

Abstract
The vast majority of primary brain tumors derive from glial cells and are collectively called gliomas. While, they share some genetic mutations with other cancers, they do present with a unique biology and have developed adaptations to meet specific biological needs. Notably, glioma growth is physically restricted by the skull, and, unless normal brain cells are destroyed, tumors cannot expand. To overcome this challenge, glioma cells release glutamate which causes excitotoxic death to surrounding neurons, thereby vacating room for tumor expansion. The released glutamate also explains peritumoral seizures which are a common symptom early in the disease. Glutamate release occurs via system Xc, a cystine-glutamate exchanger that releases glutamate in exchange for cystine being imported for the synthesis of the cellular antioxidant GSH. It protects tumor cells from endogenously produced reactive oxygen and nitrogen species but also endows tumors with an enhanced resistance to radiation- and chemotherapy. Pre-clinical data demonstrates that pharmacological inhibition of system Xc causes GSH depletion which slows tumor growth and curtails tumor invasion in vivo. An Food and Drug Administration approved drug candidate is currently being introduced into clinical trials for the treatment of malignant glioma.
Keywords: brain tumor, cell migration, glia, glutamate, ion channel, ion transport
Gliomas
Malignant gliomas comprise a heterogeneous group of primary CNS tumors that are believed to originate from glial cells or their progenitor cells. Approximately 18 000 new cases are reported annually and these account for over 12 000 deaths in the USA (Jemal et al. 2006). Although the majority of gliomas present in patients over the age of 55, gliomas are the most common solid cancer in children (Maher and Raffel 2004). Unfortunately, tumor incidence has increased steadily over the past 30 years (Jemal et al. 2006) and the prognosis remains dismal (Azizi and Miyamoto 1998). Effective therapies are lacking thus limiting treatment to radiation therapy and, where possible, surgery (Butowski et al. 2006). Average survival following diagnosis can be up to 5 years but, more commonly, patients diagnosed with high-grade glioma succumb to the disease within just 6-12 months (Huncharek and Muscat 1998).
Tumors generally form only from growth competent cells, hence, in the brain, primary tumors arise typically from glial cells and cells associated with the vasculature and meninges. These tumors are collectively called gliomas (Kleihues et al., 1995) with the majority believed to originate from astrocytes, or their progenitor cells. Astrocytes retain the ability to divide throughout life and respond to acute injury and neurological disease through proliferation and the formation of scar tissue (Little and O’Callagha 2001). These scar-associated astrocytes eventually divide and differentiate, leaving a tenacious tissue barrier. By contrast, glial-derived tumors fail to differentiate thereby maintaining unrestrained growth. Events that trigger malignant transformation of glial cells into gliomas are poorly understood. Genetic alterations in oncogenes [v-src, murine-double-minute-2 (MDM-2), cyclin-dependent kinase 4 (CDK-4), and epidermal growth factor receptor (EGFR)] and tumor suppressor genes (p53, p16, p15, and RB1) are common characteristics (Louis 1994; Von Deimling et al. 1995; Maher et al. 2001; Shapiro 2001) of the transformation of glial progenitor cells into tumorforming cells in mice (Holland et al. 1998).
Gliomas share many fundamental biological alterations with other cancers. For example, they frequently show amplification of a constitutively active epidermal growth factor receptor that enhances tumor growth in the absence of exogenous growth factors (Tang et al. 1997). Tumor suppressor genes such as p16 and p53 are frequently mutated (Von Deimling et al. 1995). The induction of new blood vessels, or neovascularization, is a typical feature of many tumors and also characterizes high-grade gliomas (Plate and Risau 1995). Despite these commonalities with other tumors, gliomas exhibit unique biological traits and have developed adaptations to support their unique biology. One of these physiological adaptations will be illustrated in this paper.
Physical constraints to tumor growth
Essentially all systemic tumors grow within soft tissue and are not physically limited in their growth. By contrast, primary brain tumors can only expand within the bony cavity of the cranium which ultimately limits tumor growth. Eighty-five percent of cranial volume is occupied by brain tissue with the remaining 15% filled with CSF. Ultimately, this space becomes insufficient for tumor expansion, necessitating the destruction and removal of normal brain tissue. Early in the disease progression tumors can be seen to push into the fluid spaces and often displace brain tissue, leading for example to a shift in midline structures as illustrated in Fig. 1. It should be noted that the brain, like any body tissue, is incompressible given its cellular constituents are predominantly composed of water. However, text books and reviews frequently but incorrectly remark on the ability of growing tumors to compress the brain. One may refer more accurately to this phenomenon as a displacement of tissue into fluid-filled spaces, notably the ventricles. Figure 1 provides several examples illustrating how different sized tumors integrate into normal brain by displacing the surrounding brain tissue. In each instance, however, the characteristic gyrations of the cortical brain surface remains essentially unaltered and without evidence of compression. Hence, in each instance tumors have vacated the space into which they grew rather than simply pushing it aside. Surprisingly, little attention had been paid to the mechanism(s) whereby gliomas destroy normal brain tissue as they grow.
