

Abstract
The mechanisms by which mammalian epidermal stem cells cease to proliferate and embark upon terminal differentiation are still poorly understood. By conditionally ablating two highly expressed transcription factors, AP-2α and AP-2γ, we unmasked functional redundancies and discovered an essential role for AP-2s in the process. In vivo and in vitro, AP-2 deficiency is accompanied by surprisingly minimal changes in basal gene expression but severely perturbed terminal differentiation and suppression of additional transcription factors and structural genes involved. In dissecting the underlying molecular pathways, we uncover parallel pathways involving AP-2 and Notch signaling, which converge to govern CCAAT/enhancer binding protein genes and orchestrate the transition from basal proliferation to suprabasal differentiation. Finally, we extend the striking similarities in compromising either Notch signaling or AP-2α/AP-2γ in developing skin to that in postnatal skin, where all hair follicles and sebaceous gland differentiation are also repressed and overt signs of premalignant conversion emerge.
Introduction
In mouse, skin begins as a single layer of surface ectoderm from which the adult epidermis and its appendages develop. These cells are marked by expression of keratins 5 and 14 (K5 and K14; Byrne et al., 1994). As mesenchyme populates the skin, localized interactions occur, initiating specialized morphogenetic programs that lead to hair follicle (HF) morphogenesis and epidermal stratification. By embryonic day (E) 14.5, epidermis is two cell layers thick that are characterized by K5/K14 in the basal layer and K1/K10 in suprabasal (spinous) cells (Fuchs and Green, 1980; Byrne et al., 1994). By E16.5–17.5, several layers of spinous cells are capped by emergence of a morphologically distinct granular layer, marked by expression of late stage differentiation markers, including transglutaminases, loricrin, and filaggrin (Rothnagel et al., 1987). Just before birth, epidermal maturation concludes, as outermost cells lose their cellular organelles, transglutaminases are activated, and lipid bilayers are extruded to sandwich the dead stratum corneum layers. Postnatally, the stratum corneum provides a functional barrier to protect animals from dehydration and infection (Segre, 2006).
Adult epidermis undergoes constant turnover and repair by maintaining within the basal layer a resident population of stem cells that are able to both proliferate and execute the suprabasal terminal differentiation program. Homeostasis is maintained by a continual flux of basal cells departing from their proliferative compartment, committing to terminally differentiate, moving outward, and being sloughed from the skin surface (Blanpain and Fuchs, 2006). The balance between epidermal growth and differentiation must be intricately controlled, and many key changes regulating this transition take place at the basal to suprabasal juncture. Transcription factors CCAAT/enhancer binding protein (C/EBP) α and β (Nerlov, 2007) as well as Notch receptor signaling have been implicated in governing cell fate within spinous cells (Maytin et al., 1999; Zhu et al., 1999; Dai and Segre, 2004; Okuyama et al., 2004; Blanpain et al., 2006).
Since early discoveries of AP-2 transcription factor binding sites in promoters of many epidermally expressed genes, AP-2 proteins have been implicated in epidermal biology (Leask et al., 1990; Snape et al., 1991; Byrne et al., 1994; Zeng et al., 1997; Maytin et al., 1999; Sinha et al., 2000; Luo et al., 2002, 2005; Panteleyev et al., 2003; Vernimmen et al., 2003; Koster et al., 2006; Wang et al., 2006). The functions of AP-2α and AP-2γ have been postulated to be active in mouse surface ectoderm by E9.5, i.e., before K5 and K14 are induced (Byrne et al., 1994; Koster et al., 2006). How AP-2 proteins function in epidermis has been less clear, nor is much known about the regulation of these transcription factors. Both basal and suprabasal genes possess AP-2 binding sites, and both positive and negative activities have been observed in vitro, depending on the particular AP-2, gene, and cell type used in the study (Byrne et al., 1994; Sinha et al., 2000; Dai and Segre, 2004; Eckert et al., 2005; Koster et al., 2006; Nagarajan et al., 2008).
To date, the best genetic evidence in support of a major role for AP-2s in vertebrate epidermal biology stems from AP-2 morpholino knockdowns in Xenopus laevis oocytes, where neurogenesis becomes promoted at the expense of epidermal fate (Luo et al., 2002). Difficulties in dissecting AP-2 functions in mammalian skin have been compounded by their broad expression and physiological importance to many tissues, often resulting in embryonic or early neonatal lethality when targeted for ablation. Individual conditional targeting of AP-2α and more recently AP-2γ, two abundantly expressed AP-2 family members in skin, has been only modestly informative. K14-Cre conditional targeting of AP-2α resulted in normal epidermal development (Wang et al., 2006), as expected because AP-2α null mice survive until birth (Zhang et al., 1996). Postnatally, epidermis lacking AP-2α also appeared morphologically normal, although a target gene, EGF receptor (EGFR), was elevated in spinous layers. Abnormalities were only unmasked when skin was stimulated by EGFR ligands, resulting in excessive localized hyperproliferation. Together, these results hinted that without AP-2α, even though epidermal development and homeostasis are maintained, the balance may be precarious (Wang et al., 2006). Similarly, Sox2-Cre ablation of AP-2γ resulted in a transient developmental delay in epidermal stratification, but by birth AP-2γ null epidermis appeared morphologically and biochemically normal (Guttormsen et al., 2008). Documentation and further dissection of the possible role for AP-2γ in epidermis were not possible because the mice had neural tube closure and osteogenesis defects and died soon after parturition.
By conditionally removing functional redundancies of AP-2 family members in skin, we've now uncovered and characterized previously masked but critical functions of AP-2 transcription factors in epidermal differentiation and barrier function acquisition. Our findings provide major new insights to our understanding of AP-2 in skin development and differentiation and provide an unexpected link between Notch and AP-2s that governs in the early switch in epidermal terminal differentiation.
Results
Morphological and physiological defects in epidermis of mice lacking AP-2α and AP-2γ
By microarray and real-time PCR, AP-2α and AP-2γ are the most abundantly expressed AP-2 genes in mouse skin (Wang et al., 2006). Antibodies monospecific for AP-2α and AP-2γ (Wang et al., 2006) localized these proteins to the nuclei of cells within basal and spinous layers (Fig. 1 A). Expression was particularly strong at the basal to suprabasal juncture, but extended throughout most of the transcriptionally active layers. Both AP-2s were also detected in HFs, and expression encompassed not only epithelial cells but also mesenchymal dermal papillae (DP; Fig. 1 A, arrows).
Figure 1.

Targeted ablation of AP-2α and AP-2γ in mouse skin. γ-cKO, α-cKO, and DcKO mice were compared with WT littermates. (A) Immunolocalization of AP-2α and AP-2γ in P0 skins. Color coding of markers is according to secondary antibodies used in detection; nuclei were counterstained with DAPI (blue). Solid white lines denote skin surface. Dotted lines denote dermoepidermal boundary. Note that DP (arrows) also express AP-2γ. Bars, 20 μm. (B) Immunoblot analyses with monospecific antibodies and real-time PCR on mRNAs were performed on P0 epidermis, enzymatically separated from the dermis/HFs. Molecular mass of proteins is shown. PCR primer sets were specific for the AP-2α and AP-2γ sequences targeted for deletion. DcKO real-time PCR values were consistently below the level of detection from three or more independent experiments. (C) P0 mutant mice and their WT littermates. (D and E) Histology and ultrastructure of P0 epidermis. Vertical bars in D represent epidermis thickness. Boxed areas in E are magnified in bottom panels. BL, basal layer; Sp, spinous layer; Gr, granular layer; SC, stratum corneum; der, dermis; KF, keratin filament; KG, keratohyalin granules. Bars, 10 μm. (F) Barrier assay, as determined by penetration of blue dye.
Because AP-2s share a common DNA binding sequence and can operate as either hetero- or homodimers, the substantially overlapping expression patterns of AP-2α and AP-2γ raised the possibility that these two family members may at least partially overlap in their functions. To explore this possibility, we used K14-Cre, which is active in all skin epithelia with proliferative potential, to generate mice conditionally deleted for AP-2γ (Guttormsen et al., 2008). We then mated these mice to our previously generated K14-Cre/AP-2αlox/lox mice (Nelson and Williams, 2004; Wang et al., 2006) to produce mice with conditional loss of both AP-2 genes.
As judged by immunodetection, immunoblot analyses, and real-time PCR, AP-2α and AP-2γ expression were efficiently extinguished in epidermis of newborn K14-Cre/AP-2αlox/loxAP-2γlox/lox double conditional knockout (DcKO) mice (Fig. 1, A and B). As expected from K14 promoter specificity, AP-2γ remained intact in DP as well as in epithelial cells of the few precocious guard HFs initiated before K14 promoter activity (Fig. 1 A and not depicted).
