

Abstract
The recent discovery of positive allosteric modulators (PAMs) for G-protein coupled receptors (GPCRs) open new possibilities to control a number of physiological and pathological processes. Understanding the mechanism of action of such compounds will provide new information on the activation process of these important receptors. Within the last 10 years, a number of studies indicate that GPCRs can form dimers, but the functional significance of this phenomena remains elusive. Here we used the metabotropic glutamate receptors as a model since these receptors, for which PAMs have been identified, are constitutive dimers. We used the quality control system of the GABAB receptor to generate mGlu dimers in which a single subunit binds a PAM. We show that one PAM per dimer is sufficient to enhance receptor activity. Such a potentiation can still be observed if the subunit unable to bind the PAM is also made unable to activate G-proteins. However, the PAM acts as a non competitive antagonist when it binds in the subunit that cannot activate G-proteins. These data are consistent with a single heptahelical domain reaching the active state per dimer during receptor activation.
Keywords: Actins; chemistry; Allosteric Site; Binding, Competitive; Cell Line; Cell Membrane; metabolism; Dimerization; Enzyme-Linked Immunosorbent Assay; Fluorescence Resonance Energy Transfer; GTP-Binding Proteins; chemistry; Humans; Inositol Phosphates; chemistry; Models, Biological; Mutagenesis, Site-Directed; Plasmids; metabolism; Point Mutation; Protein Binding; Protein Conformation; Receptors, G-Protein-Coupled; chemistry; physiology; Receptors, GABA-B; chemistry; physiology; Receptors, Metabotropic Glutamate; chemistry; Recombinant Fusion Proteins; chemistry; Time Factors
INTRODUCTION
G-protein coupled receptors (GPCRs) constitute the largest family of membrane proteins. These receptors allow a wide variety of extracellular signals ranging from photons to large glycoproteins to generate intracellular responses by activating heterotrimeric G-proteins (1). Although first considered as monomeric proteins, a growing number of studies are pointing out that GPCRs likely form dimers (see (2,3) for review). This phenomena is involved in the different stages of the GPCR “life”, including regulation of plasma membrane targeting, internalization and recycling. However the functional importance of dimerization in G-protein activation remains elusive.
Within the large family of GPCRs, those of the class C constitute a good model to study the relevance of GPCR dimerization in G-protein activation since all are constitutive dimers, either homodimers linked by a disulfide bridge (4,5), or obligatory heterodimers (6–10). This class of GPCRs includes the receptors for the main neurotransmitters, glutamate and GABA, the calcium-sensing receptor, the receptors for sweet and umami taste plus several pheromone receptors (11). Like any other GPCRs, these receptors possess a heptahelical domain (HD) involved in G-protein activation. However, they possess a large extracellular domain (ECD) where agonists bind. Dimerization of Class-C GPCRs is a prerequisite for their function (12). Indeed agonist binding in the ECD stabilizes its closed state (13–16), and leads to a change in the relative orientation of the two ECDs within the dimer. This major change in conformation of the dimer of ECDs is assumed to be associated with a conformational change in the dimer of HDs, leading to its activation (12,17).
Positive allosteric modulators (PAMs) constitute a new class of synthetic ligands acting on GPCRs which have been discovered within the last years. In contrast to full agonists, PAMs are either devoid of agonist activity or act as very partial agonists, but significantly enhance the potency and/or efficacy of agonists. Such molecules offer an excellent alternative to agonists in therapeutics (18–21), as illustrated with cinacalcet, the first GPCR PAM now on the market (22). A number of such compounds have been identified for class C GPCRs and have been shown to bind in a site equivalent to the retinal binding pocket of opsins (23–25). Recently, we reported that PAMs act as full agonists on either mGlu or GABAB receptors deleted of their large ECD (26,27). This indicates that PAMs can stabilize the active state of the HD, an effect inhibited by the ECD in the absence of agonist.
In the present study, we have examined whether one or two PAM(s) is/are required per mGlu dimer to enhance the effect of agonists. Using a system which allows us to control the subunit composition within a defined dimer (28,29), our results demonstrate that only one PAM is sufficient. Moreover, our work brings new data supporting the proposal that only one HD within these homodimeric receptors can reach an active conformation, further demonstrating the asymmetric functioning of the dimer of HDs. This represents an additional evidence for dimeric GPCRs activating a single heterotrimeric G-protein at a time.
EXPERIMENTAL PROCEDURES
Materials
L-Quisqualic acid was purchased from Tocris Cookson (Bristol; UK). Glutamate-pyruvate transaminase (GPT) was purchased from Roche (Basel, Switzerland). Culture medium, fœtal calf serum (FCS) and other products used for cell culture were purchased from GIBCO-BRL-Life Technologies, Inc. (Cergy Pontoise, France). Myo-inositol (23.4 Ci/mol) was purchased from Amersham (Saclay, France).Diphenylacetylcarbamic acid ethyl ester (Ro01-6128) (24) and 9H-xanthene-9-carboxylic acid (2-isopropyl-2H-tetrazol-5-yl)-amide (XITA) (30) were synthesized in house. 3,3′-difluorobenzaldazine (DFB) was synthesized as already described (27).