Fig. 1.
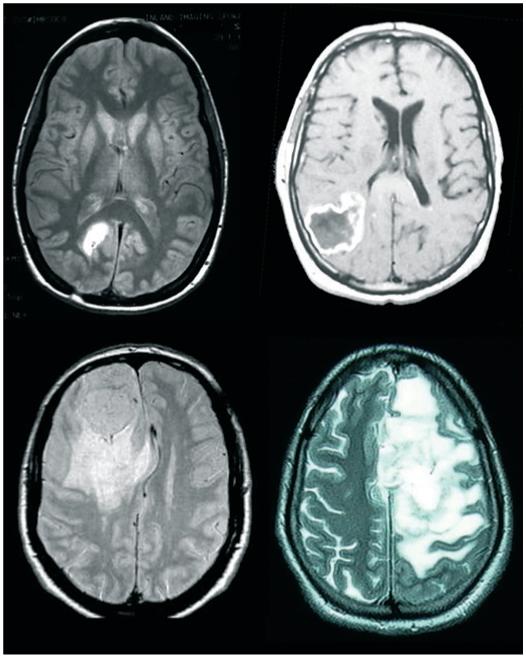
Gliomas destroy normal brain as the tumor expands. Examples of patient images from four different patients that are presented with malignant gliomas, each illustrating the fact that normal brain tissue was displaced to accommodate the tumors expansion.
To fill this void, recent studies have examined this question and collectively suggest that glioma cells, at the growth margins of an expanding tumor, release the neurotransmitter glutamate into adjacent brain tissue at concentrations that are sufficient to cause neuronal cell death (Behrens et al. 2000; Chung et al. 2005). Glutamate is the major excitatory neurotransmitter in the brain and is normally restricted to the synaptic and perisynaptic space of glutamateric synapses. Here, the transmitter binds to post-synaptic neuronal glutamate receptors and mediates fast signal transmission. In this restricted synaptic space glutamate rises to millimolar concentrations, yet its concentration in the extracellular space is typically very low, i.e. around 1 μM, to prevent the erroneous activation of extrasynaptic glutamate receptors. Prolonged activation of neuronal glutamate receptors, particularly NMDA receptors, can trigger a sustained influx of Ca2+ which readily overwhelms intracellular Ca2+ regulatory processes ultimately leading to Ca2+-mediated cell death (Choi 1988). This process, introduced several decades ago (Olney et al. 1971) and termed excitotoxicity, is now believed to be a final common death pathway in many neurological diseases including acute trauma, ischemic stroke, amyotrophic lateral scelerosis (ALS) and Alzheimer’s disease (Choi 1988; Lipton and Rosenberg 1994). Because of the vulnerability of the brain to excessive glutamate, protective mechanisms evolved in the form of Na+-dependent transport systems that maintain extracellular glutamate at harmless micromolar concentrations (Danbolt 2001). These transporters are predominantly expressed on astrocytes (Danbolt 2001) and include the excitatory amino acid transporters 1 and 2 (EAAT1 and 2) formerly known as glial amino acid transporter and glutamate transporter-1 (Danbolt 2001). Their role in neuroprotection has been conclusively demonstrated through knockout studies in which a loss of glutamate uptake by glial cells resulted in widespread neurodegeneration and lethal epilepsy (Rothstein et al. 1996; Tanaka et al. 1997).