As we had observed previously with AP-2α conditional knockout (cKO) mice (Wang et al., 2006), AP-2γ cKO animals developed normally (Fig. 1 C). At birth, AP-2γ cKO mice were phenotypically indistinguishable from their wild-type (WT) counterparts, and these mice showed no obvious abnormalities as they aged. In contrast, although AP-2α/γ DcKO offspring were born at expected Mendelian ratios, pups died within 24 h after birth. DcKO animals were less than half the size of single cKO mice and exhibited signs of severe dehydration (Fig. 1 C). Notably, DcKO pups lacked visible whiskers (Fig. 1 C).
Histological analyses revealed additional abnormalities (Fig. 1 D). In contrast to epidermis from either of the singly targeted mice, the three discrete stages of terminal differentiation—spinous, granular, and stratum corneum layers—were all noticeably diminished in DcKO epidermis. Ultrastructurally, spinous cells lacked dense networks of keratin filament bundles that typify this early stage of differentiation. In granular layers, keratohyalin granules were morphologically abnormal (Fig. 1 E, bottom).
Because animals deteriorated rapidly after birth, appeared to be severely dehydrated at death, and exhibited gross perturbations in terminal differentiation, we surmised that the barrier function might be compromised. As judged by dye exclusion assays, single cKO embryos paralleled the WT kinetics of barrier acquisition (Fig. 1 F; Wang et al., 2006). Barrier acquisition in DcKO animals, however, was not complete in E18.5 DcKO embryos, revealing a pattern more closely resembling that of E17.5 WT embryos (Fig. 1 F). Because K14 promoter is strongly active in oral epithelia, this too was likely to contribute to their perinatal death.
Molecular defects in early terminal differentiation in epidermis lacking AP-2α and AP-2γ
Further analyses revealed that abnormalities visible in newborn DcKO epidermis were already present in developing embryos. By E17.5, DcKO epidermis showed few signs of granular or stratum corneum formation, which contrasted markedly with either WT or single AP-2γ or AP-2α skins (Fig. 2 A and not depicted). Additionally, cells in the outermost layers of DcKO epidermis still possessed nuclei and other organelles (Fig. 2 A, arrows), which are typically lost in the normal terminal differentiation program.
Figure 2.

Embryonic suppression of terminal differentiation in vivo and in vitro upon loss of AP-2s. (A) Histology and immunofluorescence of the epidermis from E17.5 γ-cKO, DcKO, and WT skins. Note parakeratosis in DcKO epidermis (arrows). Bars, 20 μm. (B and E) Real-time PCR on mRNAs from embryonic epidermis and cultured 1°MKs. DcKO and α-cKO levels are expressed as fold changes versus WT (set to 1). Error bars represent standard deviation for three or more independent experiments. Cultures were in 50 μM Ca2+ (differentiation-restricted) media. (C) α6 BrdU FACS was performed on cells from epidermis of E18.5 mice pulsed 4 h with BrdU before analyses. Asterisk denotes significant difference (P < 0.05) by Student's t test. (D) Anti-active caspase 3 staining and quantification. Total number of cells counted in E18.5 epidermis are shown along with number of cells scoring positive for Cas3. (F) At 0 h, 1.5 mM Ca2+ was added to induce terminal differentiation. mRNAs were isolated at indicated time points and real-time PCR was performed. Lor, loricrin; Fil, filaggrin.
Analogous to the morphological defects, molecular deviations began at the basal to spinous transition and extended throughout metabolically active stages of terminal differentiation. DcKO epidermis displayed reduced immunolabeling for markers of both spinous (K1) and granular (involucrin and filaggrin) layers (Fig. 2 A). As judged by real-time PCR analyses, the defects in terminal differentiation were at the level of gene expression and were global, encompassing many epidermal genes encoding major structural proteins, including spinous keratins, cornified envelope proteins, and transglutaminase cross-linking proteins used in barrier formation (Fig. 2 B). These molecular defects became even more pronounced as DcKO embryos came to term (Fig. S1, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200804030/DC1).
In contrast to marked perturbations in terminal differentiating cells, the basal layer of DcKO embryos appeared largely unaffected morphologically and biochemically. K5/K14 was still mainly confined to the innermost basal layer, and K5/K14 mRNA levels appeared comparable between WT and DcKO embryonic skin (Fig. 2, A and B). This said, FACS quantifications of short-term BrdU incorporation revealed an ∼2× reduction in overall number of labeled basal cells in embryonic DcKO versus WT skin (Fig. 2 C). This defect did not appear to be attributable to alterations in apoptosis, as antibodies against active caspase 3 did not detect signs of apoptosis in either DcKO or WT epidermis (Fig. 2 D).
The consequences of AP-2α and AP-2γ loss in vivo were recapitulated when primary mouse epidermal keratinocytes (1°MK) were propagated in vitro. In low Ca2+ media, double knockout (DKO) and WT 1°MK expressed comparable levels of basal keratin mRNAs (Fig. 2 E). In contrast, even though low Ca2+ media is only weakly permissive for terminal differentiation, this was largely abrogated in DKO 1°MK, which expressed only 10–25% mRNA levels of K1, K10, Tgm1, and Tgm3 compared with their WT counterparts (Fig. 2 E). These global defects were not observed in AP-2α single KO cells, where only K1 mRNA appeared to be decreased.
When calcium levels were increased to promote terminal differentiation, differences in gene expression between DKO and WT 1°MK became profound (Fig. 2 F). Expression of both early (K1 and K10) and late (Loricrin and Filaggrin) genes of terminal differentiation were dramatically suppressed in DKO keratinocytes in vitro. These observations suggest that the impairment in epidermal barrier function reflects an intrinsic and fundamental flaw in the ability of DKO keratinocytes to transcriptionally regulate terminal differentiation. Additional studies presented in the following paragraph further strengthen this notion.
The molecular aberrations caused by loss of AP-2α and AP-2γ go beyond the normal program of epidermal terminal differentiation
The biochemical changes observed upon loss of AP-2 proteins in epidermis went beyond perturbations in the normal program of terminal differentiation. Perhaps most striking in this regard was the aberrant expression of K8 protein and mRNA in suprabasal layers of DcKO embryonic skin (Fig. 3, A and B). K8 is normally synthesized by transient surface periderm cells that disappear as embryonic epidermis matures (Fuchs, 2007). Ultrastructurally, however, no signs of periderm cells were evident in E17.5 DcKO embryos, and K8 was clearly in the keratinocytes that despite their paucity of keratin filaments were morphologically similar to spinous cells. Again, these perturbations were intrinsic to keratinocytes and observed in vitro as well as in vivo (Fig. 3 B).
Figure 3.

Ectopic suprabasal genes are expressed in the absence of AP-2α and AP-2γ. (A) Immunofluorescence of K6 and K8 in skins from E17.5 embryos. Dotted lines denote dermoepidermal boundary. Bars, 20 μm. (B) Real-time PCR of mRNAs from epidermis and 1°MK as indicated. DcKO levels are expressed as changes versus WT and error bars represent standard deviation for three or more independent experiments.
In agreement with these data, K18, the normal periderm partner for K8, was not induced in DcKO spinous cells (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200804030/DC1), whereas K6 mRNA and protein were (Fig. 3, A and B; and Fig. S2 B). K6 is a keratin that is typically only induced in normal spinous cells during normal wound healing, although it is also seen in a variety of cellular stress and/or hyperproliferative situations. Interestingly, K6 mRNA levels were ∼2× lower in DKO versus WT cells (Fig. 3 B), supporting the notion that the induction of K6 expression in DcKO skin represents cellular stress. These additional atypical perturbations seen in DcKO epidermis were largely absent in singly targeted skins, with the exception of a few K6-positive cells within AP-2γ DcKO epidermis (Fig. 3 A).
In summary, loss of both AP-2α and AP-2γ unveiled marked defects in epidermal terminal differentiation that were hidden in singly targeted animals. Although otherwise largely unaffected, the hypoproliferation displayed by embryonic DcKO epidermis could contribute to the reduction in terminally differentiating layers of newborn epidermis.
Repression of C/EBP transcription factors in part accounts for the marked defects arising from loss of AP-2α and AP-2γ
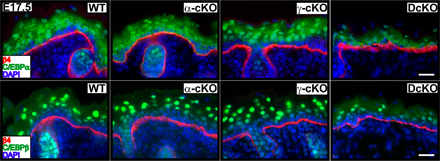
The reduction in K10 mRNA was intriguing, given prior in vitro studies that implicated the C/EBPs in regulating K10 gene expression (Maytin et al., 1999) and in inducing K1 and K10 expression when overexpressed (Zhu et al., 1999). C/EBPα and C/EBPβ are both expressed in mouse epidermis and consensus binding sites for these factors have been identified in both K1 and K10 promoters (Mack et al., 2005). Given the surprising and global repression of terminal differentiation-specific genes resulting from dual loss of AP-2α and AP-2γ, we wondered whether the status of C/EBP expression might also be altered.