Plasmids, mutagenesis and construction of chimeric mGlu receptors
In order to perform ELISA or FRET experiments, a hemagglutinin (HA) or a myc tags were introduced in the N-terminal end, right after the signal peptide of the different receptors. The construction of the N-terminal HA- or Myc- tagged rat mGlu1 or mGlu5a, pRKG5a-NHA, have been already described (6,31). As previously reported for different mGlu (32,33) or GABAB receptors (6), the presence of these tags did not modify the expression or the pharmacological properties of the receptor. The mGlu1C1, mGlu5C1, mGlu1C2 and mGlu5C2 chimera have been obtained by replacing the C-terminus of HA- or Myc-mGlu1 and HA- or Myc-mGlu5a by that of GABAB1a and GABAB2, respectively (28,29). The mGlu5 mutant sensitive to Ro01-6128, mGlu5Ro, has been obtained by introducing 3 point mutations in mGlu5: P654S and S657V in the third transmembrane segment and L743V in the fifth transmembrane segment (24). Mutations were obtained using the Quick-Change® strategy (Stratagene) and all constructions were verified by DNA sequencing.
Cell culture and transfection
HEK293 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% FCS and antibiotics (Penicillin and Streptomycin, 100 U/ml final). Cells were transiently transfected by electroporation as described elsewhere (34). To avoid any influence of glutamate in the assay medium released by the cells, the high affinity glutamate transporter EAAC1 was also co-transfected with the receptor.
Cell surface quantification by ELISA
Cell surface and total cell expression of N-terminal HA-tagged receptors was quantified by ELISA as described elsewhere (27). Transfected cells were first fixed with paraformaldehyde, then permeabilized or not (in order to measure total or cell surface expression, respectively) using Triton X100 and incubated for 30 min with rat monoclonal anti-HA antibody (clone 3F10, Roche). Cells were then incubated with a secondary goat antibody conjugated to peroxydase (Jackson ImmunoResearch, West Grove, PA) which was further detected and quantified by chemiluminescence using Supersignal West Femto (Pierce, Rockford, IL) and a Wallac Victor2 luminescence counter (Molecular devices, St Gregoire, France).
Time-resolved fluorescence resonance energy transfer (TR-FRET) analysis
Protein-protein interaction at the surface of living cells was analyzed by TR-FRET. These experiments are based on the use of specific antibodies against each partner and labeled either with Eu3+-cryptate or with AlexaFluor 647 which allows the measurement of a FRET signal in a time-resolved manner if both fluorophores are in close proximity. TR-FRET experiment were performed as previously described (35). Briefly, HEK293 cells expressing the indicated HA- or Myc-tagged receptor subunits were labeled with monoclonal anti-HA (12CA5) and/or anti-Myc (9E10; American Type Culture Collection no. CRL-1729) carrying either Eu3+-Cryptate PBP or AlexaFluor 647 (provided by Cis Bio International Research). Fluorescence of Eu3+-cryptate and TR-FRET signal were measured at 620 and 665 nm, respectively, 50 μs after excitation at 337 nm, using a RubyStar fluorimeter (BMG Labtechnologies, Champigny-sur-Marne, France). In parallel, the total fluorescence emitted at 682 nm by the bound Alexa647 antibodies was quantified after excitation at 640 nm using an Analyst™ reader (Molecular Devices). The FRET signal was measured as ΔR using the following equation: where (R665/620)POS is the ratio of the 665 nm signal over that at 620 nm measured in the presence of both antibodies, and (R665/620)NEG is the same ratio measured in the absence of the acceptor-labeled antibody.
Functionnal assays
Inositol phosphate (IP) accumulation and measurement of intracellular Ca2+ mobilization in transfected cells were performed in 96-well microplates as already described (27). The dose-response curves were fitted using the GraphPad Prism program and the following equation: where EC50 is the concentration of the compound necessary to obtain the half maximal effect and n is the Hill coefficient.
RESULTS
The aim of the present study was to determine whether one PAM is sufficient to enhance the effect of agonist on a mGlu dimer or if two PAMs (one per monomer) are required. To answer this question, we first aimed at generating a mGlu5 dimer in which only one subunit is sensitive to the mGlu1 PAM Ro01-6128.
Construction of a mGlu5 triple mutant (mGlu5R0) sensitive to Ro01-6128
Ro01-6128 and DFB are known specific PAMs of mGlu1 and mGlu5, respectively (24,27,36). Accordingly, the EC50 value of quisqualate (a potent mGlu1 and mGlu5 agonist) on mGlu1 was significantly decreased in the presence of Ro01-6128, but not in the presence of DFB (Fig. 1A). Conversely, DFB significantly decreased the EC50 value of quisqualate on mGlu5 while Ro01-6128 had no effect (Fig. 1B). In order to have access to a mGlu5 receptor sensitive to the mGlu1 PAM, 3 point mutations were introduced into the mGlu5 HD according to Knoflach et al. (24). As previously described, this mGlu5Ro mutant was sensitive to Ro01-6128, although the increase in agonist potency is not as large as that measured on mGlu1, but retained its sensitivity to DFB (Fig. 1C).
Fig. 1. Effect of positive allosteric modulators on mGlu1, mGlu5 and mGlu5Ro quisqualate-induced activity.
