Treg populations can be selected by simply isolating CD4+CD25+ cells (58). Importantly, rapamycin was not required to maintain Foxp3-expressing, functional CB Tregs. While these findings could reflect fundamental growth differences between CB and peripheral blood Tregs, they more likely indicate that if sufficiently pure populations of Tregs can be obtained from any source, costimulatory pathways such as OX40 and 4-1BB will promote Treg expansion, and rapamycin might not be required to check T effector replication. However, in peripheral blood derived Treg populations containing T effectors, costimulatory pathways other than CD28 promote the expansion of T effectors more efficiently than they promote the expansion of Tregs and therefore are not optimal for the reproducible expansion of Tregs .
In contrast to other costimulatory molecules, CD28 costimulation proves necessary for peripheral blood Treg expansion, as enriched Treg populations are unable to maintain their Treg phenotype (Foxp3 and naïve T cell marker expression) or suppressive activity in the absence of CD28 costimulation, even when expanded in the presence of rapamycin. The CD28-dependent signaling pathways that promote Treg expansion are currently unknown. However, the inability of ICOS costimulation to promote Treg expansion provides some clues. In CD4 T cells, both CD28 and ICOS costimulation induce similar genetic programs that transition resting T cells to activated effectors. However, differential regulation of several transcripts, including IL-2, ICOS, IL-9, MAL, and MYO1F is observed, suggesting the existence of both overlapping and distinct pathways (69). One shared pathway is PI3K activation (53) which likely rules this pathway out as being solely required for Treg expansion. Additionally, Singer and colleagues have identified a proline-rich sequence within the murine CD28 cytoplasmic tail that is crucial for thymic Treg differentiation (23). It will be interesting to determine if this motif is required for human Treg expansion ex vivo and if so, what signaling molecules are recruited to this motif.
In all of our culture experiments using enriched peripheral blood Tregs (n > 20) we found that CD28 costimulation in the presence of rapamycin resulted in the expansion of functional Tregs. However, in the absence of rapamycin, CD28 costimulation did not consistently yield functional Tregs. There has been some debate as to the mechanism(s) by which rapamycin maintains the Treg phenotype in culture. One study indicated that in the presence of rapamycin Tregs preferentially expand at the expense of T effectors (15) whereas another study suggested that rapamycin temporally imparts Treg-like activity to effector cells, and upon rapamycin removal, the cells revert to an effector phenotype (20). Resolving this controversy is crucial, because if rapamycin only produces “pseudo Tregs”, then its therapeutic utility is suspect. Our data help clarify this issue. We previously demonstrated that Foxp3 expression induces pim2 (21), a serine/threonine kinase that mediates rapamycin resistance (70). Thus, Tregs are programmed to preferentially expand in the presence of rapamycin. In the present study, we observed that CD4 T cells, when expanded in the presence of rapamycin, exhibited suppressive activity in vitro (Fig 5). However, when the function of these cells was assayed in an in vivo mouse model, these cells failed to prevent xenogeneic-GVHD. We interpret these data to indicate that rapamycin can induce a suppressive phenotype, albeit transient, in effector T cells. These observations highlight the dangers of relying solely on in vitro suppression assays to measure Treg activity. Thus, our data show that rapamycin can simultaneously select for Tregs and confer a transient Treg-like phenotype upon T effector cells. An important implication of this dual effect of rapamycin is that Tregs must be sufficiently purified (∼60% Foxp3 expressing cells) prior to initiating culture. Insufficient purity may result in the predominance of rapamycin-induced “pseudo Tregs” within the culture.