To explore this possibility, we first used immunofluorescence microscopy with monospecific antibodies against C/EBPα and C/EBPβ (Fig. 4 A and Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200804030/DC1). In WT E17.5 and E18.5 back skins, strong nuclear immunolabeling for both C/EBPα and C/EBPβ was observed in multiple suprabasal epidermal layers. In comparably aged DcKO back skins, however, nuclear suprabasal labeling was diminished. Some immunolabeling persisted in basal DcKO cells, but even in WT epidermis, basal labeling was always cytoplasmic rather than nuclear. Together, these findings suggested that C/EBPα and C/EBPβ's role in transcriptional regulation is primarily in terminally differentiating cells, where loss of AP-2α and AP-2γ compromised its expression, and hence its function.
Figure 4.

C/EBP gene expression is regulated by AP-2α and AP-2γ. (A) Immunofluorescence with monospecific C/EBPα and C/EBPβ antibodies in E18.5 skin. Dotted lines denote dermoepidermal boundary. Bars, 20 μm. (B) Immunoblots of protein lysates from 1°MK cultured under 50-μM or 1.5-mM Ca2+ conditions. The C/EBPα antibody detects both Cα p42 and Cα p30 isoforms; the C/EBPβ antibody detects LAP* and LAP isoforms. β-Actin is used as a loading control. (C) Analyses of the effects of AP-2 ablation on C/EBP gene expression and promoter activity. Real-time PCRs with primers specific for C/EBPα and C/EBPβ were performed on mRNAs from in vivo epidermis and 1°MK. DcKO levels are expressed as changes versus WT and error bars represent standard deviation for three or more independent experiments. (bottom right) Luciferase assay of C/EBPα promoter activity in 1°MK in high Ca2+ media. 1.5 kb of C/EBPα promoter plus 5′-UTR sequences were used to drive firefly luciferase expression (pC/EBPα-pGL3; Tang et al., 1997). Fold change represents pC/EBPα-pGL3 firefly luciferase activity divided by basal level of pGL3 firefly luciferase activity, with cytomegalovirus Renilla luciferase activity as the standardized internal control for transfection efficiency. Three independent experiments performed in duplicate are represented. Error bars represent standard deviations. (D) Effects of ectopic expression of AP-2α, C/EBPα, or C/EBPβ in DKO and WT 1°MK. Where indicated (vector), cells were either transduced 36 h with an IRES-GFP empty vector (control) or an IRES-GFP retroviral expression vector encoding the transcription factor indicated. Transduced cells were purified by FACS, mRNAs were isolated, and real-time PCRs were performed with the primers indicated.
These in vivo results were also corroborated with studies in vitro. As judged by immunoblot analyses of protein lysates from 1°MK cultured in the presence of low Ca2+, both isoforms of C/EBPα, Cα p42 and Cα p30 (Calkhoven et al., 2000), were barely detectable when AP-2α and AP-2γ were absent but readily detected in WT lysates (Fig. 4 B). Even when Ca2+ levels were elevated, Cα p42 levels were >5× reduced, and Cα p30 was not detected in the DKO lysates (Fig. 4 B). Studies with antibodies that recognize both the LAP* and LAP isoforms of C/EBPβ (Calkhoven et al., 2000) also corroborated the results obtained by immunofluorescence microscopy (Fig. 4 B), although at low Ca2+, only the LAP isoform was detectable in WT cells.
Real-time PCR revealed that the reduction in C/EBP proteins occurring upon loss of AP-2 factors was reflected at least in part at the mRNA level (Fig. 4 C). This was true both for skin in vivo and for 1°MK in culture (Fig. 4 C). As judged by a C/EBPα luciferase reporter assay, C/EBPα promoter activity was diminished nearly 10-fold when AP-2 proteins were absent (Fig. 4 C). Within the 1.4-kb C/EBPα promoter fragment plus its 0.1-kb 5′-UTR, there are multiple consensus AP-2 binding sites. Whether AP-2 is a direct transcriptional activator of the C/EBP genes is an intriguing issue but one beyond the scope of this study.
Like C/EBP, AP-2 proteins have also been implicated in regulating terminal differentiation-specific genes (Snape et al., 1991; Luo et al., 2003, 2005). To dissect their relative importance, we therefore tested the consequences to two early terminal differentiation genes, K1 and K10, when AP-2 and C/EBP activities were independently restored in AP-2 DKO keratinocytes. For this purpose, we retrovirally infected cells with transgenes expressing either GFP (control vector) or, additionally, transcription factor AP-2α, C/EBPα, or C/EBPβ. Infected cells were enriched by FACS purification based on GFP positivity.
The results of these experiments are shown in Fig. 4 D. When AP-2α was reexpressed, C/EBPα levels were restored in their entirety, confirming that loss of AP-2 underlies this change. Correspondingly, expression of both K1 and K10 genes was also enhanced by AP-2 reexpression. Interestingly, however, this was also the case when C/EBPα or C/EBPβ expression was restored in AP-2 null 1°MK. C/EBPβ was more effective than either AP-2α and/or C/EBPα, but all three had appreciable effects on both K10 and K1 expression. These findings support the conclusion that in part, the dramatic alterations in expression of genes encoding structural proteins of terminal differentiation are attributable to concomitant loss of expression of additional key transcriptional regulators resulting from loss of AP-2α and AP-2γ. Moreover, in contrast to the previous notion that AP-2s might function basally to suppress C/EBP (Maytin et al., 1999), our studies support the view that AP-2 factors participate positively in suprabasal transactivation of C/EBPα and C/EBPβ and in subsequent coordinate orchestration of terminal differentiation by AP-2s and C/EBPs in epidermis.
Defects in downstream effectors of Notch signaling in the absence of AP-2α and AP-2γ
The marked defects in terminal differentiation that originated at the basal to spinous layer transition in mice lacking AP-2α and AP-2γ resembled that caused by loss of RBP/J, the obligate canonical Notch signaling partner (Blanpain et al., 2006). Given that Notch signaling is also known to play a key role not only in K1 and K10 expression but also in governing terminal differentiation (Lowell et al., 2000; Okuyama et al., 2004; Blanpain et al., 2006; Estrach et al., 2007), we wondered whether there might be a functional link between Notch signaling, AP-2 transcription, and induction of the terminal differentiation program in epidermal cells.
To test this possibility, we transduced AP-2 DKO 1°MK with a retroviral vector encoding the Notch intracellular domain (NICD), which functions as a transcriptional coactivator for the RBP/J DNA binding protein (Artavanis-Tsakonas et al., 1999; Iso et al., 2003; Lai, 2004). As expected from prior studies with WT 1°MK (Okuyama et al., 2004; Blanpain et al., 2006), expression of NICD in our control (WT) cultures in low Ca2+ media (differentiation restricted) increased mRNA levels of K1 and K10 but not K14 (Fig. 5 A, green bars). Notably, however, this effect was nearly abolished when the same vector was introduced into AP-2 DKO keratinocytes (Fig. 5 A, orange bars). These findings suggested a role for AP-2 transcription factors in mediating the effects of Notch-dependent spinous differentiation.
Figure 5.

Notch signaling is active but cannot induce terminal differentiation in the absence of AP-2α and AP-2γ. (A) Real-time PCR analyses of WT and DKO 1°MK transduced with either an IRES-GFP empty vector (control) or an IRES-GFP retroviral vector encoding the NICD. After 36 h, transduced cells were FACS enriched and K14, K1, and K10 mRNA levels were measured. Fold change upon NICD induction was compared with the control vector. (B) Same experiment as in A, but documenting the mRNA levels of Hes1, Hey1, and RBP/J. (C) Same experiment as in A, but showing that AP-2 mRNA levels are only modestly affected by activated Notch. (D) RBP/Jlox/lox 1°MK were cultured in duplicate in low Ca2+ medium, and then transduced with either an adenovirus expressing a cytomegalovirus-Cre recombinase (Cre) or cytomegalovirus-GFP (control). At t = 2 d, one set of transduced cells were switched to high Ca2+ to induce differentiation. FACS was performed at t = 4 d and mRNAs were subjected to real-time PCR analyses with the primers indicated. Error bars represent standard deviation for three or more independent experiments.
To determine whether AP-2 might be required to mediate Notch signaling, we repeated the experiments, this time evaluating whether two NICD/RBP/J target genes (Hes1 and Hey1) known to be expressed in keratinocytes could be activated in the absence of AP-2. As shown in Fig. 5 B, NICD expression in AP-2 null 1°MK had little or no effect on downstream activation of these canonical Notch target genes. Interestingly, AP-2 DKO cells showed a mild decrease in RBP/J mRNA, whose levels were not affected by NICD expression (Fig. 5 B). Based on these data, the function of AP-2α and AP-2γ in blocking Notch-induced spinous gene expression appeared to be both independent of Hes1 and Hey1 and also independent of the ability of Notch signaling to form and activate the bipartite transcription complex composed of NICD and RBP/J.