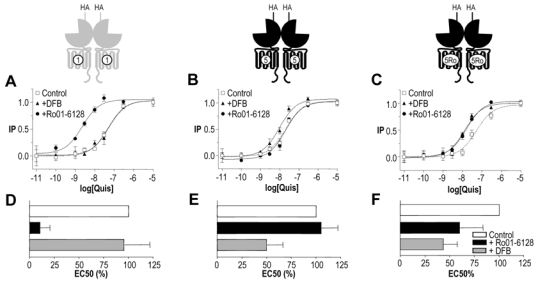
A–C. Effect of increasing doses of quisqualate on IP production by mGlu1, mGlu5 or mGlu5Ro receptor in the absence (open squares) or in presence of Ro01-6128 (black circles) or DFB (black triangles). Values are means ± S.E.M. of triplicate determinations from a representative experiment. D–F. Effect of Ro01-6128 and DFB on EC50 of quisqualate on mGlu1, mGlu5 or mGlu5Ro. EC50 values are normalized to that measured in the absence of modulator. Values are mean ± S.E.M. of n = 10, 8 and 7 experiments respectively.
Generation of mGlu5Ro:mGlu5 heterodimers
By co-expressing mGlu5Ro and mGlu5 is the same cells, one is expecting that about 50% of the surface receptors correspond to the mGlu5Ro:mGlu5 heterodimer. In order to largely increase the proportion of this receptor combination, we took advantage of the quality control system of the GABAB receptor. Indeed, in this heterodimeric receptor, the GABAB1 subunit is retained in the endoplasmic reticulum unless its cytoplasmic ER retention signal is masked by the C-terminal tail of the associated GABAB2 subunit (7). We previously reported that this quality control system can be transferred to mGlu receptors by swapping their C-terminal tail by that of GABAB1 (C1) or GABAB2 (C2) (28,29). Moreover, our previous data demonstrated that the G-protein coupling efficacies of mGlu receptors bearing either the C1 or C2 tail were equivalent.
Here, we swapped the C-terminal tail of mGlu5Ro by that of GABAB1. This mGlu5RoC1 subunit does not reach the cell surface, unless co-expressed with mGlu5C2 that possesses the C-terminal tail of GABAB2, as shown by ELISA performed on intact cells (Fig. 2A–B) and functional analysis (data not shown) (28,29). The presence of the HA-mGlu5RoC1:myc-mGlu5C2 dimers at the cell surface was verified by a HTRF approach (35), using anti-HA and anti-myc antibodies labeled with Europium-cryptate and AlexaFluor®647, respectively (Fig. 2C). No such a HTRF signal was measured in cells expressing HA-mGlu5 and myc-V2 receptors despite a similar expression level of these two receptors, demonstrating the specificity of this assay (data not shown)(28). Moreover, while a clear HTRF signal was obtained in cells expressing HA-mGlu5 using anti-HA antibodies labeled with either the donor or acceptor fluorophores, no such signal could be detected in cells co-expressing HA-mGlu5RoC1 and myc-mGlu5C2 (Fig. 2C). This demonstrates the absence of HA-mGlu5RoC1 homodimers at the cell surface.
Fig. 2. Cell surface expression, dimerization and pharmacology of mGlu5RoC1:mGlu5C2 chimera receptors.

A. Cartoon depicting the expected cell distribution of HA-mGlu5RoC1 and Myc-mGlu5C2 cotransfected in the same cell. PM stands for Plasma Membrane and ER for Endoplasmic reticulum while HA- or Myc-indicates that the receptor possesses either a HA- or a Myc-tag at its N-terminal extremity. B. Percentage of receptor reaching the cell surface as determined by anti-HA ELISA on intact versus permeabilized cells. C. Protein-protein interaction as detected by TR-FRET. Anti-HA and/or anti-Myc antibodies carrying either Eu3+-Cryptate PBP or AlexaFluor 647 were used in these experiments. Values are means ± S.E.M. of triplicate determinations from a representative experiment. D. Effect of Ro01-6128 and DFB on EC50 of quisqualate on mGlu5RoC1:mGlu5C2. EC50 are normalized to 100% in control conditions. Values are mean ± S.E.M. of n = 10 experiments and are expressed as percentage of the EC50 value measured in the absence of modulator.
Although mGlu5C2 dimers are expected to reach the cell surface in cells expressing both this subunit and mGlu5RoC1, the proportion of mGlu5C2 homodimers can be maintained to a minimum by transfecting the cells with a higher proportion of mGlu5RoC1 plasmid (29).
Ro01-6128 acts as a positive allosteric modulator of the mGlu5RoC1:mGlu5C2 dimer
In cells co-expressing mGlu5RoC1 and mGlu5C2, both Ro01-6128 and DFB significantly enhanced the potency of quisqualate (Fig. 2D). Since in these cells only the mGlu5RoC1:mGlu5C2 combination and mGlu5C2 homodimers reach the cell surface, the potentiation observed with Ro01-6128 can only results from its action on the mGlu5RoC1:mGlu5C2 heterodimers. This indicates that a single PAM per dimer can lead to a clear enhancement of agonist potency. Of interest, the potentiation observed with Ro01-6128 is similar to that measured on homodimeric mGlu5Ro, suggesting that a single PAM is sufficient for the full enhancement of agonist action on the dimer. In agreement with this possibility, DFB that binds in both mGlu5RoC1 and mGlu5C2 subunit displays a similar effect as Ro01-6128 in cells expressing these two subunits (Fig. 2D).
