

Abstract
Over the past two decades it has become apparent that essentially all living cells express voltage-activated ion channels. While the role of ion channels for electrical signaling between excitable cells is well known, their function in non-excitable cells is somewhat enigmatic. Research on cancer cells suggests that certain ion channels, K+ channels in particular, may be involved in aberrant tumor growth and channel inhibitors often lead to growth arrest. An unsuspected role for K+ and Cl- channels has now been documented for primary brain tumors, glioma, where the concerted activity of these channels promotes cell invasion and the formation of brain metastasis. Specifically, Ca2+-activated K+ (BK) channels colocalize with ClC-3 Cl- channels to the invading processes of these tumor cells. Upon a rise in intracellular Ca2+, these channels activate and release K+ and Cl- ions together with obligated water causing a rapid shrinkage of the leading process. This in turn facilitates the invasion of the cell into the narrow and tortuous extracellular brain spaces. The NKCC1 cotransporter accumulates intracellular Cl- to unusually high concentrations, thereby establishing an outward directed gradient for Cl- ions. This allows glioma cells to utilize Cl- as an osmotically active anion during invasion. Importantly, the inhibition of Cl- channels retards cell volume changes, and, in turn, compromises tumor cell invasion. These findings have led to the clinical evaluation of a Cl- channel blocking peptide, chlorotoxin, in patients with malignant glioma. Data from this clinical trial shows remarkable tumor selectivity for chlorotoxin. The experimental therapeutic was well tolerated and is now evaluated in a multi-center phase II clinical trial. A similar role for Cl- and K+ channels is suspected in other metastatic cancers, and lessons learned from studies of gliomas may pave the way towards the development of novel therapeutics targeting ion channels.
Keywords: glioma, invasion, cell migration, chlorotoxin, experimental therapeutic, glia
Approximately half of the human brain is occupied by glial cells. These are generally viewed as support cells and include oligodendrocytes, microglial cells and astrocytes. Microglia are immune-competent cells in the nervous system. Oligodendrocytes form the myelin sheet that insulates fast conducting axons. Astrocytes aid normal brain function by maintaining extracellular ion and neurotransmitter homeostasis. Importantly, unlike neurons, astrocytes retain the ability to divide throughout life and can respond to various forms of brain insults by forming glial scars that seal off injured brain. This process, often termed reactive gliosis, is believed to involve the dedifferentiation and proliferation of astrocytes or their progenitor cells and possibly other cells of glial lineage. While in the context of brain injury cell proliferation appears to be a well-regulated and potentially beneficial process, the malignant transformation of glial cells leads to uncontrolled growth and the formation of primary brain tumors. Hence the intrinsic ability of glial cells to divide makes them a likely source for cancers originating within the brain, an organ in which most other cells are post mitotic and will hence not give rise to cancerous growth.
The transformation of glial cells to malignancy is not well understood. As with many cancers, mutations in tumor suppressor genes such as P53 and amplification of oncogenes such as EGFR are common and well documented (1). Yet no single genetic cause has been identified and studies examining risk factors have been largely elusive (2).
All cancerous glial cells are collectively called gliomas (3). The vast majority belong to the astrocytic lineage and these include astrocytomas and glioblastomas, the most malignant of the gliomas. The World Health Organization (WHO) classifies gliomas on a scale of I-IV with one being least malignant, four being the most (4). Criteria for this classification include nuclear atypia, evidence for mitosis, neovascularization and eventually tissue necrosis. Grade IV glioblastomas typically present with all these histopathological features.
Approximately 18,000 new gliomas are diagnosed each year in the U.S. alone and over 60% belong to the most malignant grade IV glioblastomas (5). These tumors are untreatable and patients survive less than 12 months on average (6). Even lower grade astrocytomas frequently progress towards a higher grade and hence carry a similarly dismal prognosis. Surgery remains the only treatment modality that provides some benefit, yet the invasive nature of these tumors prevents complete surgical resection in most instances (7).