The lack of a unique cell surface marker to identify Tregs and a clear understanding of how they function to control immune responses in vivo has hindered the development of Treg therapies. While Foxp3 is the best marker, it has limitations. For one, it is a transcription factor and there is currently no way to measure its expression in live cells. Secondly, especially in humans, Foxp3 expression has not always correlated with suppressive activity. Upon T cell activation Foxp3 is transiently induced (71-73) and this induction does not result in suppressive activity (74). More striking is the ability of TGF-β treated human T effectors to express high levels Foxp3 for sustained periods of time yet have absolutely no suppressive activity (75,76). This disconnect between Foxp3 expression and suppressive activity may reflect specific modifications of the Foxp3 promoter (77) or the higher order ensembles Foxp3 forms with other molecules within the cell (78). In any case, the definition of a Treg remains a functional one. Here, we show that in a xenogeneic-GVHD mouse model provides a robust measurement of Treg activity, as it accurately distinguishes between “pseudo Tregs” (control bulk CD4 T cells expanded with rapamycin) and true Tregs. It should be noted again that no rapamycin was given to the mice, suggesting that functional Tregs expanded with rapamycin retain a suppressive phenotype in the absence of rapamycin.
In conclusion, our data indicate that CD28 provides an essential signal for mature Tregs, extending its role previously demonstrated in the mouse (4). Human enriched peripheral blood Tregs can be expanded over 1000-fold within three weeks of ex vivo culture in the presence of rapamycin by two stimulations using cell-based aAPCs that deliver CD28 costimulation. These cells retain potent in vitro and in vivo suppressive function and are an attractive population of cells for the prevention and perhaps treatment of GVHD and autoimmunity in humans.
Acknowledgments
We thank the CFAR Immunology Core for providing apheresis product; Cory Waters and Tim Fong for helpful suggestions and Chanelle Case, Kathleen Haines and Ben Paramonte for technical assistance in maintaining the cell-based aAPCs, Dr. Gwenn Danet-Desnoyers, Tony Secreto, and the rest of the Stem Cell and Xenograft Core staff for maintaining the mouse colony and assistance with the GVHD model.
Supported by JDRF Collaborative Center for Cell Therapy, a sponsored research grant from Becton, Dickinson & Company, R01CA105216, R01AI057838, R37HL56067, P01CA670493, and a translational grant (# 6220) from the Leukemia and Lymphoma Society.
References
- 1.Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, Shimizu J, Takahashi T, Nomura T. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev. 2006;212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x. [DOI] [PubMed] [Google Scholar]
- 2.Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- 3.Bluestone JA, Abbas AK. NATURAL VERSUS ADAPTIVE REGULATORY T CELLS. Nat Rev Immunol. 2003;3:253–257. doi: 10.1038/nri1032. [DOI] [PubMed] [Google Scholar]
- 4.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 5.Szanya V, Ermann J, Taylor C, Holness C, Fathman CG. The Subpopulation of CD4+CD25+ Splenocytes That Delays Adoptive Transfer of Diabetes Expresses LSelectin and High Levels of CCR7. J Immunol. 2002;169:2461–2465. doi: 10.4049/jimmunol.169.5.2461. [DOI] [PubMed] [Google Scholar]