One possible explanation consistent with the results at hand is that Notch signaling is responsible for AP-2 expression, which would place AP-2s downstream of Notch. If so, then expression of NICD in low Ca2+ medium should result in elevated AP-2 mRNA levels in WT keratinocytes. Whereas NICD stimulated ∼2.5-fold elevation in AP-2γ expression, AP-2α mRNA levels were relatively unaffected (Fig. 5 C). Given our findings that AP-2α and AP-2γ are functionally redundant, these differences did not seem sufficient to account for the dramatic effects of loss of AP-2 function on NICD-mediated changes in expression of basal to spinous genes.
To gain further insights into the relation between Notch signaling and AP-2s, we examined the consequences of RBP/J loss of function on AP-2 gene expression. Given the deleterious long-term effects of RBP/J ablation, we used a GFP-tagged adenovirus expressing Cre recombinase to target ablation in 1°MK cultures of RBP/Jlox/lox cells. 48 h after ablation, FACS was used to isolate Cre-infected cells by GFP positivity. Cre activity led to efficient ablation of RBP/J gene expression, as indicated by the corresponding down-regulation in K1 and Hes1 mRNAs in Cre-targeted RBP/Jlox/lox 1°MK (Fig. 5 D). However, loss of RBP/J did not dramatically alter AP-2α or AP-2γ expression in 1°MK cultured in either low or high Ca2+ medium (Fig. 5 D). Samples taken 5 d after Cre transduction showed similar results, decreasing the possibility that large amounts of residual RBP/J protein remained (unpublished data). Together, these data indicate that AP-2α and AP-2γ act independently of canonical Notch signaling but converge through some common downstream mechanism that governs epidermal differentiation.
Notch signaling regulates expression of C/EBPα
Given our identification of C/EBPs as downstream targets of AP-2 transcriptional activities, and based on our results thus far, we turned our attention to the possibility that canonical Notch signaling might contribute to epidermal differentiation by regulating C/EBP expression. To test this hypothesis, we examined the consequences of RBP/J ablation on C/EBPα mRNA levels. As shown in Fig. 6 A, Cre-induced RBP/J ablation markedly reduced C/EBPα gene expression in 1°MK grown in both low and high Ca2+ culture conditions (Fig. 6 A). This was further confirmed at the protein level, where decreases in both p42 and p30 isoforms of C/EBPα were observed upon Cre-induced RBP/J ablation (Fig. 6 B). Conversely, elevated Notch signaling resulting from NICD expression in low Ca2+ WT cells gave an approximately sixfold increase in C/EBPα transcription, whereas this induction was largely abolished in DKO keratinocytes (Fig. 6 C).
Figure 6.

Regulation of C/EBPα gene expression by canonical Notch signaling pathway. Real-time PCR (A) and immunoblot analyses (B) of C/EBPα gene expression in control vector or Cre-infected RBP/Jlox/lox (R KO) 1°MK cultured in low or high Ca2+ media. (C) Same experiment as in A, except NICD-induced C/EBPα gene expression is measured. All experiments were performed three or more times, error bars represent standard deviation, and quantifications and details were as described in Fig. 2. (D) Real-time PCR and immunoblot analyses of C/EBPα gene expression in E18.5 WT versus K14-Cre/RBP/Jlox/lox cKO (R cKO) epidermis in vivo. (E) C/EBPα immunofluorescence. Bars, 20 μm. (F) WT and DKO keratinocytes were infected with lentivirus harboring either an empty vector (control) or a vector expressing a shRNA targeting ∼60% RBP/J knockdown. Infected cells were selected with puromycin and real-time PCR analyses were performed on these cells cultured at high Ca2+ conditions.
These effects observed in vitro were recapitulated in vivo, as judged by both real-time PCR and immunoblot analyses of epidermal mRNAs and proteins, respectively, from E18.5 K14-Cre/RBP/Jlox/lox cKO (Fig. 6 D, R cKO) embryos and their WT littermates. Similar to AP-2 DcKO animals, back skins of RBP/J cKO E18.5 animals also displayed markedly diminished nuclear C/EBPα immunolabeling of suprabasal cells compared with their WT counterparts (Fig. 6 E).
Finally, because full knockout of RBP/J blocks keratinocyte growth, we used lentivirus to express a short hairpin RNA (shRNA) that produced >60% of RBP/J mRNA inhibition in 1°MK. Interestingly, RBP/J knockdown resulted in a further reduction of C/EBPα mRNA expression in AP-2α/AP-2γ DKO cells (Fig. 6 F). In contrast, RBP/J knockdown did not lead to further reduction of K1 mRNA, whose expression thus seems to rely on both AP-2 and Notch. Together these data point to the view that AP-2α and AP-2γ act synergistically with the canonical Notch pathway to regulate C/EBP transcription factors that in turn contribute at least partially to governance of the basal to spinous transition.
More parallels between Notch signaling and AP-2 transcription: hyperproliferation and squamous metaplasia in postnatal skin lacking AP-2α and AP-2γ
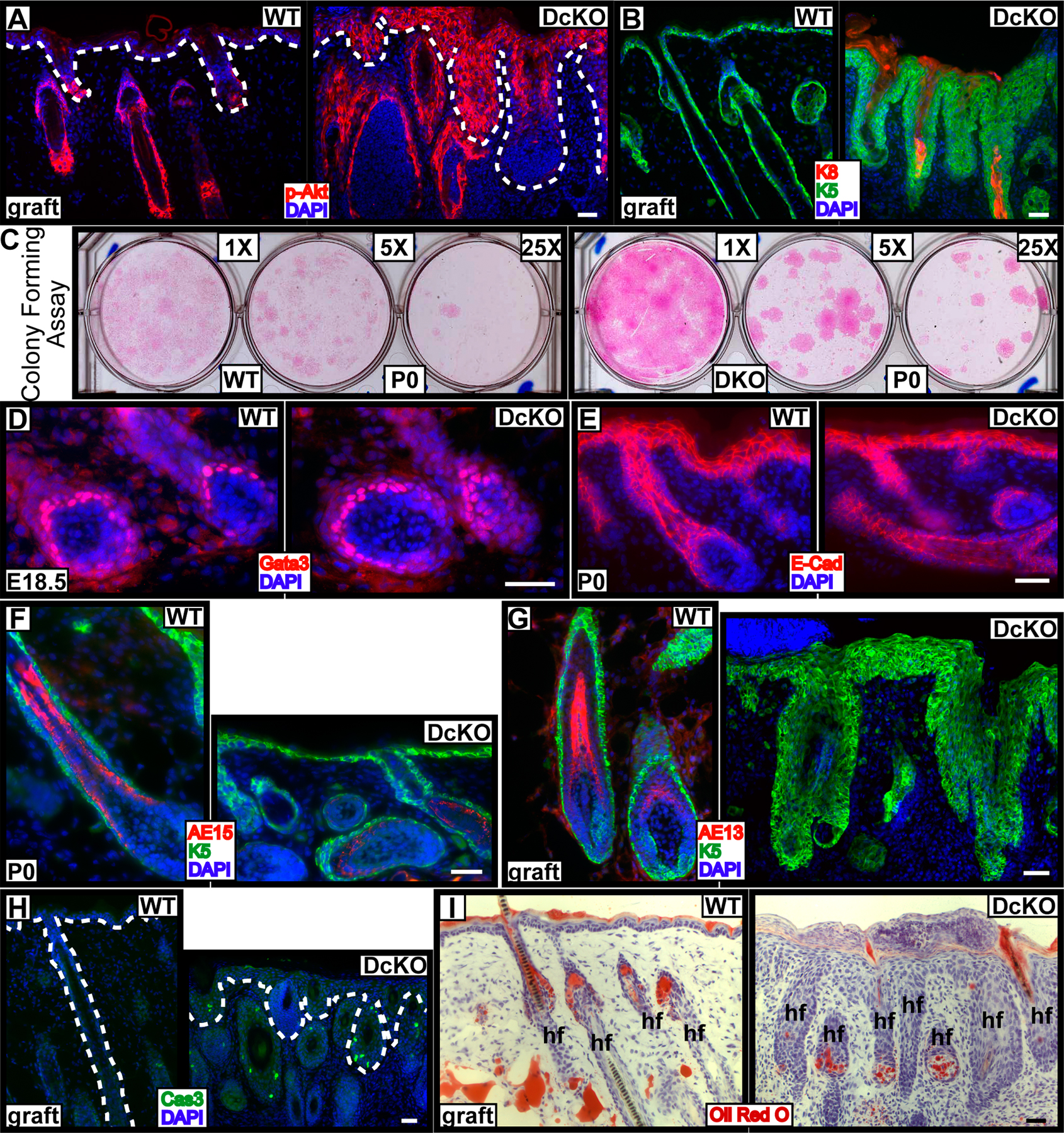
Given the parallel and yet converging pathways of Notch signaling and AP-2 expression and the phenotypic similarities between conditionally targeted Notch and AP-2 loss-of-function mutations in skin development, we wondered whether this link might persist in adulthood. Previous studies on Notch signaling revealed that despite impaired epidermal proliferation in embryonic skin lacking RBP/J, engrafted postnatal skin is associated with hyperproliferation and impaired terminal differentiation (Blanpain et al., 2006). This is also seen when Notch is partially compromised in skin (Nicolas et al., 2003; Pan et al., 2004; Lee et al., 2007). To pursue this parallel further, we engrafted doubly targeted AP-2α and AP-2γ null skin to the backs of Nude mice.