The Unique Biology of Gliomas
Gliomas are unique cancers that have adapted their biology to meet unique demands. For example, gliomas are the only cancers that grow in a confined space, the cranium, and hence cannot expand unless space is vacated for the tumors growth. Indeed, only 15% of the cranial volume is fluid-filled and tumor expansion into this ventricular space is common. However, this space is ultimately insufficient to accommodate the growing tumor. To overcome these spatial constraints, gliomas actively kill neurons in the peritumoral regions through the release of high concentrations of the neurotransmitter glutamate (8). Prolonged activation of NMDA receptors causes peritumoral seizures and has been shown to cause exciototoxic neuronal cell death (9), which has been implicated as the final death pathway in many neurological diseases including stroke, ALS and other neurodegenerative diseases (10). Tumors appear to have adopted the same mechanism of cell killing to vacate room for their growth, yet they synthesize and release glutamate quite differently. Research along this vein has opened new treatment strategies targeting glutamate transporters (11) and shown promising results in preclinical studies (12). This topic was recently reviewed (13) and will not be further discussed here.
A second unique aspect, explored in greater depth in this article, pertains to the unique way in which gliomas spread and form metastasis. Unlike other cancers that spread hematogenously, gliomas do not to disseminate through the vasculature. Their failure to extravasate into blood vessels explains why gliomas are rarely—if ever—found outside the brain and hence do not form metastasis in other organ sites. They do, however, spread quite effectively within the brain and spinal cord. Quite commonly, at the time of diagnosis, tumor cells have diffusely invaded the surrounding brain and may have already crossed to the other brain hemisphere.
Cell Invasion
As they spread and form metastasis, glioma cells migrate through the narrow extracellular brain spaces often following the path of nerve fibers or blood vessels. Figure 1 shows examples of tumor cells visualized as they are actively invading. The association with blood vessels is readily visible in Figure 1A where EGFP expressing glioma cells are shown by confocal microscopy in cortical brain slices. Even in the absence of blood flow, cells readily seek out blood vessels and use them as substrate for migration. Signals involved in this process are not known. In all likelihood a favorable extracellular matrix substrate contributes to this attraction. As illustrated in Figure 1B, which shows an electron micrograph of an invading glioma cell within normal brain, invading cells commonly assume an elongated spindle-shaped morphology that suggests that cells have shrunk to fit into the narrow spaces within the brain. As indicated in the cartoon in Figure 1C, this shrinkage may affect the entire cell or just the leading cell processes. Research discussed below suggests that glioma cells have developed an unusual ability to undergo such volume changes and that they express specific ion channels and transporters that facilitate adjustments in their shape.
Figure 1.

Invading glioma cells in situ. (A) Confocal images of invading D54MG cell stably expressing EGFP adhere to blood vessels. Cells often show an elongated wedge-shaped appearance, A, bottom. (B) The elongated shape is quite apparent in an electronmicrograph that captured an invading glioma cell extending between normal brain cells. (C) Cell shrinkage requires efflux of water which is energetically driven out of the cell through the concerted secretion of Cl- and K+ through ion channels. Glutamate is shown as possible motogenic stimulus acting via AMPA-R to raise intracellular Ca2+ which may in turn activate Ca2+-activated BK channels. A color version of this figure is available in the online version of the journal.
Hypothesized Role of Glioma Ion Channels in Cell Invasion
To discuss the novel concept that ion channels may aid tumor cell migration, it is advantageous to introduce a working model for which supporting evidence will be provided in the following paragraphs. Such a model is depicted in Figure 1C and illustrates that cell shrinkage requires cytoplasmic water content to be reduced by the efflux of salt and obligated water. The energetic driving force for this process is proposed to be the electrochemical driving force for Cl- and K+ which are both accumulated intracellularly through the NKCC1 and Na+/K+-ATPase transporters respectively. A concerted opening of K+ and Cl- channels will then cause the efflux of KCl and obligated water. As illustrated in the cartoon, the activation of these channels may be a result of intracellular Ca2+ increases for example caused by the activation of Ca2+-permeable AMPA receptors (12). Moreover, this model predicts that the inhibition of either of the underlying channels or transporters would compromise the process of cell invasion.