Glioma cells release glutamate and kill peritumoral neurons by an excitoxic mechanism
The first evidence that glial-derived tumor cells release glutamate rather than remove it from the extracellular space came from a transport study that directly compared normal and malignant glial cells (Ye et al. 1999). This study used glioma cell lines established from patients as well as acute patient biopsy tissues showing a lack of expression of the most prominent astrocytic glutamate transporter EAAT2 (glutamate transporter-1), a finding that was recently corroborated by microarray analysis of glioma tissues (de Groot et al. 2005). Staining of tissue biopsies with EAAT2 antibodies clearly demarcates the tumor boundaries where non-malignant astrocytes prominently express EAAT2, yet gliomas lack its expression (Fig. 2a). During embryonic development, astrocytes also express EAAT1 (glial amino acid transporter) and many cultured astrocytes maintain expression of this related member of the EAAT family of Na+-dependent transporters (Ullensvang et al. 1997). Interestingly, EAAT1 is found mislocalized to the nucleus of gliomas in brain biopsies from glioma patients (Ye et al. 1999). This failure to express functional glutamate transporters results in significant reductions in glutamate uptake, representing at best 5% of that observed in normal astrocytes. More surprising was the converse finding that these tumor cells actually release glutamate. In the presence of extracellular cystine, a monolayer of cultured glioma cells is able to raise extracellular glutamate concentrations in a culture flask 500-fold within just a few hours (Fig. 2b). When hippocampal or cortical neurons are placed in the vicinity of gliomas, neurons undergo NMDA-dependent excitotoxic cell death (Fig. 2c) (Ye and Sontheimer 1999). Further molecular, pharmacological, and ion-dependence studies ultimately led to the identification of system Xc,a cystine-glutamate exchanger (Ye et al. 1999), as the principal protein responsible for glutamate release from glioma cells (Ye and Sontheimer 1999). Once released into the peritumoral space (Ye and Sontheimer 1999; Takano et al. 2001), glutamate causes widespread excitotoxic death of peritumoral neurons (Ye and Sontheimer 1999). We and others have proposed that this form of excitotoxic neuronal cell death allows the tumor to expand into the vacated space (Takano et al. 2001). In a more recent study discussed below (Lyons et al. 2007), we suggest that the released glutamate may also serve as a trophic factor promoting glioma cell invasion and the formation of tumor metastasis. It is worth mentioning that glutamate has been shown to more broadly influence the biology of malignant and non-malignant glial cells. For example, activation of metabotropic, G-protein coupled glutamate receptors stimulates DNA synthesis and activates mitogen-activated protein kinases in astrocytes (Schinkmann et al. 2000). In glioma cells, Ca2+ influx through alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors stimulates cell proliferation through phosphorylation of AKT (Ishiuchi et al. 2007). Moreover, in areas of tissue inflammation, glutamate activates microglial cells (Biber et al. 2007) which would be expected to contribute to the inflammatory milieu around a growing tumor. These additional effects of glutamate will undoubtedly contribute to the evolution of the disease and warrant further study.
Fig. 2.
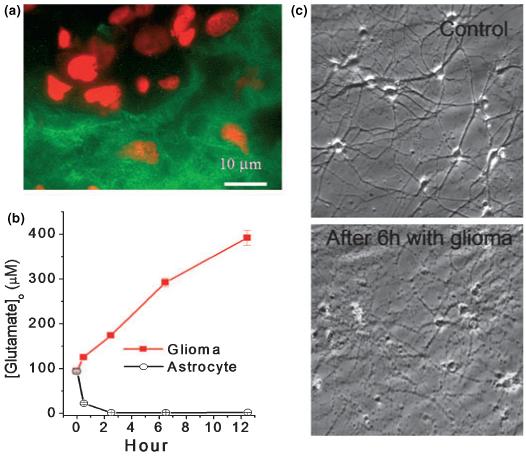
Gliomas release glutamate. (a) Gliomas in vivo do not express the GLT-1 glutamate transporter (green) while the surrounding astrocytes do. The GLT-1 staining demarcates the tumor boundaries. Red staining is propridium iodine to label cell nuclei. (b) Glioma cells release glutamate as determined by sampling medium in a 70-cm3 flask as a function of time. By contrast non-malignant astrocytes remove glutamate when challenged with 100 μM at the start. (c) Time-laps video microscopy shows pronounced neuronal cell death when hippocampal neurons are placed in the vicinity of glioma cells yet without touching them (sandwich culture). With permission from: (Ye and Sontheimer 1999).
Glutamate release is mediated by the cystine-glutamate antiporter system Xc
As indicated above, the Na+ independent release of glutamate occurs via the system Xc cystine glutamate antiporter. This electroneutral, Na+ independent amino acid transporter (Sato et al. 1999) exchanges cystine for glutamate (GLU) (Ye et al. 1999) and is up-regulated under conditions of oxidative stress (Kim et al. 2001). Molecular cloning identified this transporter as a dimeric protein composed of a catalytic (xCT) and a regulator subunit (4F2hc). The transporter is found in both normal and malignant human brain by western blot (Fig. 3a) but appears universally up-regulated in glioma tissues. Indeed, being responsible for the cellular synthesis of GSH and hence cellular redox regulation, it is not surprising that system Xc is regulated by oxidative stress and nitric oxide (Dun et al. 2006) presumably via the activation of transcription factor NF-E2-related factor which regulate xCT transcription via binding to an antioxidant response element (Sun et al. 2005). In normal glia cells, system Xc does not account for a significant glutamate release as any glutamate release through the antiporter in conjunction with cystine uptake is quickly removed through reuptake by EAAT2 which has a much higher capacity for Glu transport and a high affinity of extracellular Glu. This concept is schematized in Fig. 3c. In turn, the lack of reuptake mechanisms in gliomas explains a progressive accumulation of glutamate in the vicinity of the tumor which inflicts excitotoxic injury on adjacent neurons as schematized in Fig. 3d.