By 12–14 d after engraftment, WT skin grafts are typically in the final stages of recovering from a woundlike response, and by 21–45 d, grafts display normal epidermal homeostasis (Blanpain et al., 2004; Claudinot et al., 2005). Distinct from WT, DcKO grafts appeared rough and flaky, both at day 13 and 45 after grafting (Fig. 7 A). Additionally, the hair coat appeared sparse and disorderly. Histological and immunological analyses of d27 DcKO grafts revealed a grossly thickened epidermis accompanied by dramatic impairment in expression of K1, filaggrin, and the cornified envelope protein loricrin (Fig. 7, B–E). In contrast to embryonic DcKO epidermis, engrafted DcKO epidermis displayed overt hyperproliferation, with suprabasal expansion of the basal markers K5 and β4 integrin, ectopic expression of K6, and suppression of normal differentiation markers from all three different layers (Fig. 7, C–G). Thus, as in embryonic skin development, postnatal AP-2α and AP-2γ DcKO skin grafts resembled those of RBP/J cKO grafts.
Figure 7.

Loss of AP-2α and AP-2γ blocks rather than delays differentiation in postnatal skin: similarities to loss of Notch function. Skins from E18.5 WT and DcKO mice were engrafted onto Nude mice. (A) Gross appearance at indicated days after grafting. (B–G) Histology and immunofluorescence of frozen skin sections from 27-d grafts. Arrows in C denote K1 in a few degenerating HFs. Dotted lines denote dermoepidermal boundary. Bars, 40 μm.
Whereas postnatal skin from mice singly targeted for AP-2γ appeared normal, we previously found that as mice that were singly targeted for AP-2α aged, they healed their wounds but displayed signs of epidermal hyperproliferation in mechanically traumatized skin regions (Wang et al., 2006). Because this was accompanied by elevated EGFR and Akt signaling (Wang et al., 2006), we tested and found similar up-regulation of this signaling pathway in the grafted DcKO skins (Fig. S4 A, available at http://www.jcb.org/cgi/content/full/jcb.200804030/DC1). The hyperproliferative epidermis of DcKO skin differed significantly from that of AP-2α cKO skin: it was uniform, rapidly developing, and sustained and exhibited K8 and other biochemical abnormalities found only upon additional targeting for AP-2γ (Fig. S4 B).
When taken together, the most striking defect shared by both E17.5 and engrafted DcKO skin was the profound repression of terminal differentiation. Given this commonality, we surmise that the hyperproliferative state of the engrafted but not the E17.5 DcKO skin is likely secondary and attributable to an ill-fated rescue response to a defective epidermal barrier and terminal differentiation program. That said, it is also possible that AP-2s and Notch play dual roles in epidermal proliferation and differentiation, acting to promote either event depending on cellular context. Klf4 is an example of a transcription factor with such dual roles, acting as either tumor suppressor or oncogene depending on context (Rowland and Peeper, 2006).
Defects in HF differentiation in AP-2α and AP-2γ DcKO skin
A final similarity between loss of AP-2 factors versus RBP/J came from examining the consequences of gene targeting to HF differentiation. Loss of AP-2s did not appear to affect overall numbers of HFs. Moreover, E18.5 DcKO guard HFs still expressed markers of HF maturation: companion layer (K6+), IRS (Gata3+), and hair shaft precursor (AE15+; Fig. S2 B and Fig. S4, D and F). That said, guard hair development initiates early and perhaps precedes strong K14 promoter activity (Vasioukhin et al., 1999). In contrast, newborn DcKO animals did not form visible whiskers (Fig. 1 C), implying a defect in HF differentiation when AP-2α and AP-2γ are missing.
To explore further the possible importance of AP-2s in HF differentiation, we engrafted E18.5 skins onto the backs of Nude mice immunocompromised and defective in HF morphogenesis. In WT grafts, hair formation was already visible at day 13, when the bandages were removed. In contrast, DcKO grafts displayed no visible hairs even 45 d after grafting (Fig. 7 A). Histological examinations revealed large degenerating cysts, densely filled with small cells of hyperchromatic nuclei (Fig. 7 B). These cysts lacked hair shaft structures and displayed a paucity of hair differentiation markers, such as AE13 and Gata3, but aberrant expression of K8 and increased active caspase 3 (Fig. S4, B, G, and H; and not depicted). Oil Red O staining was largely missing in the epithelium of DcKO grafted skins, indicating that sebaceous gland differentiation was also impaired in the absence of AP-2 (Fig. S4 I).
Discussion
Dissecting how AP-2 functions in epidermal biology has awaited unmasking the layers of functional redundancy that are caused by the multiplicity of AP-2 factors expressed in skin. By conditionally targeting the two most abundantly expressed AP-2 genes, we've discovered that AP-2 factors play a prominent role in the master regulatory switch at the basal–suprabasal juncture. The central participation of AP-2 transcription in the molecular trigger that coaxes basal epidermal cells to initiate terminal differentiation adds a newfound physiologically significance to this family of transcription factors in epidermal development and homeostasis. The additional marked defects in sebaceous gland and HF differentiation that arise from loss of AP-2α and AP-2γ further unveil and expand the importance of these proteins in repressing multiple differentiation lineages within the skin.
Surprisingly, despite striking perturbations in terminal differentiation-specific gene expression arising from loss of AP-2α and AP-2γ, we did not observe marked alterations in basal gene expression at least in development and homeostasis. This was both surprising and unexpected based on single AP-2α or AP-2γ null phenotypes and on the presence of functional AP-2 sites within the K14 and K5 promoters (Leask et al., 1990; Byrne et al., 1994; Sinha et al., 2000). It is possible that other AP-2 family members expressed in skin might function in regulating basal fate (Sinha et al., 2000). Additionally, based on our discovery that K14/K5 is expanded suprabasally in engrafted skins that lack AP-2α and AP-2γ, it is tempting to speculate that suprabasal K14/K5 might be a component of a hyperproliferative feedback mechanism triggered when the terminal differentiation process is perturbed.
Other major functions unveiled from our double loss of function are previously masked roles for AP-2α and AP-2γ in HF maturation and sebaceous gland differentiation. HFs were dramatically altered in their angling, morphology, and differentiation in AP-2 DcKO animals. In addition, expression of terminal differentiation-specific genes for HF was markedly suppressed. The degeneration of HFs into cystlike structures was accompanied by elevated levels of apoptosis, suggesting that AP-2α and AP-2γ also likely operate at the transition between proliferation and differentiation of HF progenitors and maintain a normal HF differentiation program.
One of the most striking features of skin lacking AP-2α and AP-2γ was its resemblance to skin compromised for Notch signaling through loss of RBP/J. This was true not only for epidermis, but also HFs and sebaceous glands, all known to be regulated by Notch signaling (Okuyama et al., 2004; Pan et al., 2004; Blanpain and Fuchs, 2006). Moreover, the parallels extended to the gross hyperproliferation and premalignant states observed postnatally (Nicolas et al., 2003; Blanpain et al., 2006; Lee et al., 2007).
Although no such connections between AP-2 and Notch have previously been described in vertebrates, our results were intriguing in light of genetic studies that placed the single Drosophila melanogaster AP-2 gene downstream of Notch signaling in joint and leg development (Kerber et al., 2001). Similar to what we observed in mouse skin, fly joints harboring loss- or gain-of-function mutations in either dAP-2 or Notch exhibited similar phenotypes. However, in this fly tissue, a linear pathway was suggested, based on the ability of activated Notch to induce dAP-2 expression (Kerber et al., 2001). The loss of dAP-2 did not affect epidermis (Monge et al., 2001), although it is notable that fly epidermis exists as only a single layer of cells that secrete their cuticle rather than undergo a terminal differentiation program to form a barrier to the body surface.