Generation of heterodimeric mGlu1:mGlu5 receptors
In order to further confirm that a single PAM per dimer can potentiate the activity of these receptors, we aimed at generating a receptor dimer in which each subunit can be targeted by a specific PAM. To that aim, we examined whether a mGlu1:mGlu5 heterodimer could be generated in cells co-transfected with HA-mGlu1C1 and Myc-mGlu5C2 or with HA-mGlu5C1 and Myc-mGlu1C2.
As shown in Fig. 3A, while mGlu1C1 and mGlu5C1 expressed alone are retained in the ER, mGlu5C2 and mGlu1C2 allow the surface targetting of mGlu1C1 and mGlu5C1, respectively. As described above, the presence of HA-mGlu1C1:myc-mGlu5C2 and HA-mGlu5C1:myc-mGlu1C2 heterodimers at the cell surface was clearly demonstrated with the HTRF approach using anti-HA and anti-Myc antibodies labeled with donor or acceptor fluorophores (Fig. 3B). In addition, mGlu1C1 or mGlu5C1 homodimers could not be detected with this technique in these same cells.
Fig. 3. Cell surface expression, dimerization and pharmacology of mGlu1C1:mGlu5C2 and mGlu5C1:mGlu1C2 heterodimers.
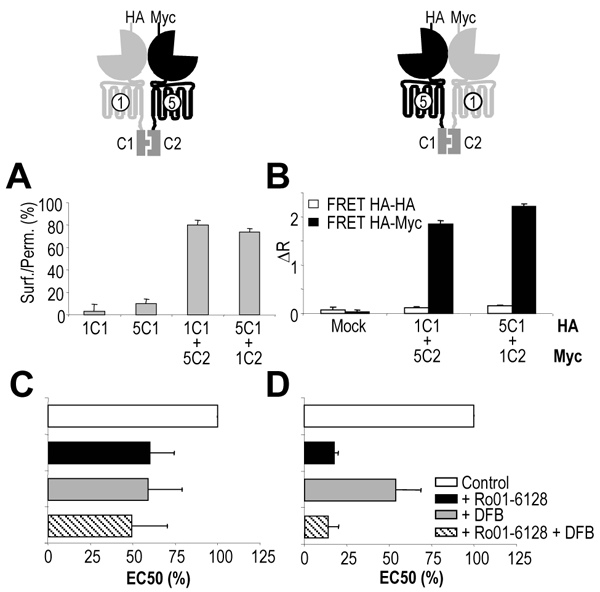
A. Percentage of receptor reaching the cell surface as determined by anti-HA ELISA on intact and permeabilized cells. B. Protein-protein interaction as detected by TR-FRET using anti-HA and/or anti-Myc antibodies carrying either Eu3+-Cryptate PBP or AlexaFluor 647. Values are means ± S.E.M. of triplicate determinations from a representative experiment. C, D. Effect of Ro01-6128 and DFB on EC50 of quisqualate on mGlu1C1:mGlu5C2 and mGlu5C1:mGlu1C2. EC50 values are expressed as percentage of the EC50 value measured in the absence of modulator. Values are mean ± S.E.M. of n = 10 and 5 experiments respectively.
Since no mGlu1:mGlu5 heterodimers have been reported with the wild-type subunits (4), the replacement of the C-terminal tails of these receptors by those of GABAB1 and GABAB2 is sufficient to allow the formation of such heterodimers.
Heterodimeric mGlu1:mGlu5 receptors are positively modulated by both Ro01-6128 and DFB
As shown in Fig. 3, both the mGlu1 and mGlu5 PAMs potentiated the effect of quisqualate in cells co expressing mGlu1C1 and mGlu5C2 (Fig. 3C) as well as in cells co-expressing mGlu5C1 and mGlu1C2 (Fig. 3D), as indicated by the increase in agonist potency (decrease of the EC50). In cells expressing mGlu1C1 and mGlu5C2, the potentiation by Ro01-6128 can only result from the effect of this PAM on the mGlu1C1:mGlu5C2 heterodimer, since this receptor combination is the only one at the cell surface containing a Ro01-6128 binding site. Similarly, the effect of DFB on cells expressing mGlu5C1 and mGlu1C2 can only result from the enhancement of the agonist potency at the mGlu5C1:mGlu1C2. This further demonstrates that a single PAM per dimer is sufficient to enhance agonist potency at these receptors.
Of interest, when both PAMs were added simultaneously, no significant further potentiation was observed, suggesting that a single PAM per dimer is sufficient for the full enhancement of the receptor dimers activity (Fig. 3C,D).
Generation of heterodimeric mGlu receptors in which one subunit is unable to activate G proteins
Because our data demonstrate that one PAM is sufficient to positively modulate the activation of mGlu dimers, we next aimed at examining the effect of PAMs on receptor dimers in which one subunit is unable to activate G-proteins. We previously reported that a point mutation (F781P) in the third (i3) intracellular loop of mGlu1 receptors prevents their ability to activate G-proteins (29,37). Such a mutation was introduced in myc-mGlu1C2 and the corresponding mutant named myc-mGlu1XC2. This mutant receptor was correctly trafficked to the cell surface but no longer activate PLC (data not shown) (28,29). MGlu1XC2 allowed the plasma membrane insertion of HA-mGlu5C1 and the presence of HA-mGlu5C1:myc-mGlu1XC2 heterodimers at the cell surface was confirmed by the large HTRF signal (data not shown). Moreover, the co-expression of these subunits leads to the formation of functional receptors (Fig. 4). Such functional response can only results from the heteromeric C1:C2 combination, since the C2 homodimers do not activate G-proteins because of the mutation in their i3 loop, and the C1 dimers are retained inside the cell.