Fig. 3.

Cystine-glutamate exchange in gliomas. (a) Glioma cell lines and acute biopsies highly express the catalytic (xCT) and regulatory (4F2hc) subunit of the system Xc cystine glutamate exchanger. (b) this transporter is responsible for the release of glutamate from the tumor into the surrounding brain. (c) In normal glia, glutamate released via system Xc is taken back up by the Na+-dependent GLT-1 transporter. (d) In gliomas, the loss of this reuptake via GLT-1 causes a buildup of glutamate and excitotoxic injury. (e) The main purpose of the system Xc transporter is to supply cysteine for the production of the cellular antioxidant GSH.
Although the initial studies investigating glutamate release from gliomas were based on cultured cells (Ye and Sontheimer 1999) and animal models bearing experimental gliomas (Takano et al. 2001), these findings have now been corroborated in patients with malignant gliomas. One of these studies used microdialysis (Roslin et al. 2003) to directly sample peritumoral fluids in fully ambulatory glioma patients; the other used spectroscopic magnetic resonance imaging to detect glutamate (Rijpkema et al. 2003). Both studies confirm significant increases in peritumoral glutamate in all glioma patients examined. This finding explains why up to 80% of all glioma patients present with peritumoral seizures, many of them during early, often less malignant stages of the disease (Oberndorfer et al. 2002). The glutamate released from gliomas would be expected to activate neuronal glutamate receptors that underlie such seizures in the vicinity of the tumor and likely precede excitotoxic cell death.
Glutamate release is an obligatory byproduct of GSH synthesis
While the excitotoxic elimination of peritumoral neurons may be of potential benefit to the growing tumor, it is unlikely that this pathway evolved with the primary purpose to kill neurons. Instead, it is more likely that glutamate release occurs as an obligatory byproduct of cystine uptake by these tumor cells (Fig. 3b and e). Cystine is an essential precursor for the biosynthesis of the cellular antioxidant GSH, a tripeptide of glutamate, cysteine (the reduced form of cystine) and glycine. The supply of cysteine is essential for GSH biosynthesis making cellular uptake of cystine rate limiting for overall GSH production. GSH levels are increased in gliomas and may make these tumors more resistant to oxidative stress and exposure to certain chemotherapeutic drugs (Iida et al. 1997). Importantly, radiation damage which is mediated by hydroxyl radicals (Knuutila 1984) is also reduced by GSH (Simone et al. 1983) and hence elevated GSH levels confer radiation resistance to tumors (Mitchell et al. 1989).
Therapeutic targeting of system Xc-mediated glutamate release to contain tumor growth
If system Xc represent the major uptake pathway for cystine and release pathway for glutamate, one would expect that its inhibition should interfere with tumor growth in two ways. First, limiting cystine uptake should starve the cells of GSH making the cells more susceptible to oxidative challenges such as radiation treatment. Second, the inhibition of glutamate release should curtail seizures and may contain tumor expansion. To examine these possibilities, two pharmacological inhibitors have been identified as suitable drug candidates. These are S-4CPG, a phenylgycin derivative and agonist for metabotrobic glutamate receptors (Ye and Sontheimer 1999), and sulfasalazine (Chung et al. 2005), a drug clinically used to treat chronic inflammatory bowl disease. Both drugs inhibit cystine uptake and glutamate release at 100-250 μM drug concentration (Chung et al. 2005). Importantly, a 24 h exposure to either drug causes the near complete depletion of intracellular GSH (Fig. 4a) and in turn leads to growth arrest of gliomas (Chung et al. 2005). The growth retarding effect is entirely because of a depletion of GSH as cells can be rescued from growth arrest with a membrane permeant, GSH ester. As sulfasalazine is a Food and Drug Administration approved drug that can be readily given to patients, pre-clinical studies examining its growth retarding effect have mainly focused on this compound. Specifically, severe combined immunodeficient (scid) mice with implanted human gliomas were treated twice daily by i.p. injection of sulfasalazine for up to 10 weeks and tumor response was monitored by bioluminescence imaging as well as quantitative stereology on histopathological specimens (Chung et al. 2005). As illustrated in Fig. 4, sulfasalazine reduced tumor volume by 60-80%. These studies were carried out with sulfasalazine doses comparable with those taken by patients suffering from Crohn’s disease for which an adequate safety profile is well established. These findings suggest a rapid path to clinical translation and the author’s home institution is in the process of initiating a clinical study to evaluate oral sulfasalazine in a phase I clinical study for patients with malignant glioma.