In mouse skin, the existence of multiple AP-2 and Notch signaling pathway members seems to have added several additional and interesting layers of complexities to what appears to be an ancient regulatory mechanism. As in fly joints, where loss of dAP-2 did not perturb Notch's ability to activate its target genes (big brain and E(spl); Kerber et al., 2001), Notch signaling was still intact in AP-2 DKO mouse keratinocytes, as indicated by the ability of NICD to induce its classical target genes Hes1 and Hey1. We also observed a severalfold induction of AP-2γ by NICD expression. However, AP-2α showed little or no change in response to NICD, and neither of these AP-2 factors exhibited appreciable down-regulation when RBP/J expression was ablated in keratinocytes. Rather, the inability of Notch to elicit its full effects on epidermal terminal differentiation in mice lacking these AP-2 factors appeared to be rooted at least in part in the loss of C/EBP transcription, which in turn works concomitantly with AP-2 factors to orchestrate a complex program of gene expression necessary to produce the epidermal barrier. Although our data strongly support a parallel relationship, the slightly reduced expression of RBP/J in AP-2 DKO keratinocytes raises the possibility that cross talk does exist between AP-2 and Notch, and this could impact C/EBP as well as other downstream gene expression.
Although exploration into the mechanisms underlying AP-2 and epidermal appendage formation is beyond the scope of the present study, it is intriguing that both C/EBPα and C/EBPβ display strong nuclear localization in sebocytes (Bull et al., 2002; unpublished data), and both C/EBPs and AP-2s also function in adipocyte differentiation and lipid metabolism (Jiang et al., 1998; Otto and Lane, 2005). This opens the possibility that AP-2 transcription factors and Notch signaling may also converge on C/EBPs to regulate differentiation and lipid metabolism of these lineages. In contrast, neither C/EBPα nor C/EBPβ was detected in the differentiating layers of HFs (Bull et al., 2002; unpublished data), suggesting that some other downstream targets mediate the effects of AP-2 transcription factors and Notch signaling in HF lineage differentiation.
Concluding remarks
Nearly two decades after their initial discovery in epidermis and the subsequent widespread occurrence of AP-2 binding sites in epidermal promoters, major functions for AP-2 factors in mammalian skin epithelium are starting to emerge. Although further layers of redundancy are likely to unravel additional interesting roles for AP-2s in skin biology, the loss of AP-2α and AP-2γ has already unmasked a hitherto unexpected link between AP-2 transcription factors and Notch signaling in a mammalian tissue. Previously only observed in Drosophila (Kerber et al., 2001), this surprising connection between Notch and AP-2 in a mammalian tissue now highlights the existence of an ancient molecular link that over evolution has been elaborated on greatly. Given the widespread involvement of both Notch signaling and AP-2 expression in many cell types and tissues, these findings merit further investigation in the future.
Based on the evolutionary and/or tissue-specific differences already noted between the action of dAP-2 (Kerber et al., 2001) and mammalian AP-2α and AP-2γ (this study) and the findings that AP-2s can act as either transcriptional repressors or activators (Pfisterer et al., 2002), it is both plausible and likely that additional complexities in transcriptional activities of AP-2s and their tissue- and differentiation stage–dependent contexts will emerge as more studies are conducted.
Finally, it is interesting to speculate that the mechanism underlying the hyperproliferative feedback pathway that seems to operate when the epidermal barrier is compromised may involve a precariously elevated EGFR signaling sensor that arises from differences in AP-2 levels/expression. If true, this notion would help to explain the confusing and often opposing correlations in expression levels of AP-2 family members in human cancers (Pellikainen and Kosma, 2007).
Materials and methods
Mice
K14-Cre/AP-2αlox/lox cKO (α-cKO) mice were generated as described in Wang et al. (2006). AP-2γlox/lox mice were crossed with K14-Cre mice to generate K14-Cre/AP-2γlox/lox cKO (γ-cKO) mice (Vasioukhin et al., 1999). α-cKO and γ-cKO mice were then mated to generate K14-Cre/AP-2αlox/loxAP-2γlox/lox DcKO mice.
Immunostaining and immunoblots
Histology and immunofluorescence were performed as described previously (Wang et al., 2006). For immunohistochemistry, tissues were fixed in formaldehyde and then dehydrated and embedded in paraffin. Antigen unmasking was performed in Retriever 2100 for 20 min (PickCell Laboratories BV). Antibodies and dilutions used were as follows: AP-2α (mouse, 1:100; Millipore), K5 (chicken, 1:500; Fuchs Laboratory), AP-2γ (mouse, 1:100; Millipore), K1 (rabbit, 1:250; Fuchs Laboratory), Involucrin (rabbit, 1:200; Covance), Filaggrin (rabbit, 1:250; Covance), BrdU (rat, 1:100; Abcam), active caspase 3 (rabbit, 1:500; R&D Systems), K8 (rat, 1:1,000; Fuchs Laboratory), K6 (rabbit, 1:1,000; Fuchs Laboratory), C/EBPα (rabbit, 1:250; Santa Cruz Biotechnology, Inc.), C/EBPβ (rabbit, 1:250; Santa Cruz Biotechnology, Inc.), integrin β4 chain (rat, 1:250; BD Biosciences), Loricrin (rabbit, 1:250; Covance), K18 (rabbit, 1:50, Fuchs Laboratory), phospho-Akt (rabbit, 1:250; Cell Signaling Technology), Gata3 (mouse, 1:50; Santa Cruz Biotechnology, Inc.), AE15 (mouse, 1:50; Fuchs Laboratory), and AE13 (mouse, 1:50; Fuchs Laboratory). Secondary antibodies coupled to FITC, Alexa 488, or Texas red were obtained from Jackson ImmunoResearch Laboratories. Nuclei were labeled by DAPI for immunofluorescence.
For immunoblots, total cell lysates were prepared in lysis buffer. Samples were separated on 12% SDS-PAGE gels, transferred to nitrocellulose, and blotted overnight with the indicated antibodies.
Real-time PCR
RNAs were isolated using RNeasy Plus Mini kit (QIAGEN). RNA quality and concentration were measured by spectrophotometer (NanoDrop). Equal RNA amounts were added to reverse transcription reaction mix (Invitrogen) with oligo-dT(12) as primer. All PCR test primers flanked exon–intron boundaries to avoid misinterpretation from genomic contamination. Real-time PCRs of RNA (i.e., not reverse transcribed) were used as negative controls. Real-time PCR was conducted with a LightCycler system (LightCycler 480; Roche). Reactions were performed using the indicated primers and template mixed with the LightCycler DNA master SYBR Green kit and run for 45 cycles. Specificity of the reactions was determined by subsequent melting curve analysis. LightCycler analysis software was used to remove background fluorescence (noise band). The number of cycles needed to reach the crossing point for each sample was used to calculate the amount of each product using the 2−ΔΔCP method. Levels of PCR product were expressed as a function of GAPDH.
Primer sequences are as follows: K1F, 5′-GACACCACAACCCGGACCCAAAACTTAGAC-3′; K1R, 5′-ATACTGGGCCTTGACTTCCGAGATGATG-3′; K10F, 5′-GCCAGAACGCCGAGTACCAACAAC-3′; K10R, 5′-GTCACCTCCTCAATAATCGTCCTG-3′; FilF, 5′-GTGGCCAACAGAGAATGAG-3′; FilR, 5′-ATGATGCCCAGAACTATGTGAC-3′; Tgm1F, 5′-GCGGAGGGCTGTGGAGAAGG-3′; Tgm1R, 5′-GGGTGCGCAAACGGAAGGTG-3′; Tgm3F, 5′-GCTTGGGGGTTCGCTCTCG-3′; Tgm3R, 5′-CTGCCTACTGCCTTGGTGCTGAT-3′; K8F, 5′-TGGAGGGGGAGGAGAGCAG-3′; K8R, 5′-AAGGTTGGCCAGAGGATTAGG-3′; K6F, 5′-AGCCACCGTGGCTACAGTGC-3′; K6R, 5′- CTGGTTGATGGTGACCTCTT-3′; C/EBPαF, 5′-AGCTGGCCCCGTGAGAAAAATGA-3′; C/EBPαR, 5′-AATGGTCCCCGTGTCCTCCTATCC-3′; C/EBPβF, 5′-CTATTTCTATGAGAAAAGAGGCGTATGTAT-3′; C/EBPβR, 5′-ATTCTCCCAAAAAAGTTTATTAAAATGTCT-3′; AP-2αF, 5′-TGCGGAGAGCGAAGTC-3′; AP-2αR, 5′-GGCCTCGGTGAGATAGTTCT-3′; AP-2γF, 5′-GCCACGCGGAAGAGTATGT-3′; AP-2γR, 5′-CCCGGGTTCATGTAGGACTTA-3′; RBP/JF, 5′-AATTTCCACGCCAGTTCACAA-3′; RBP/JR, 5′-GTCCCCGCCGCCATTCA-3′.