Fig. 4. Effect of positive allosteric modulators on heterodimers in which one subunit is unable to activate G-proteins.
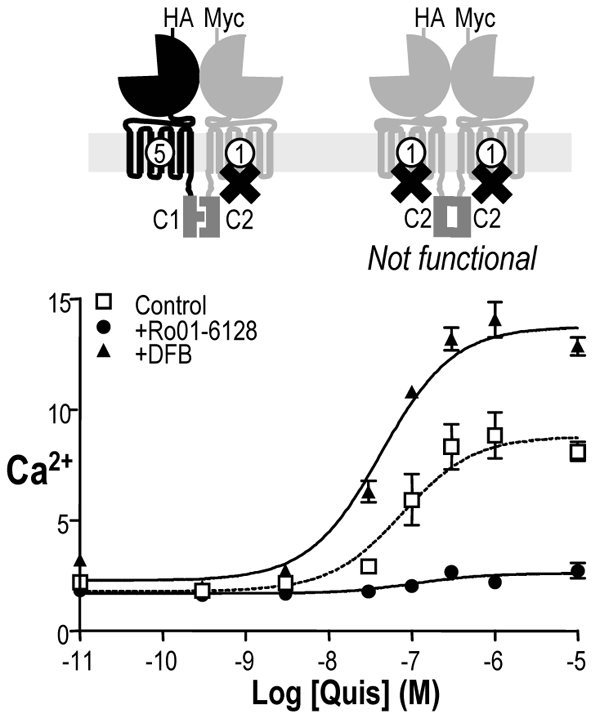
A. Effect of increasing doses of quisqualate on IP production induced by mGlu5C1:mGlu1XC2 receptor activation in the absence of modulator (open squares), in presence of Ro01-6128 (black circles) or DFB (black triangles). Values are means ± S.E.M. of n = 5 experiments.
PAMs act as non-competitive antagonists when interacting in the subunit unable to activate G-proteins
As shown in Fig. 4, the mGlu5 PAM DFB enhanced agonist activity at the mGlu5C1:mGlu1XC2 combination, indicating that potentiation can be observed when PAMs bind in the functional HD. Of interest, when the PAM binds in the HD unable to activate G-proteins, then it acts as a non competitive antagonist rather than a positive modulator. Indeed, the mGlu1 PAM Ro01-6128 decreases the maximal agonist effect on the mGlu5C1:mGlu1XC2 combination (Fig. 4). These results are not due to a particularity of Ro01-6128 itself since similar results were obtained using XITA, another mGlu1 selective PAM (data not shown).
DISCUSSION
We previously demonstrated that PAMs of class C GPCRs act by stabilizing the active conformation of the HD (26,27). By using the quality control system of the GABAB receptor incorporated in mGlu receptor subunits, we generated receptor dimers containing a single binding site for a PAM. Using this approach, we show here that a single PAM per dimer is sufficient to fully potentiate receptor activity, further demonstrating the asymmetric functioning of the dimer of HDs within this class of GPCRs.
To generate mGlu dimers possessing a single site for a PAM, we first mutated the mGlu5 receptor to make it sensitive to the mGlu1 PAM Ro01-6128. In agreement with published data the three mutations introduced in TM3 and TM5 were sufficient to allow Ro01-6128 to enhance agonist potency on the mGlu5 receptor. However, these three mutations did not prevent the effect of the mGlu5 selective PAM DFB, indicating that the residues responsible for the selectivity of Ro01-6128 and DFB are not at the same position in the mGlu1 and mGlu5 HD. Accordingly, either Ro01-6128 and DFB bind at different sites, or they bind differently in a similar binding pocket. By examining in more details the divergence between mGlu1 and mGlu5 receptors at the level of the proposed Ro01-6128 binding pocket, additional positions can be identified that may possibly be involved in the selective action of DFB.
As previously reported, the exchange of the C-terminal tail of mGlu receptors by that of either GABAB1 (C1) or GABAB2 (C2), allows to largely increase the proportion of receptor dimers composed of two defined subunits at the cell surface. Indeed, the receptors possessing the C1 tail reach the cell surface only when associated with the receptor chimera containing the C2 tail. Such heterodimers can be detected at the cell surface with the HTRF approach, and, because a disulfide bridge linked the subunits of a mGlu dimer, such heterodimers are not expected to dissociate at the cell surface. Indeed, no homodimers of subunits bearing the C1 tail can be detected with the HTRF technology. Of interest, this approach also allows the formation of mGlu1:mGlu5 heterodimers. This is in contrast with what has been reported with the full length wild-type subunits (4). This suggests that the C-terminal tails of these receptors play a major role in their selective interaction. In agreement with this proposal, mGlu1 and mGlu5 receptors mostly differ at the level of their C-terminal tails. Whereas the sequence identity between the ECDs and HDs of these two receptors is 75%, it is less than 30% for the C-terminal tails. Further supporting the involvement of the C-terminal tail in the control of mGlu receptor dimer formation, the mGlu 1a and mGlu1b receptor splice variants that differ in their C-terminal tail only, have been shown not to form heterodimers (38). However, we cannot exclude the possibility that the formation of the mGlu1:mGlu5 dimer is the consequence of a strong coiled-coil interaction between the GABAB1 and GABAB2 C-tails.