Fig. 4.

Inhibition of system Xc causes GSH depletion and reduces tumor size in vivo. (a) Sulfasalazine, which inhibits system Xc, causes a dose- and time-dependent reduction in GSH levels. (b) Intraperitoneal injection, twice daily, of 8 mg/kg sulfasalazine causes a 75% reduction in tumor volume in tumor bearing animals. (c) A representative comparison of control, saline injected to sulfasalazine treated animal at 30 days treatment. With permission from (Chung et al. 2005).
As a second approach, clinical studies may seek to inhibit the glutamate receptors targeted on neurons and glioma cells, with neuronal NMDA receptor being the primary candidate. Although NMDA-receptor antagonists have had mixed results in the treatment of stroke (Lipton 2004), they may represent a viable treatment modality in advanced stages of brain cancer warranting further study. Use-dependent glutamate receptor antagonists such as Amantadine have been explored with promising indication toward reducing aberrant activation of neuronal NMDA receptors in clinical studies for stroke (Lipton 2004), Parkinson’s disease (Verhagen et al. 1998) and, based on animal studies (Takano et al. 2001), may indeed represent a viable treatment strategy for gliomas.
Glutamate stimulates glioma metastasis
As discussed above, the unusual ability of glioma cells to invade the normal brain presents a huge clinical challenge. Consequently, a great deal of research has been devoted toward gaining a better understanding of the underlying biology. This research has identified a multitude of factors and adaptations which include a preference for certain substrate for cell migration (Demuth and Berens 2004). Indeed, invading gliomas can synthesize their own unique extracellular matrix as they invade (Zamecnik 2005). These cells interact with extracellular matrix through integrin receptors (Gladson 1999), also involved in the dynamic attachment/detachment of focal adhesion sites. Some of these dynamic changes appear to correlate with changes in intracellular Ca2+ (Manning et al. 2000) and intracellular Ca2+ oscillations frequently correlate with cell migration (Maghazachi 2000). These have also been observed in migratory glioma cells (Bordey et al. 2000). Ca2+ oscillations can be triggered by a number of extracellular ligands including growth factors (Bryant et al. 2004), lysophosphatidic acid (Manning et al. 2000) and neurotransmitters such as acetylcholine (Komuro and Rakic 1996) and glutamate (Kim et al. 1994). Glutamate is of particular interest in this context as studies on cerebellar granule cells suggest an absolute requirement for glutamate-mediated Ca2+ oscillations in neuronal migration (Komuro and Rakic 1996). Here, the activation of NMDA receptor causes the transient influx of Ca2+ which in turn regulates the rate and velocity of granule cell migration (Komuro and Rakic 1998). Although glioma cells express receptors for glutamate, unlike granule cells, they do not express NMDA receptor. Instead, they express a subclass of AMPA receptors which is Ca2+ permeable (Lyons et al. 2007). These receptors lack the GluR2 subunit which is required to prevent Ca2+ permeation (Hollmann et al. 1991) resulting in Ca2+ permeable AMPA receptor. To demonstrate a role for these receptors in glioma migration, Ishiuchi et al. (2002) transfected the GluR2 subunit into glioma cells using a viral transfection system. This rendered glioma cells unable to respond to glutamate with Ca2+ oscillations and upon implantation into a host animal brain these glioma cells failed to invade. Hence glutamate acts to promote cell migration through activating Ca2+ entry. Although glioma cells utilize a different receptor system, they appear to recapitulate the same underlying biology that is observed in early development in neuronal migration. Interestingly, oligodendrocytes progenitor cells which may be a non-malignant precursor for gliomas (Dai et al. 2001) have recently been shown to engage AMPA-receptor activation as they migrate. Here, glutamate binding to AMPA receptor causes the formation of an alphav integrin/proteolipid protein (PLP)/neurotransmitter receptor protein complex that reduces binding to the extracellular matrix thereby enhancing oligodendrocytes progenitor cells migration (Gudz et al. 2006).