Electron microscopy
Tissues were fixed for >1 h in 2% glutaraldehyde, 4% formaldehyde, and 2 mM CaCl2 in 0.05 M sodium cacodylate buffer, and then processed for epon embedding. Samples were visualized with a transmission electron microscope (Tecnai 12-G2; FEI Company).
Barrier function assay
Barrier assays were performed as described previously (Kaufman et al., 2003). In the absence of a functional epidermal barrier, the X-gal solution penetrates the skin, and an endogenous β-galactosidase–like activity catalyzes production of a blue precipitate.
FACS analyses
FACS analyses of keratinocytes from embryonic mouse back skin were performed as described previously (Blanpain et al., 2006). Cell cycle analyses were performed using the BrdU Flow kit (BD Biosciences). FACS purification of transduced keratinocytes was performed on a FACS Vantage SE system equipped with FACS DiVa software (BD Biosciences). Cells were gated for single events and viability, and then sorted according to their GFP expression.
Cell culture and reporter assays
Mouse keratinocytes were grown in E medium (15% serum and 50 μM CaCl2), and calcium switch experiments were performed by adding calcium chloride at a 1.5-mM final concentration (Wang et al., 2006). Retroviral transfection experiments were performed on cells grown in E-low Ca2+ media, and cultures were analyzed for gene expression 36 h later.
For colony-forming assays, equal numbers of keratinocytes isolated from postnatal day 0 (P0) back skin were serial diluted by 5× and plated onto consecutive wells of 6-well plates seeded with mitomycin-treated 3T3 fibroblasts in rich media. After 14 d, colony number and size were visualized with 1% rhodamine B (Sigma-Aldrich).
Reporter assays were performed as described previously (Wang et al., 2006). We used the dual-glo luciferase assay kit to monitor both the firefly luciferase activity driven by the C/EBPα promoter plus 5′-UTR sequence and Renilla luciferase under the control of a constitutive promoter to control for transfection efficiency. Results were expressed as a ratio of firefly to Renilla luciferase activities.
Plasmids and constructs
The C/EBPα reporter construct was a gift from D. Lane (Johns Hopkins University, Baltimore, MD). Retroviral murine stem cell virus construct encoding AP-2α was described in Wang et al. (2006). Retroviral (murine stem cell virus) construct encoding C/EBPα was obtained by inserting C/EBPα cDNA digested out of construct #10470123 (American Type Culture Collection) into murine stem cell virus. Retroviral construct encoding C/EBPβ was a gift from J. Friedman (The Rockefeller University, New York, NY). The lentiviral construct encoding a shRNA targeting RBP/J was obtained from Sigma-Aldrich.
Image acquisition and manipulation
The histology and immunofluorescent images were taken by an Axioskop 2 mot plus microscope (Carl Zeiss, Inc.). The objectives used were 20×/0.5 Plan-NEOFLUAR ∞/0.17 and 40×/1.3 oil Plan-NEOFLUAR ∞/0.17 (Carl Zeiss, Inc.). The images were taken at room temperature in antifade as an imaging medium for immunofluorescent images and 80% glycerol for hematoxylin and eosin images. The fluorochromes used were FITC, rhodamine red X, and DAPI. A camera (SPOT RT slider; Diagnostic Instruments, Inc.) and Metamorph 6 software (MDS Analytical Technologies) were used to acquire the pictures. Photoshop 6.0 (Adobe) software was used for contrast and brightness adjustment. The Immunoblot images were acquired with an Alphaimager (Alpha Innotech) and Photoshop 6.0 was used for contrast and brightness adjustment.
Online supplemental material
Fig. S1 shows that conditional loss of AP-2α and AP-2γ suppresses terminal differentiation in E18.5 and P0 epidermis. Fig. S2 shows absence of K18, but ectopic K6 expression in E18.5 and P0 DcKO epidermis. Fig. S3 shows diminished C/EBPα and C/EBPβ immunostaining in E17.5 DcKO but not in α-cKO or γ-cKO epidermis. Fig. S4 shows that AP-2α and AP-2γ are necessary for terminal differentiation within HFs. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200804030/DC1.
Supplementary Material
Acknowledgments
We are grateful for the C/EBPα reporter construct from Daniel Lane and the C/EBPβ retroviral expression vector from Jeffrey Friedman. We thank Lisa Polak and Nicole Stokes for their expert assistance in skin engraftments; Svetlana Mazel for flow cytometry; and Valerie Horsley, Haiying Zhang, and other members of the Fuchs Laboratory for their thoughtful suggestions and advice.
E. Fuchs is an Investigator of the Howard Hughes Medical Institute. This work was supported by the National Institutes of Health grants NIH-RO1-AR31737 (E. Fuchs) and DE12728 (T. Williams).
Abbreviations used in this paper: 1°MK, primary mouse epidermal keratinocytes; C/EBP, CCAAT/enhancer binding protein; cKO, conditional knockout; DcKO, double cKO; DKO, double knockout; DP, dermal papilla; E, embryonic day; EGFR, EGF receptor; HF, hair follicle; NICD, Notch intracellular domain; P0, postnatal day 0; shRNA, short hairpin RNA; WT, wild type.
References
- Artavanis-Tsakonas, S., M.D. Rand, and R.J. Lake. 1999. Notch signaling: cell fate control and signal integration in development. Science. 284:770–776. [DOI] [PubMed] [Google Scholar]
- Blanpain, C., and E. Fuchs. 2006. Epidermal stem cells of the skin. Annu. Rev. Cell Dev. Biol. 22:339–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain, C., W.E. Lowry, A. Geoghegan, L. Polak, and E. Fuchs. 2004. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 118:635–648. [DOI] [PubMed] [Google Scholar]
- Blanpain, C., W.E. Lowry, H.A. Pasolli, and E. Fuchs. 2006. Canonical notch signaling functions as a commitment switch in the epidermal lineage. Genes Dev. 20:3022–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull, J.J., S. Muller-Rover, C.M. Chronnell, R. Paus, M.P. Philpott, and I.A. McKay. 2002. Contrasting expression patterns of CCAAT/enhancer-binding protein transcription factors in the hair follicle and at different stages of the hair growth cycle. J. Invest. Dermatol. 118:17–24. [DOI] [PubMed] [Google Scholar]
- Byrne, C., M. Tainsky, and E. Fuchs. 1994. Programming gene expression in developing epidermis. Development. 120:2369–2383. [DOI] [PubMed] [Google Scholar]
- Calkhoven, C.F., C. Muller, and A. Leutz. 2000. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 14:1920–1932. [PMC free article] [PubMed] [Google Scholar]
- Claudinot, S., M. Nicolas, H. Oshima, A. Rochat, and Y. Barrandon. 2005. Long-term renewal of hair follicles from clonogenic multipotent stem cells. Proc. Natl. Acad. Sci. USA. 102:14677–14682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, X., and J.A. Segre. 2004. Transcriptional control of epidermal specification and differentiation. Curr. Opin. Genet. Dev. 14:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert, D., S. Buhl, S. Weber, R. Jager, and H. Schorle. 2005. The AP-2 family of transcription factors. Genome Biol. 6:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrach, S., R. Cordes, K. Hozumi, A. Gossler, and F.M. Watt. 2007. Role of the Notch ligand Delta1 in embryonic and adult mouse epidermis. J. Invest. Dermatol. 128:825–832. [DOI] [PubMed] [Google Scholar]
- Fuchs, E. 2007. Scratching the surface of skin development. Nature. 445:834–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, E., and H. Green. 1980. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell. 19:1033–1042. [DOI] [PubMed] [Google Scholar]
- Guttormsen, J., M.I. Koster, J.R. Stevens, D.R. Roop, T. Williams, and Q.A. Winger. 2008. Disruption of epidermal specific gene expression and delayed skin development in AP-2 gamma mutant mice. Dev. Biol. 317:187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso, T., L. Kedes, and Y. Hamamori. 2003. HES and HERP families: multiple effectors of the Notch signaling pathway. J. Cell. Physiol. 194:237–255. [DOI] [PubMed] [Google Scholar]