Using this approach, we show that a single PAM per dimer can augment the potency of agonists. This is nicely illustrated by the increase in quisqualate potency by Ro01-6128 in cells co-expressing mGlu5RoC1 and mGlu5C2, as well as in cells co-expressing mGlu1C1 and mGlu5C2. Indeed, at the surface of these cells, only the C1:C2 heterodimers possess a binding site for Ro01-6128, and, moreover, a single site per dimer. Similarly, the mGlu5 selective PAM, DFB, increases agonist potency in cells expressing mGlu5C1 and mGlu1C2. Of interest, the potentiating effect of a single PAM per dimer is similar to that obtained with receptor dimers in which two PAMs can theoretically bind per dimer. This suggests that a single PAM per dimer is sufficient for the full enhancement. In agreement with this proposal, the presence of both mGlu1 and mGlu5 PAMs in cells expressing mGlu1:mGlu5 heterodimers did not further increase the potentiation obtained with the most efficient PAM applied alone.
Because PAMs have been shown to stabilize the active conformation of the HD of either mGlu5 (6) or GABAB2 (26) receptors, and since a single PAM per dimer appears sufficient for the full activity, one may propose that a single HD per dimer is turned into an active conformation during activation of these dimeric receptors. This is consistent with our previous study showing that two antagonists interacting in the HD are required to inhibit activity of mGlu dimers (29). This, plus other data described below nicely fit with the proposal that the dimer of HD functions in an asymmetrical way, a single HD being activated at a time. As illustrated in Fig. 5 using the mGlu1:mGlu5 heterodimer as an example, activation of this dimeric receptor with agonists leads to only one of the HD, either that of mGlu1 or that of mGlu5, in an active state. Accordingly, binding of Ro01-6128 should favor the activation of the mGlul HD, whereas DFB should favor the activation of the mGlu5 HD. This is consistent with the enhancing effect of a PAM acting in mGlu5 when the mGlu1 HD is made unable to activate G-proteins by a point mutation in the i3 loop. Conversely, this model nicely explains the non-competitive action of PAMs acting in the subunit bearing the i3 loop mutation. Indeed, by binding in this subunit, the PAM is expected to favor the formation of the active conformation of this subunit, and so to prevent activation of the other HD, leading to a smaller response. Further supporting this conclusion, an inverse agonist binding in the HD mutated in the i3 loop favors the activation of the associated subunit, and as such acts as a enhancer, as previously described (29).
Fig. 5. Asymmetric activation mechanism of a dimer of mGlu.
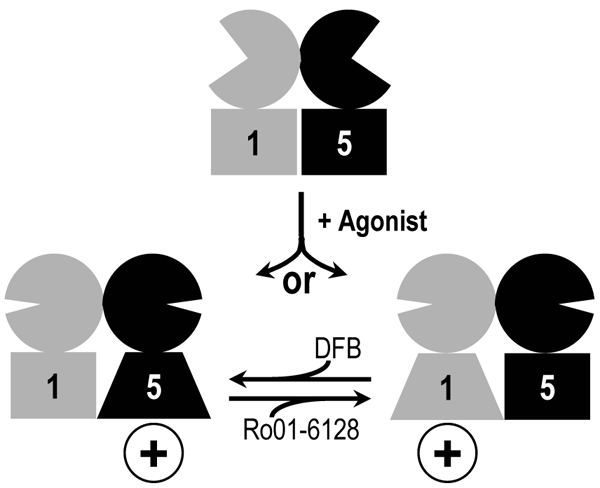
Schematic representation of the activation of a mGlu1:mGlu5 heterodimer and the effect of specific mGlu1 and mGlu5 positive allosteric modulators: Ro01-6128 and DFB, respectively. Agonist binding induces the closure of extracellular domains that triggers the activation of a single heptahelical domain or the other. The binding of a single positive allosteric modulator is sufficient to potentiate the dimer activity by further stabilizing the active state of the mGlu1 subunit (for Ro01-6128) or mGlu5 (for DFB).
Taken together, our data strongly support the asymmetric functioning of the dimer of HDs in mGlu receptors that we previously proposed (29). Such an observation is surprising considering 1) that the mGlu receptors are homodimers, and as such symmetrical complexes, and 2) that the dimer of ECDs functions in a symmetrical way, the activation of both ECDs being required for full activation of the receptor (28). Accordingly, one has to imagine some hindrance at the level of the dimer of HDs that prevents both HDs to reach an active state at a time. This may result from some specificity of the dimer interface at that level of the receptor dimer. However, one attractive possibility is that the heterotrimeric G-protein is responsible for this effect, since this protein is not symmetric. Although highly speculative, this idea is supported by recent data on the class A GPCRs that show that a single heterotrimeric G-protein interacts with a dimer of HDs, the alpha subunit contacting a first HD via its C-terminal end, whereas the second HD contacts both the N-terminal alpha helix of the alpha subunit as well as the beta-gamma dimer (3,39–41).