In the case of neurons, the source for glutamate acting in a migration-promoting way (i.e. as a ‘motogen’) is unknown. In gliomas, glutamate is release from the tumor cells themselves and may function in a dual capacity. It clears the way for the tumor growth, and it may activate AMPA receptors on the same or neighboring glioma cells to initiate the necessary Ca2+ signal for promoting cell motility and invasion. Indeed, recent studies support this hypothesis (Lyons et al. 2007), suggesting that glioma cells respond preferentially to glutamate released via system Xc. As illustrated in Fig. 5a, inhibition of glutamate release via system Xc using sulfasalazine inhibits Ca2+ responses and also reduces Transwell glioma migration (Fig. 5b). The fact that migration could be restored when glutamate was added back into the system in the continued presence of the system Xc inhibitor sulfasalazine (Fig. 5b) supports a motogenic function for glutamate. Moreover, the inhibition of AMPA receptors with Jolo-Tx, a specific inhibitor of Ca2+-permeable AMPA receptors, prevented migration stimulated by glutamate clearly implicating this receptor as essential in glioma invasion. Together this data supports the hypothesis that glutamate serves a dual purpose in glioma biology: (i) by acting as an excitotoxin, it clears space for tumor expansion thereby promoting tumor growth; and (ii) by acting as a motogen, it enhances the motility and consequently promotes tumor invasion.
Fig. 5.

Glioma cells respond to glutamate as they invade. (a) Ca2+ imaging shows oscillatory changes in intracellular Ca2+ if cells are maintained in the presence of extracellular cystine (Cys), stimulating glutamate release via the cystine-glutamate exchanger. Ca2+ oscillations are abolished if cystine-glutamate exchange is inhibited with sulfasalazine (SAS). (b) Inhibition of glutamate release by SAS also inhibits Transwell migration, as does blockade of Ca2+ permeable AMPA receptors with GYKI or JSTx. Data represent normalized cell number as percentage of untreated control cells. Importantly SAS inhibition is overcome by exogenous addition of glutamate. (c) Glioma cells that express enhanced green fluorescent protein were imaged as they invaded cortical brain slices in situ. Cells typically migrate along blood vessel where they display ‘chain migration’ [a and c with permission from (Lyons et al. 2007)].
Glioma cells often display chain migration whereby many cells follow a lone leader (Fig. 5c). It is conceivable that the leading cell recruits its followers through the targeted (paracrine) release of glutamate as it migrates. These findings have important therapeutic implications as inhibition of either AMPA receptor on gliomas or the autocrine glutamate from glioma via system Xc should prevent glioma metastasis. As alluded to before, glutamate receptor antagonists may represent a viable approach to reduce glioma invasion and warrant further study. The inhibition of glutamate release from gliomas using sulfasalazine has not been previously considered as a glioma therapeutic but now appears to be a plausible strategy to curtail glioma invasion.
Epilogue
As these examples illustrate, glioma cells utilize transporters and transmitter receptors in ways to enhance their unique biology. They appear to co-opt the function of these proteins which are normally involved in cell-cell signaling or to maintain a homeostatic brain environment. They use them to enhance their own ability to grow and invade normal brain. Unexpectedly, amino acid transporters may be considered viable and much needed therapeutic targets for glioma and possibly many other cancers.
Acknowledgements
The author is grateful for the continued supported by grants from the National Institutes of Health RO1 NS-31234, RO1 NS-52634, RO1 NS-36692, and P50-CA97247.
Abbreviation used
- AMPA
alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate
- EAAT
excitatory amino acid transporter
References
- Azizi SA, Miyamoto C. Principles of treatment of malignant gliomas in adults: an overview. J. Neurovirol. 1998;4:204–216. doi: 10.3109/13550289809114520. [DOI] [PubMed] [Google Scholar]
- Behrens PF, Langemann H, Strohschein R, Draeger J, Hennig J. Extracellular glutamate and other metabolites in and around RG2 rat glioma: an intracerebral microdialysis study. J. Neurooncol. 2000;47:11–22. doi: 10.1023/a:1006426917654. [DOI] [PubMed] [Google Scholar]
- Biber K, Neumann H, Inoue K, Boddeke HW. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H, Trouslard J. Muscarinic activation of BK channels induces membrane oscillations in glioma cells and leads to inhibition of cell migration. J. Membr. Biol. 2000;176:31–40. doi: 10.1007/s00232001073. [DOI] [PubMed] [Google Scholar]