- 6.Tarbell KV, Petit L, Zuo X, Toy P, Luo X, Mqadmi A, Yang H, Suthanthiran M, Mojsov S, Steinman RM. Dendritic cell-expanded, islet-specific CD4+ CD25+ CD62L+ regulatory T cells restore normoglycemia in diabetic NOD mice. J. Exp. Med. 2007;204:191–201. doi: 10.1084/jem.20061631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.PICCIRILLO CA, TRITT M, SGOUROUDIS E, ALBANESE A, PYZIK M, HAY V. Control of Type 1 Autoimmune Diabetes by Naturally Occurring CD4+CD25+ Regulatory T Lymphocytes in Neonatal NOD Mice. Ann NY Acad Sci. 2005;1051:72–87. doi: 10.1196/annals.1361.048. [DOI] [PubMed] [Google Scholar]
- 8.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In Vitro-expanded Antigen-specific Regulatory T Cells Suppress Autoimmune Diabetes. J. Exp. Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lundsgaard D, Holm TL, Hornum L, Markholst H. In vivo control of diabetogenic T-cells by regulatory CD4+CD25+ T-cells expressing Foxp3. Diabetes. 2005;54:1040–1047. doi: 10.2337/diabetes.54.4.1040. [DOI] [PubMed] [Google Scholar]
- 10.Kohm AP, Carpentier PA, Anger HA, Miller SD. Cutting Edge: CD4+CD25+ Regulatory T Cells Suppress Antigen-Specific Autoreactive Immune Responses and Central Nervous System Inflammation During Active Experimental Autoimmune Encephalomyelitis. J Immunol. 2002;169:4712–4716. doi: 10.4049/jimmunol.169.9.4712. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, Weiner HL. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int. Immunol. 2004;16:249–256. doi: 10.1093/intimm/dxh029. [DOI] [PubMed] [Google Scholar]
- 12.Mottet C, Uhlig HH, Powrie F. Cutting Edge: Cure of Colitis by CD4+CD25+ Regulatory T Cells. J Immunol. 2003;170:3939–3943. doi: 10.4049/jimmunol.170.8.3939. [DOI] [PubMed] [Google Scholar]
- 13.June CH, Blazar BR. Clinical application of expanded CD4+25+ cells. Semin. Immunol. 2006;18:78–88. doi: 10.1016/j.smim.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Bluestone JA, Thomson AW, Shevach EM, Weiner HL. What does the future hold for cell-based tolerogenic therapy? Nat Rev Immunol. 2007;7:650–654. doi: 10.1038/nri2137. [DOI] [PubMed] [Google Scholar]
- 15.Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J. Immunol. 2007;178:320–329. doi: 10.4049/jimmunol.178.1.320. [DOI] [PubMed] [Google Scholar]
- 16.Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J. Immunol. 2006;177:8338–8347. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- 17.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–4748. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 18.Coenen JJ, Koenen HJ, van RE, Hilbrands LB, Joosten I. Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood. 2006;107:1018–1023. doi: 10.1182/blood-2005-07-3032. [DOI] [PubMed] [Google Scholar]
- 19.Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective Survival of Naturally Occurring Human CD4+CD25+Foxp3+ Regulatory T Cells Cultured with Rapamycin. J Immunol. 2007;178:320–329. doi: 10.4049/jimmunol.178.1.320. [DOI] [PubMed] [Google Scholar]
- 20.Valmori D, Tosello V, Souleimanian NE, Godefroy E, Scotto L, Wang Y, Ayyoub M. Rapamycin-Mediated Enrichment of T Cells with Regulatory Activity in Stimulated CD4+ T Cell Cultures Is Not Due to the Selective Expansion of Naturally Occurring Regulatory T Cells but to the Induction of Regulatory Functions in Conventional CD4+ T Cells. J Immunol. 2006;177:944–949. doi: 10.4049/jimmunol.177.2.944. [DOI] [PubMed] [Google Scholar]