Acknowledgments
The authors wish to thanks Dr. Mohamed Ayoub and Dr. Ashley Brady for critical reading of the manuscript. We are thankful to Drs. Ivo Stary and Daniela Frankova for the synthesis of Ro0l-6128. This work was supported by grants from the CNRS, the Action Concertée Incitative “Molécules et Cibles Thérapeutiques” of the French ministry of research and technology, the fondation Paul Hamel, the comité Parkinson from the Fondation de France, Addex Pharmaceuticals and the European Community (grant LSHB-CT-200-503337) to JPP and by the Grant Agency of Czech Republic (GACR 301/03/1183 and 301/03/H095), Grant Agency Academy of Science of Czech Republic (KJB5039402) and AVOZ 50390512 to JB. CG was supported by a fellowship from the Fondation pour la Recherche Médicale, JK by a bourse de Docteur Ingénieur from the CNRS, and DM was supported by both CisBio International and the French government (CIFRE fellowship).
Footnotes
The abbreviations used are: PAM, positive allosteric modulator; GPCR, G-protein coupled receptor; mGlu, metabotropic glutamate receptor; GABAB, gamma amino butyric acid receptor B; HD, heptahelical domain; ECD, extracellular domain; GPT, glutamate pyruvate transaminase; DMEM, Dulbecco’s modified Eagle’s medium; FCS, fetal calf serum; Ro0l-6128, Diphenylacetylcarbamic acid ethyl ester; XITA, 9H-xanthene-9-carboxylic acid (2-isopropyl-2H-tetrazol-5-yl)-amide; DFB, difluorobenzaldazine; HA, hemaglutinin A, HEK293, human embryonic kydney 293; TR-FRET, time resolved-fluorescence resonance energy transfer; HTRF, homogeneous time resolved fluorescence; IP, inositol phosphate.
References
- 1.Bockaert J, Pin JP. EMBOJ. 1999;18:1723–1729. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terrillon S, Bouvier M. EMBO Rep. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park PS, Filipek S, Wells JW, Palczewski K. Biochemistry. 2004;43:15643–15656. doi: 10.1021/bi047907k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romano C, Yang WL, O’Malley KL. J Biol Chem. 1996;271:28612–28616. doi: 10.1074/jbc.271.45.28612. [DOI] [PubMed] [Google Scholar]
- 5.Tsuji Y, Shimada Y, Takeshita T, Kajimura N, Nomura S, Sekiyama N, Otomo J, Usukura J, Nakanishi S, Jingami H. J Biol Chem. 2000;275:28144–28151. doi: 10.1074/jbc.M003226200. [DOI] [PubMed] [Google Scholar]
- 6.Galvez T, Duthey B, Kniazeff J, Blahos J, Rovelli G, Bettler B, Prézeau L, Pin JP. EMBO J. 2001;20:2152–2159. doi: 10.1093/emboj/20.9.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Margeta-Mitrovic M, Jan YN, Jan LY. Neuron. 2000;27:97–106. doi: 10.1016/s0896-6273(00)00012-x. [DOI] [PubMed] [Google Scholar]
- 8.Nelson G, Chandrashekar J, Hoon MA, Feng L, Zhao G, Ryba NJ, Zuker CS. Nature. 2002;416:199–202. doi: 10.1038/nature726. [DOI] [PubMed] [Google Scholar]
- 9.Nelson G, Hoon MA, Chandrashekar J, Zhang Y, Ryba NJP, Zuker CS. Cell. 2001;106:381–390. doi: 10.1016/s0092-8674(01)00451-2. [DOI] [PubMed] [Google Scholar]
- 10.Xu H, Staszewski L, Tang H, Adler E, Zoller M, Li X. Proc Natl Acad Sci USA. 2004;101:14258–14263. doi: 10.1073/pnas.0404384101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pin J-P, Galvez T, Prezeau L. Pharmacol Ther. 2003 doi: 10.1016/s0163-7258(03)00038-x. in press. [DOI] [PubMed] [Google Scholar]
- 12.Pin JP, Kniazeff J, Goudet C, Bessis AS, Liu J, Galvez T, Acher F, Rondard P, Prezeau L. Biol Cell. 2004;96:335–342. doi: 10.1016/j.biolcel.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 14.Tsuchiya D, Kunishima N, Kamiya N, Jingami H, Morikawa K. Proc Natl Acad Sci USA. 2002;99:2660–2665. doi: 10.1073/pnas.052708599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bessis AS, Rondard P, Gaven F, Brabet I, Triballeau N, Prézeau L, Acher F, Pin J-P. Proc Natl Acad Sci (USA) 2002;99:11097–11102. doi: 10.1073/pnas.162138699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kniazeff J, Saintot P-R, Goudet C, Liu J, Charnet A, Guillon G, Pin J-P. J Neurosci. 2004;24 doi: 10.1523/JNEUROSCI.3141-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tateyama M, Abe H, Nakata H, Saito O, Kubo Y. Nat Struct Mol Biol. 2004;11:637–642. doi: 10.1038/nsmb770. [DOI] [PubMed] [Google Scholar]