- Bryant JA, Finn RS, Slamon DJ, Cloughesy TF, Charles AC. EGF activates intracellular and intercellular calcium signaling by distinct pathways in tumor cells. Cancer Biol. Ther. 2004;3:1243–1249. doi: 10.4161/cbt.3.12.1233. [DOI] [PubMed] [Google Scholar]
- Butowski NA, Sneed PK, Chang SM. Diagnosis and treatment of recurrent high-grade astrocytoma. J. Clin. Oncol. 2006;24:1273–1280. doi: 10.1200/JCO.2005.04.7522. [DOI] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system [review] Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J. Neurosci. 2005;25:7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog. Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J. Neurooncol. 2004;70:217–228. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- Dun Y, Mysona B, Van Ells T, Amarnath L, Ola MS, Ganapathy V, Smith SB. Expression of the cystine-glutamate exchanger (Xc-) in retinal ganglion cells and regulation by nitric oxide and oxidative stress. Cell Tissue Res. 2006;324:189–202. doi: 10.1007/s00441-005-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladson CL. The extracellular matrix of gliomas: modulation of cell function. J. Neuropathol. Exp. Neurol. 1999;58:1029–1040. doi: 10.1097/00005072-199910000-00001. [DOI] [PubMed] [Google Scholar]
- de Groot JF, Liu TJ, Fuller G, Yung WK. The excitatory amino acid transporter-2 induces apoptosis and decreases glioma growth in vitro and in vivo. Cancer Res. 2005;65:1934–1940. doi: 10.1158/0008-5472.CAN-04-3626. [DOI] [PubMed] [Google Scholar]
- Gudz TI, Komuro H, Macklin WB. Glutamate stimulates oligodendrocyte progenitor migration mediated via an alphav integrin/myelin proteolipid protein complex. J. Neurosci. 2006;26:2458–2466. doi: 10.1523/JNEUROSCI.4054-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA - gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Huncharek M, Muscat J. Treatment of recurrent high grade astrocytoma; results of a systematic review of 1,415 patients. Anticancer Res. 1998;18:1303–1311. [PubMed] [Google Scholar]
- Iida M, Sunaga S, Hirota N, Kuribayashi N, Sakagami H, Takeda M, Matsumoto K. Effect of glutathione-modulating compounds on hydrogen-peroxide-induced cytotoxicity in human glioblastoma and glioma cell lines. J. Cancer Res. Clin. Oncol. 1997;123:619–622. doi: 10.1007/s004320050115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiuchi S, Tsuzuki K, Yoshida Y, et al. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002;8:971–978. doi: 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- Ishiuchi S, Yoshida Y, Sugawara K, et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J. Neurosci. 2007;27:7987–8001. doi: 10.1523/JNEUROSCI.2180-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics. CA Cancer J. Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- Kim WT, Rioult MG, Cornell-Bell AH. Glutamate-induced calcium signaling in astrocytes. Glia. 1994;11:173–184. doi: 10.1002/glia.440110211. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kanai Y, Chairoungdua A, Cha SH, Matsuo H, Kim DK, Inatomi J, Sawa H, Ida Y, Endou H. Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochim. Biophys. Acta. 2001;1512:335–344. doi: 10.1016/s0005-2736(01)00338-8. [DOI] [PubMed] [Google Scholar]
- Kleihues P, Soylemezoglu F, Schaüble B, Scheithauer BW, Burger PC. Histopathology, classification, and grading of gliomas. Glia. 1995;15:211–221. doi: 10.1002/glia.440150303. [DOI] [PubMed] [Google Scholar]
- Knuutila S. Role of free radicals in genetic damage (mutation) Med. Biol. 1984;62:110–114. [PubMed] [Google Scholar]
- Komuro H, Rakic P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron. 1996;17:275–285. doi: 10.1016/s0896-6273(00)80159-2. [DOI] [PubMed] [Google Scholar]
- Komuro H, Rakic P. Orchestration of neuronal migration by activity of ion channels, neurotransmitter receptors, and intracellular Ca2+ fluctuations. J. Neurobiol. 1998;37:110–130. [PubMed] [Google Scholar]
- Lipton SA. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx. 2004;1:101–110. doi: 10.1602/neurorx.1.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders [see comments] [review] [89 refs] N. Engl. J. Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Little AR, O’Callagha JP. Astrogliosis in the adult and developing CNS: is there a role for proinflammatory cytokines? Neurotoxicology. 2001;22:607–618. doi: 10.1016/s0161-813x(01)00032-8. [DOI] [PubMed] [Google Scholar]
- Louis DN. The p53 gene and protein in human brain tumors. J. Neuropathol. Exp. Neurol. 1994;53:11–21. doi: 10.1097/00005072-199401000-00002. [DOI] [PubMed] [Google Scholar]
- Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67:9463–9471. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghazachi AA. Intracellular signaling events at the leading edge of migrating cells. Int. J. Biochem. Cell Biol. 2000;32:931–943. doi: 10.1016/s1357-2725(00)00035-2. [DOI] [PubMed] [Google Scholar]
- Maher CO, Raffel C. Neurosurgical treatment of brain tumors in children. Pediatr. Clin. North Am. 2004;51:327–357. doi: 10.1016/S0031-3955(03)00206-2. [DOI] [PubMed] [Google Scholar]
- Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- Manning TJ, Jr, Parker JC, Sontheimer H. Role of lysophosphatidic acid and rho in glioma cell motility. Cell Motil. Cytoskeleton. 2000;45:185–199. doi: 10.1002/(SICI)1097-0169(200003)45:3<185::AID-CM2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Mitchell JB, Cook JA, DeGraff W, Glatstein E, Russo A. Glutathione modulation in cancer treatment: will it work? Int. J. Radiat. Oncol. Biol. Phys. 1989;16:1289–1295. doi: 10.1016/0360-3016(89)90301-5. [DOI] [PubMed] [Google Scholar]
- Oberndorfer S, Schmal T, Lahrmann H, Urbanits S, Lindner K, Grisold W. The frequency of seizures in patients with primary brain tumors or cerebral metastases. An evaluation from the Ludwig Boltzmann Institute of Neuro-Oncology and the Department of Neurology, Kaiser Franz Josef Hospital, Vienna. Wien. Klin. Wochenschr. 2002;114:911–916. [PubMed] [Google Scholar]
- Olney JW, Ho OL, Rhee V. Cytotoxic effects of acidic and sulphur containing amino acids on the infant mouse central nervous system. Exp. Brain Res. 1971;14:61–76. doi: 10.1007/BF00234911. [DOI] [PubMed] [Google Scholar]
- Plate KH, Risau W. Angiogenesis in malignant gliomas. Glia. 1995;15:339–347. doi: 10.1002/glia.440150313. [DOI] [PubMed] [Google Scholar]
- Rijpkema M, Schuuring J, van der MY, van der GM, Bernsen H, Boerman R, van der KA, Heerschap A. Characterization of oligodendrogliomas using short echo time 1H MR spectroscopic imaging. NMR Biomed. 2003;16:12–18. doi: 10.1002/nbm.807. [DOI] [PubMed] [Google Scholar]
- Roslin M, Henriksson R, Bergstrom P, Ungerstedt U, Bergenheim AT. Baseline levels of glucose metabolites, glutamate and glycerol in malignant glioma assessed by stereotactic microdialysis. J. Neurooncol. 2003;61:151–160. doi: 10.1023/a:1022106910017. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- Schinkmann KA, Kim TA, Avraham S. Glutamate-stimulated activation of DNA synthesis via mitogen-activated protein kinase in primary astrocytes: involvement of protein kinase C and related adhesion focal tyrosine kinase. J. Neurochem. 2000;74:1931–1940. doi: 10.1046/j.1471-4159.2000.0741931.x. [DOI] [PubMed] [Google Scholar]
- Shapiro JR. Genetics of brain neoplasms. Curr. Neurol. Neurosci. Rep. 2001;1:217–224. doi: 10.1007/s11910-001-0021-y. [DOI] [PubMed] [Google Scholar]
- Simone G, Tamba M, Quintiliani M. Role of glutathione in affecting the radiosensitivity of molecular and cellular systems. Radiat. Environ. Biophys. 1983;22:215–223. doi: 10.1007/BF01323711. [DOI] [PubMed] [Google Scholar]
- Sun X, Erb H, Murphy TH. Coordinate regulation of glutathione metabolism in astrocytes by Nrf2. Biochem. Biophys. Res. Commun. 2005;326:371–377. doi: 10.1016/j.bbrc.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat. Med. 2001;7:1010–1015. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tang P, Steck PA, Yung WK. The autocrine loop of TGF-alpha/EGFR and brain tumors. J. Neurooncol. 1997;35:303–314. doi: 10.1023/a:1005824802617. [DOI] [PubMed] [Google Scholar]
- Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC. Differential developmental expression of the two rat brain glutamate transporter proteins GLAST and GLT. Eur. J. Neurosci. 1997;9:1646–1655. doi: 10.1111/j.1460-9568.1997.tb01522.x. [DOI] [PubMed] [Google Scholar]
- Verhagen ML, Del Dotto P, van den MP, Fang J, Mouradian MM, Chase TN. Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology. 1998;50:1323–1326. doi: 10.1212/wnl.50.5.1323. [DOI] [PubMed] [Google Scholar]
- Von Deimling A, Louis DN, Wiestler OD. Molecular pathways in the formation of gliomas. Glia. 1995;15:328–338. doi: 10.1002/glia.440150312. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383–4391. [PubMed] [Google Scholar]
- Ye ZC, Rothstein JD, Sontheimer H. Compromised glutamate transport in human glioma cells: reduction-mislocalization of sodium-dependent glutamate transporters and enhanced activity of cystine-glutamate exchange. J. Neurosci. 1999;19:10767–10777. doi: 10.1523/JNEUROSCI.19-24-10767.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamecnik J. The extracellular space and matrix of gliomas. Acta Neuropathol. (Berl.) 2005;110:435–442. doi: 10.1007/s00401-005-1078-5. [DOI] [PubMed] [Google Scholar]