- 21.Basu S, Golovina T, Mikheeva T, June CH, Riley JL. Cutting edge: foxp3-mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. J Immunol. 2008;180:5794–5798. doi: 10.4049/jimmunol.180.9.5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bour-Jordan H, Salomon BL, Thompson HL, Szot GL, Bernhard MR, Bluestone JA. Costimulation controls diabetes by altering the balance of pathogenic and regulatory T cells. J. Clin. Invest. 2004;114:979–987. doi: 10.1172/JCI20483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat. Immunol. 2005;6:152–162. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- 24.Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, Zheng XX, Strom TB, Bluestone JA. Cutting Edge: CD28 Controls Peripheral Homeostasis of CD4+CD25+ Regulatory T Cells. J Immunol. 2003;171:3348–3352. doi: 10.4049/jimmunol.171.7.3348. [DOI] [PubMed] [Google Scholar]
- 25.Lyddane C, Gajewska BU, Santos E, King PD, Furtado GC, Sadelain M. Cutting Edge: CD28 Controls Dominant Regulatory T Cell Activity during Active Immunization. J Immunol. 2006;176:3306–3310. doi: 10.4049/jimmunol.176.6.3306. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Amarnath S, Chen W. Requirement of CD28 signaling in homeostasis/survival of TGF-beta converted CD4+CD25+ Tregs from thymic CD4+ Tregs from thymic CD4+CD25- single positive T cells. Transplantation. 2006;82:953–964. doi: 10.1097/01.tp.0000232330.46688.37. [DOI] [PubMed] [Google Scholar]
- 27.Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, Little S, Havlir DV, Richman DD, Gruener N, Pape G, Waters A, Easterbrook P, Salio M, Cerundolo V, McMichael AJ, Rowland-Jones SL. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8:379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 28.Ruprecht CR, Gattorno M, Ferlito F, Gregorio A, Martini A, Lanzavecchia A, Sallusto F. Coexpression of CD25 and CD27 identifies FoxP3+ regulatory T cells in inflamed synovia. J Exp. Med. 2005;201:1793–1803. doi: 10.1084/jem.20050085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang ZZ, Novak AJ, Ziesmer SC, Witzig TE, Ansell SM. CD70+ non-Hodgkin lymphoma B cells induce Foxp3 expression and regulatory function in intratumoral CD4+ CD25 T cells. Blood. 2007;110:2537–2544. doi: 10.1182/blood-2007-03-082578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004;22:531–562. doi: 10.1146/annurev.immunol.21.120601.141122. [DOI] [PubMed] [Google Scholar]
- 31.Niedbala W, Cai B, Liu H, Pitman N, Chang L, Liew FY. Nitric oxide induces CD4+CD25+ Foxp3 regulatory T cells from CD4+CD25 T cells via p53, IL-2, and OX40. Proceedings of the National Academy of Sciences. 2007;104:15478–15483. doi: 10.1073/pnas.0703725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kroemer A, Xiao X, Vu MD, Gao W, Minamimura K, Chen M, Maki T, Li XC. OX40 Controls Functionally Different T Cell Subsets and Their Resistance to Depletion Therapy. J Immunol. 2007;179:5584–5591. doi: 10.4049/jimmunol.179.8.5584. [DOI] [PubMed] [Google Scholar]
- 33.Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Li XC. OX40 costimulation turns off Foxp3+ TREGS. Blood. 2007;110:2501–2510. doi: 10.1182/blood-2007-01-070748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.So T, Croft M. Cutting Edge: OX40 Inhibits TGF-beta- and Antigen-Driven Conversion of Naive CD4 T Cells into CD25+Foxp3+ T cells. J Immunol. 2007;179:1427–1430. doi: 10.4049/jimmunol.179.3.1427. [DOI] [PubMed] [Google Scholar]
- 35.Ito T, Wang YH, Duramad O, Hanabuchi S, Perng OA, Gilliet M, Qin FX, Liu YJ. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc. Natl. Acad. Sci. U. S. A. 2006;103:13138–13143. doi: 10.1073/pnas.0603107103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J. Exp. Med. 2008;205:825–839. doi: 10.1084/jem.20071341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi BK, Bae JS, Choi EM, Kang WJ, Sakaguchi S, Vinay DS, Kwon BS. 4-1BB-dependent inhibition of immunosuppression by activated CD4+CD25+ T cells. J Leukoc Biol. 2004;75:785–791. doi: 10.1189/jlb.1003491. [DOI] [PubMed] [Google Scholar]