- 18.May LT, Christopoulos A. Curr Opin Pharmacol. 2003;3:551–556. doi: 10.1016/s1471-4892(03)00107-3. [DOI] [PubMed] [Google Scholar]
- 19.Goudet C, Binet V, Prézeau L, Pin JP. Drug discovery today. 2004;1:125–133. [Google Scholar]
- 20.Kew JN. Pharmacol Ther. 2004;104:233–244. doi: 10.1016/j.pharmthera.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 21.Jensen AA, Spalding TA. Eur J Pharm Sci. 2004;21:407–420. doi: 10.1016/j.ejps.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Shoback DM, Bilezikian JP, Turner SA, McCary LC, Guo MD, Peacock M. J Clin Endocrinol Metab. 2003;88:5644–5649. doi: 10.1210/jc.2002-021597. [DOI] [PubMed] [Google Scholar]
- 23.Petrel C, Kessler A, Dauban P, Dodd RH, Rognan D, Ruat M. J Biol Chem. 2004;279:18990–18997. doi: 10.1074/jbc.M400724200. [DOI] [PubMed] [Google Scholar]
- 24.Knoflach F, Mutel V, Jolidon S, Kew JN, Malherbe P, Vieira E, Wichmann J, Kemp JA. Proc Natl Acad Sci U S A. 2001;98:13402–13407. doi: 10.1073/pnas.231358298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaffhauser HJ, Rowe BA, Morales S, Chavez-Noriega LE, Yin R, Jachec C, Rao SP, Bain G, Pinkerton AB, Vernier J-M, Bristow LJ, Varney MA, Daggett LP. Mol Pharmacol. 2003;64:798–810. doi: 10.1124/mol.64.4.798. [DOI] [PubMed] [Google Scholar]
- 26.Binet V, Brajon C, Le Corre L, Acher F, Pin JP, Prezeau L. J Biol Chem. 2004;279:29085–29091. doi: 10.1074/jbc.M400930200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goudet C, Gaven F, Kniazeff J, Vol C, Liu J, Cohen-Gonsaud M, Acher F, Prezeau L, Pin JP. Proc Natl Acad Sci U S A. 2004;101:378–383. doi: 10.1073/pnas.0304699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kniazeff J, Bessis AS, Maurel D, Ansanay H, Prezeau L, Pin JP. Nat Struct Mol Biol. 2004;11:706–713. doi: 10.1038/nsmb794. [DOI] [PubMed] [Google Scholar]
- 29.Hlavackova V, Goudet C, Kniazeff J, Zikova A, Maurel D, Vol C, Trojanova J, Prézeau L, Pin JP, Blahos J. Embo J. 2005;24:499–509. doi: 10.1038/sj.emboj.7600557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jolidon S, Mutel V, Vieira E, Wichmann J. 2002/0022648 US patent. 2002
- 31.Ango F, Albani-Torregrossa S, Joly C, Robbe D, Michel JM, Pin JP, Bockaert J, Fagni L. Neuropharmacology. 1999;38:793–803. doi: 10.1016/s0028-3908(99)00005-2. [DOI] [PubMed] [Google Scholar]
- 32.Fourgeaud L, Bessis AS, Rossignol F, Pin JP, Olivo J-c, Hémar A. J Biol Chem. 2003;278:12222–12230. doi: 10.1074/jbc.M205663200. [DOI] [PubMed] [Google Scholar]
- 33.Havlickova M, Blahos J, Brabet I, Liu J, Hruskova B, Prezeau L, Pin JP. J Biol Chem. 2003;278:35063–35070. doi: 10.1074/jbc.M306555200. [DOI] [PubMed] [Google Scholar]
- 34.Brabet I, Parmentier ML, De Colle C, Bockaert J, Acher F, Pin JP. Neuropharmacology. 1998;37:1043–1051. doi: 10.1016/s0028-3908(98)00091-4. [DOI] [PubMed] [Google Scholar]
- 35.Maurel D, Kniazeff J, Mathis G, Trinquet E, Pin JP, Ansanay H. Anal Biochem. 2004;329:253–262. doi: 10.1016/j.ab.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 36.O’Brien JA, Lemaire W, Chen TB, Chang RSL, Jacobson MA, Ha SN, Lindsley CW, Schaffhauser HJ, Sur C, Pettibone DJ, Conn PJ, Williams DL., Jr Mol Pharmacol. 2003;64:731–740. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- 37.Francesconi A, Duvoisin RM. J Biol Chem. 1998;273:5615–5624. doi: 10.1074/jbc.273.10.5615. [DOI] [PubMed] [Google Scholar]
- 38.Robbins MJ, Ciruela F, Rhodes A, McIlhinney RA. J Neurochem. 1999;72:2539–2547. doi: 10.1046/j.1471-4159.1999.0722539.x. [DOI] [PubMed] [Google Scholar]
- 39.Baneres JL, Parello J. J Mol Biol. 2003;329:815–829. doi: 10.1016/s0022-2836(03)00439-x. [DOI] [PubMed] [Google Scholar]
- 40.Hamm HE. Proc Natl Acad Sci U S A. 2001;98:4819–4821. doi: 10.1073/pnas.011099798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang Y, Fotiadis D, Filipek S, Saperstein DA, Palczewski K, Engel A. J Biol Chem. 2003;278:21655–21662. doi: 10.1074/jbc.M302536200. [DOI] [PMC free article] [PubMed] [Google Scholar]