- Jiang, M.S., Q.Q. Tang, J. McLenithan, D. Geiman, W. Shillinglaw, W.J. Henzel, and M.D. Lane. 1998. Derepression of the C/EBPalpha gene during adipogenesis: identification of AP-2alpha as a repressor. Proc. Natl. Acad. Sci. USA. 95:3467–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman, C.K., P. Zhou, H.A. Pasolli, M. Rendl, D. Bolotin, K.C. Lim, X. Dai, M.L. Alegre, and E. Fuchs. 2003. GATA-3: an unexpected regulator of cell lineage determination in skin. Genes Dev. 17:2108–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerber, B., I. Monge, M. Mueller, P.J. Mitchell, and S.M. Cohen. 2001. The AP-2 transcription factor is required for joint formation and cell survival in Drosophila leg development. Development. 128:1231–1238. [DOI] [PubMed] [Google Scholar]
- Koster, M.I., S. Kim, J. Huang, T. Williams, and D.R. Roop. 2006. TAp63alpha induces AP-2gamma as an early event in epidermal morphogenesis. Dev. Biol. 289:253–261. [DOI] [PubMed] [Google Scholar]
- Lai, E.C. 2004. Notch signaling: control of cell communication and cell fate. Development. 131:965–973. [DOI] [PubMed] [Google Scholar]
- Leask, A., M. Rosenberg, R. Vassar, and E. Fuchs. 1990. Regulation of a human epidermal keratin gene: sequences and nuclear factors involved in keratinocyte-specific transcription. Genes Dev. 4:1985–1998. [DOI] [PubMed] [Google Scholar]
- Lee, J., J.M. Basak, S. Demehri, and R. Kopan. 2007. Bi-compartmental communication contributes to the opposite proliferative behavior of Notch1-deficient hair follicle and epidermal keratinocytes. Development. 134:2795–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell, S., P. Jones, I. Le Roux, J. Dunne, and F.M. Watt. 2000. Stimulation of human epidermal differentiation by delta-notch signalling at the boundaries of stem-cell clusters. Curr. Biol. 10:491–500. [DOI] [PubMed] [Google Scholar]
- Luo, T., M. Matsuo-Takasaki, M.L. Thomas, D.L. Weeks, and T.D. Sargent. 2002. Transcription factor AP-2 is an essential and direct regulator of epidermal development in Xenopus. Dev. Biol. 245:136–144. [DOI] [PubMed] [Google Scholar]
- Luo, T., Y.H. Lee, J.P. Saint-Jeannet, and T.D. Sargent. 2003. Induction of neural crest in Xenopus by transcription factor AP2alpha. Proc. Natl. Acad. Sci. USA. 100:532–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, T., Y. Zhang, D. Khadka, J. Rangarajan, K.W. Cho, and T.D. Sargent. 2005. Regulatory targets for transcription factor AP2 in Xenopus embryos. Dev. Growth Differ. 47:403–413. [DOI] [PubMed] [Google Scholar]
- Mack, J.A., S. Anand, and E.V. Maytin. 2005. Proliferation and cornification during development of the mammalian epidermis. Birth Defects Res. C. Embryo Today. 75:314–329. [DOI] [PubMed] [Google Scholar]
- Maytin, E.V., J.C. Lin, R. Krishnamurthy, N. Batchvarova, D. Ron, P.J. Mitchell, and J.F. Habener. 1999. Keratin 10 gene expression during differentiation of mouse epidermis requires transcription factors C/EBP and AP-2. Dev. Biol. 216:164–181. [DOI] [PubMed] [Google Scholar]
- Monge, I., R. Krishnamurthy, D. Sims, F. Hirth, M. Spengler, L. Kammermeier, H. Reichert, and P.J. Mitchell. 2001. Drosophila transcription factor AP-2 in proboscis, leg and brain central complex development. Development. 128:1239–1252. [DOI] [PubMed] [Google Scholar]
- Nagarajan, P., R.A. Romano, and S. Sinha. 2008. Transcriptional control of the differentiation program of interfollicular epidermal keratinocytes. Crit. Rev. Eukaryot. Gene Expr. 18:57–79. [DOI] [PubMed] [Google Scholar]
- Nelson, D.K., and T. Williams. 2004. Frontonasal process-specific disruption of AP-2alpha results in postnatal midfacial hypoplasia, vascular anomalies, and nasal cavity defects. Dev. Biol. 267:72–92. [DOI] [PubMed] [Google Scholar]
- Nerlov, C. 2007. The C/EBP family of transcription factors: a paradigm for interaction between gene expression and proliferation control. Trends Cell Biol. 17:318–324. [DOI] [PubMed] [Google Scholar]
- Nicolas, M., A. Wolfer, K. Raj, J.A. Kummer, P. Mill, M. van Noort, C.C. Hui, H. Clevers, G.P. Dotto, and F. Radtke. 2003. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 33:416–421. [DOI] [PubMed] [Google Scholar]
- Okuyama, R., B.C. Nguyen, C. Talora, E. Ogawa, A. Tommasi di Vignano, M. Lioumi, G. Chiorino, H. Tagami, M. Woo, and G.P. Dotto. 2004. High commitment of embryonic keratinocytes to terminal differentiation through a Notch1-caspase 3 regulatory mechanism. Dev. Cell. 6:551–562. [DOI] [PubMed] [Google Scholar]
- Otto, T.C., and M.D. Lane. 2005. Adipose development: from stem cell to adipocyte. Crit. Rev. Biochem. Mol. Biol. 40:229–242. [DOI] [PubMed] [Google Scholar]
- Pan, Y., M.H. Lin, X. Tian, H.T. Cheng, T. Gridley, J. Shen, and R. Kopan. 2004. gamma-secretase functions through Notch signaling to maintain skin appendages but is not required for their patterning or initial morphogenesis. Dev. Cell. 7:731–743. [DOI] [PubMed] [Google Scholar]
- Panteleyev, A.A., P.J. Mitchell, R. Paus, and A.M. Christiano. 2003. Expression patterns of the transcription factor AP-2alpha during hair follicle morphogenesis and cycling. J. Invest. Dermatol. 121:13–19. [DOI] [PubMed] [Google Scholar]
- Pellikainen, J.M., and V.M. Kosma. 2007. Activator protein-2 in carcinogenesis with a special reference to breast cancer—a mini review. Int. J. Cancer. 120:2061–2067. [DOI] [PubMed] [Google Scholar]
- Pfisterer, P., J. Ehlermann, M. Hegen, and H. Schorle. 2002. A subtractive gene expression screen suggests a role of transcription factor AP-2 alpha in control of proliferation and differentiation. J. Biol. Chem. 277:6637–6644. [DOI] [PubMed] [Google Scholar]
- Rothnagel, J.A., T. Mehrel, W.W. Idler, D.R. Roop, and P.M. Steinert. 1987. The gene for mouse epidermal filaggrin precursor. Its partial characterization, expression, and sequence of a repeating filaggrin unit. J. Biol. Chem. 262:15643–15648. [PubMed] [Google Scholar]
- Rowland, B.D., and D.S. Peeper. 2006. KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer. 6:11–23. [DOI] [PubMed] [Google Scholar]
- Segre, J.A. 2006. Epidermal barrier formation and recovery in skin disorders. J. Clin. Invest. 116:1150–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha, S., L. Degenstein, C. Copenhaver, and E. Fuchs. 2000. Defining the regulatory factors required for epidermal gene expression. Mol. Cell. Biol. 20:2543–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snape, A.M., R.S. Winning, and T.D. Sargent. 1991. Transcription factor AP-2 is tissue-specific in Xenopus and is closely related or identical to keratin transcription factor 1 (KTF-1). Development. 113:283–293. [DOI] [PubMed] [Google Scholar]
- Tang, Z., I. Treilleux, and M. Brown. 1997. A transcriptional enhancer required for the differential expression of the human estrogen receptor in breast cancers. Mol. Cell. Biol. 17:1274–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasioukhin, V., L. Degenstein, B. Wise, and E. Fuchs. 1999. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc. Natl. Acad. Sci. USA. 96:8551–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernimmen, D., D. Begon, C. Salvador, S. Gofflot, M. Grooteclaes, and R. Winkler. 2003. Identification of HTF (HER2 transcription factor) as an AP-2 (activator protein-2) transcription factor and contribution of the HTF binding site to ERBB2 gene overexpression. Biochem. J. 370:323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., D. Bolotin, D.H. Chu, L. Polak, T. Williams, and E. Fuchs. 2006. AP-2α: a regulator of EGF receptor signaling and proliferation in skin epidermis. J. Cell Biol. 172:409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Y.X., K. Somasundaram, and W.S. el-Deiry. 1997. AP2 inhibits cancer cell growth and activates p21WAF1/CIP1 expression. Nat. Genet. 15:78–82. [DOI] [PubMed] [Google Scholar]
- Zhang, J., S. Hagopian-Donaldson, G. Serbedzija, J. Elsemore, D. Plehn-Dujowich, A.P. McMahon, R.A. Flavell, and T. Williams. 1996. Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature. 381:238–241. [DOI] [PubMed] [Google Scholar]
- Zhu, S., H.S. Oh, M. Shim, E. Sterneck, P.F. Johnson, and R.C. Smart. 1999. C/EBPbeta modulates the early events of keratinocyte differentiation involving growth arrest and keratin 1 and keratin 10 expression. Mol. Cell. Biol. 19:7181–7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}