- 38.Tan JT, Whitmire JK, Murali-Krishna K, Ahmed R, Altman JD, Mittler RS, Sette A, Pearson TC, Larsen CP. 4-1BB Costimulation Is Required for Protective Anti-Viral Immunity After Peptide Vaccination. J Immunol. 2000;164:2320–2325. doi: 10.4049/jimmunol.164.5.2320. [DOI] [PubMed] [Google Scholar]
- 39.Choi BK, Kim YH, Kang WJ, Lee SK, Kim KH, Shin SM, Yokoyama WM, Kim TY, Kwon BS. Mechanisms Involved in Synergistic Anticancer Immunity of Anti-4-1BB and Anti-CD4 Therapy. Cancer Res. 2007;67:8891–8899. doi: 10.1158/0008-5472.CAN-07-1056. [DOI] [PubMed] [Google Scholar]
- 40.Diehl L, van Mierlo GJD, den Boer AT, van der Voort E, Fransen M, van Bostelen L, Krimpenfort P, Melief CJM, Mittler R, Toes REM, Offringa R. In Vivo Triggering Through 4-1BB Enables Th-Independent Priming of CTL in the Presence of an Intact CD28 Costimulatory Pathway. J Immunol. 2002;168:3755–3762. doi: 10.4049/jimmunol.168.8.3755. [DOI] [PubMed] [Google Scholar]
- 41.Elpek KG, Yolcu ES, Franke DDH, Lacelle C, Schabowsky RH, Shirwan H. Ex Vivo Expansion of CD4+CD25+FoxP3+ T Regulatory Cells Based on Synergy between IL-2 and 4-1BB Signaling. J Immunol. 2007;179:7295–7304. doi: 10.4049/jimmunol.179.11.7295. [DOI] [PubMed] [Google Scholar]
- 42.Zhang B, Maris CH, Foell J, Whitmire J, Niu L, Song J, Kwon BS, Vella AT, Ahmed R, Jacob J, Mittler RS. Immune suppression or enhancement by CD137 T cell costimulation during acute viral infection is time dependent. J Clin. Invest. 2007;117:3029–3041. doi: 10.1172/JCI32426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foell JL, ez-Mendiondo BI, Diez OH, Holzer U, Ruck P, Bapat AS, Hoffmann MK, Mittler RS, Dannecker GE. Engagement of the CD137 (4-1BB) costimulatory molecule inhibits and reverses the autoimmune process in collagen-induced arthritis and establishes lasting disease resistance. Immunology. 2004;113:89–98. doi: 10.1111/j.1365-2567.2004.01952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foell J, McCausland M, Burch J, Corriazzi N, Yan XJ, Suwyn C, O'Neil SP, Hoffmann MK, Mittler RS. CD137-mediated T cell co-stimulation terminates existing autoimmune disease in SLE-prone NZB/NZW F1 mice. Ann. N. Y. Acad. Sci. 2003;987:230–235. doi: 10.1111/j.1749-6632.2003.tb06052.x. [DOI] [PubMed] [Google Scholar]
- 45.Spence PJ, Green EA. Foxp3+ regulatory T cells promiscuously accept thymic signals critical for their development. Proceedings of the National Academy of Sciences. 2008;105:973–978. doi: 10.1073/pnas.0709071105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guiducci C, Valzasina B, Dislich H, Colombo MP. CD40/CD40L interaction regulates CD4+CD25+ T reg homeostasis through dendritic cell-produced IL-2. Eur. J Immunol. 2005;35:557–567. doi: 10.1002/eji.200425810. [DOI] [PubMed] [Google Scholar]
- 47.Kumanogoh A, Wang X, Lee I, Watanabe C, Kamanaka M, Shi W, Yoshida K, Sato T, Habu S, Itoh M, Sakaguchi N, Sakaguchi S, Kikutani H. Increased T Cell Autoreactivity in the Absence of CD40-CD40 Ligand Interactions: A Role of CD40 in Regulatory T Cell Development. J Immunol. 2001;166:353–360. doi: 10.4049/jimmunol.166.1.353. [DOI] [PubMed] [Google Scholar]
- 48.McGregor CM, Schoenberger SP, Green EA. CD154 is a negative regulator of autoaggressive CD8+ T cells in type 1 diabetes. Proceedings of the National Academy of Sciences. 2004;101:9345–9350. doi: 10.1073/pnas.0402807101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marks E, Verolin M, Stensson A, Lycke N. Differential CD28 and Inducible Costimulatory Molecule Signaling Requirements for Protective CD4+ T-Cell-Mediated Immunity against Genital Tract Chlamydia trachomatis Infection. Infect. Immun. 2007;75:4638–4647. doi: 10.1128/IAI.00465-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam KP, Coyle AJ, Kroczek RA, Hutloff A. ICOS Controls the Pool Size of Effector-Memory and Regulatory T Cells. J Immunol. 2008;180:774–782. doi: 10.4049/jimmunol.180.2.774. [DOI] [PubMed] [Google Scholar]
- 51.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412. N Engl J Med. 2006;355:1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 52.Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, Carroll RG, Riley JL, June CH. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol. Ther. 2007;15:981–988. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol. 2003;171:166–174. doi: 10.4049/jimmunol.171.1.166. [DOI] [PubMed] [Google Scholar]
- 54.Thomas AK, Maus MV, Shalaby WS, June CH, Riley JL. A cell-based artificial antigen-presenting cell coated with anti-CD3 and CD28 antibodies enables rapid expansion and long-term growth of CD4 T lymphocytes. Clin. Immunol. 2002;105:259–272. doi: 10.1006/clim.2002.5277. [DOI] [PubMed] [Google Scholar]
- 55.Maus MV, Thomas AK, Leonard DG, Allman D, Addya K, Schlienger K, Riley JL, June CH. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat. Biotechnol. 2002;20:143–148. doi: 10.1038/nbt0202-143. [DOI] [PubMed] [Google Scholar]
- 56.Duggleby RC, Shaw TN, Jarvis LB, Kaur G, Gaston JS. CD27 expression discriminates between regulatory and non-regulatory cells after expansion of human peripheral blood CD4+ CD25+ cells. Immunology. 2007;121:129–139. doi: 10.1111/j.1365-2567.2006.02550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Earle KE, Tang Q, Zhou X, Liu W, Zhu S, Bonyhadi ML, Bluestone JA. In vitro expanded human CD4+CD25+ regulatory T cells suppress effector T cell proliferation. Clin. Immunol. 2005;115:3–9. doi: 10.1016/j.clim.2005.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Godfrey WR, Spoden DJ, Ge YG, Baker SR, Liu B, Levine BL, June CH, Blazar BR, Porter SB. Cord blood CD4(+)CD25(+)-derived T regulatory cell lines express FoxP3 protein and manifest potent suppressor function. Blood. 2005;105:750–758. doi: 10.1182/blood-2004-06-2467. [DOI] [PubMed] [Google Scholar]
- 59.Porter SB, Liu B, Rogosheske J, Levine BL, June CH, Kohl VK, Wagner JE, Miller JS, Blazar BR. Suppressor function of umbilical cord blood-derived CD4+CD25+ T-regulatory cells exposed to graft-versus-host disease drugs. Transplantation. 2006;82:23–29. doi: 10.1097/01.tp.0000225824.48931.af. [DOI] [PubMed] [Google Scholar]
- 60.Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, Edinger M. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood. 2006;108:4260–4267. doi: 10.1182/blood-2006-06-027409. [DOI] [PubMed] [Google Scholar]
- 61.Li L, Godfrey WR, Porter SB, Ge Y, June CH, Blazar BR, Boussiotis VA. CD4+CD25+ regulatory T-cell lines from human cord blood have functional and molecular properties of T-cell anergy. Blood. 2005;106:3068–3073. doi: 10.1182/blood-2005-04-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, Sugamura K, Ishii N. Distinct Roles for the OX40-OX40 Ligand Interaction in Regulatory and Nonregulatory T Cells. J Immunol. 2004;172:3580–3589. doi: 10.4049/jimmunol.172.6.3580. [DOI] [PubMed] [Google Scholar]
- 63.Herman AE, Freeman GJ, Mathis D, Benoist C. CD4+CD25+ T Regulatory Cells Dependent on ICOS Promote Regulation of Effector Cells in the Prediabetic Lesion. J. Exp. Med. 2004;199:1479–1489. doi: 10.1084/jem.20040179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–244. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cantrell DA, Smith KA. Transient expression of interleukin 2 receptors. Consequences for T cell growth. J Exp. Med. 1983;158:1895–1911. doi: 10.1084/jem.158.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Levine BL, Bernstein WB, Connors M, Craighead N, Lindsten T, Thompson CB, June CH. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J Immunol. 1997;159:5921–5930. [PubMed] [Google Scholar]
- 67.Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, Basu S, Riley JL, Hancock WW, Shen Y, Saouaf SJ, Greene MI. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc. Natl. Acad. Sci. U. S. A. 2007;104:4571–4576. doi: 10.1073/pnas.0700298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith B, Kasamon YL, Miller CB, Chia C, Gocke C, Kowalski J, Tartakovsky I, Biedrzycki B, Jones RJ, Hege K, Levitsky HI. K562/GM-CSF vaccination reduces tumor burden, including achieving molecular remissions, in chronic myeloid leukemia (CML) patients (PTS) with residual disease on imatinib mesylate (IM) J Clin Oncol (Meeting Abstracts) 2006;24:6509. doi: 10.1158/1078-0432.CCR-09-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, Gregson BP, June CH, Linsley PS. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11790–11795. doi: 10.1073/pnas.162359999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fox CJ, Hammerman PS, Thompson CB. The Pim kinases control rapamycin-resistant T cell survival and activation. J Exp. Med. 2005;201:259–266. doi: 10.1084/jem.20042020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Allan SE, Passerini L, Bacchetta R, Crellin N, Dai M, Orban PC, Ziegler SF, Roncarolo MG, Levings MK. The role of 2 FOXP3 isoforms in the generation of human CD4+ Tregs. J Clin Invest. 2005;115:3276–3284. doi: 10.1172/JCI24685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walker MR, Carson BD, Nepom GT, Ziegler SF, Buckner JH. De novo generation of antigen-specific CD4+CD25+ regulatory T cells from human CD4+ Proc. Natl. Acad. Sci. U. S. A. 2005;102:4103–4108. doi: 10.1073/pnas.0407691102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walker MR, Kasprowicz DJ, Gersuk VH, Benard A, Van LM, Buckner JH, Ziegler SF. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+ J Clin Invest. 2003;112:1437–1443. doi: 10.1172/JCI19441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, Roncarolo MG, Levings MK. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int. Immunol. 2007;19:345–354. doi: 10.1093/intimm/dxm014. [DOI] [PubMed] [Google Scholar]
- 75.Horwitz DA, Zheng SG, Wang J, Gray JD. Critical role of IL-2 and TGF-beta in generation, function and stabilization of Foxp3(+)CD4(+) Treg. Eur. J Immunol. 2008;38:912–915. doi: 10.1002/eji.200738109. [DOI] [PubMed] [Google Scholar]
- 76.Shevach EM, Tran DQ, Davidson TS, Andersson J. The critical contribution of TGF-beta to the induction of Foxp3 expression and regulatory T cell function. Eur. J Immunol. 2008;38:915–917. doi: 10.1002/eji.200738111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baron U, Floess S, Wieczorek G, Baumann K, Grutzkau A, Dong J, Thiel A, Boeld TJ, Hoffmann P, Edinger M, Turbachova I, Hamann A, Olek S, Huehn J. DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3(+) conventional T cells. Eur. J Immunol. 2007;37:2378–2389. doi: 10.1002/eji.200737594. [DOI] [PubMed] [Google Scholar]
- 78.Li B, Saouaf SJ, Samanta A, Shen Y, Hancock WW, Greene MI. Biochemistry and therapeutic implications of mechanisms involved in FOXP3 activity in immune suppression. Curr. Opin. Immunol. 2007;19:583–588. doi: 10.1016/j.coi.2007.07.006. [DOI] [PubMed] [Google Scholar]