K+ and Cl- Transporters in Glioma
All prokaryotic cells maintain elevated intracellular K+ concentrations through the activity of the Na+/K+-ATPase, which transports Na+ out of the cell in exchange for K+ import. In most cells including glioma cells, intracellular K+ concentrations are around 100-150 mM whereas extracellular K+ in the brain is ∼3 mM. As a result, there is a chemical gradient for K+ to leave the cell. Due to the large number of immobile negatively charged proteins contained in a cell, and the differential distribution of K+ by the Na+/K+-ATPase, most cells also have a membrane potential that is significantly negative. Consequently all cells maintain an electrochemical K+ gradient that favors the efflux of K+. This is typically not the case for Cl- ions. Indeed, in many cells, including neurons and oligodendrocytes, [Cl-]i is around 10 mM, and is said to be at an electrochemical equilibrium, hence there exists no net driving force for Cl- to enter or leave the cell (14). In immature neurons, however, Cl- is actively accumulated by the activity of the Na+/K+/2Cl- cotransporter NKCC1 (15), which uses the inward directed Na+ gradient to accumulate Cl- against its chemical gradient to concentrations ranging from 20-140 mM (14). As a consequence, the driving force for Cl- now favors an efflux of Cl- upon opening of Cl- channels. This explains why immature neurons show a depolarizing response to GABA, caused by the efflux of negatively charged Cl-. Upon differentiation, most neurons show a change in expression of NKCC1 in favor of expression of KCC, a transporter that releases Cl- and as a result, the outward directed driving force for Cl- ions is lost and GABA responses become either hyperpolarizing or simply stabilize the resting potential (14). Interestingly, glioma cells also maintain high intracellular Cl- which we determined to be around 80-100 mM. Similar to immature neurons, this is accomplished by the activity of the NKCC1 transporters which is highly expressed in glioma tissue (16). Even at a relatively depolarized resting potential of -40 mV (17), any opening of Cl- channels causes Cl- efflux (18). Taken together, glioma cells therefore maintain ionic gradients that would favor the efflux of Cl- and K- through ion channels or transporters and hence these ion fluxes would provide the energetic force for water to leave the cells.
Ion Channels in Glioma
As a next step we must consider the pathways through which K+ and Cl- leave the cells to cause a specific regional decrease in water content. Obvious candidates are K+ and Cl- ion channels as well as the KCC transporter which moves both K+ and Cl- out of the cell, as depicted in Figure 1C.
Glioma K+ Channels
Unlike non-malignant astrocytes or oligodendrocytes, which have a very negative resting membrane potential (-80 mV), glioma cells are much more depolarized (∼-40 mV) (17). This difference appears to be primarily due to an absence of inwardly rectifying K+ channels on the cell membrane. Both astrocytes and oligodendrocytes express Kir4.1, a Cs+ and Ba2+ sensitive channel that has very high open probability at the resting membrane potential and hence confers an unusual K+ permeability on these cells (19). Kir4.1 is thought to be the pathway through which excess neuronally released K+ is taken up into glial cells thereby buffering activity dependent changes in K+ (20). Although glioma cells express Kir4.1, channels are mislocalized to the cell nucleus (17) (Fig. 2A-D) most likely due to a trafficking defect. This mislocalization may not be accidental but quite purposeful. In astrocytes Kir4.1 expression coincides with cells becoming post-mitotic or differentiated (21). Indeed, in astrocytes Kir4.1 activity must be suppressed to allow cell division. Hence one would predict that expression of Kir4.1 in glioma membranes would suppress their ability to divide. This is indeed, the case (22) as illustrated by the fact that overexpression of recombinant Kir4.1 in human glioma cells causes them to stop dividing (Fig. 2E). Hence a sequestration of functional Kir4.1 channels into compartments where they cannot participate in the plasma membrane K+ conductance appears to be essential for the unrestraint growth of gliomas.
Figure 2.
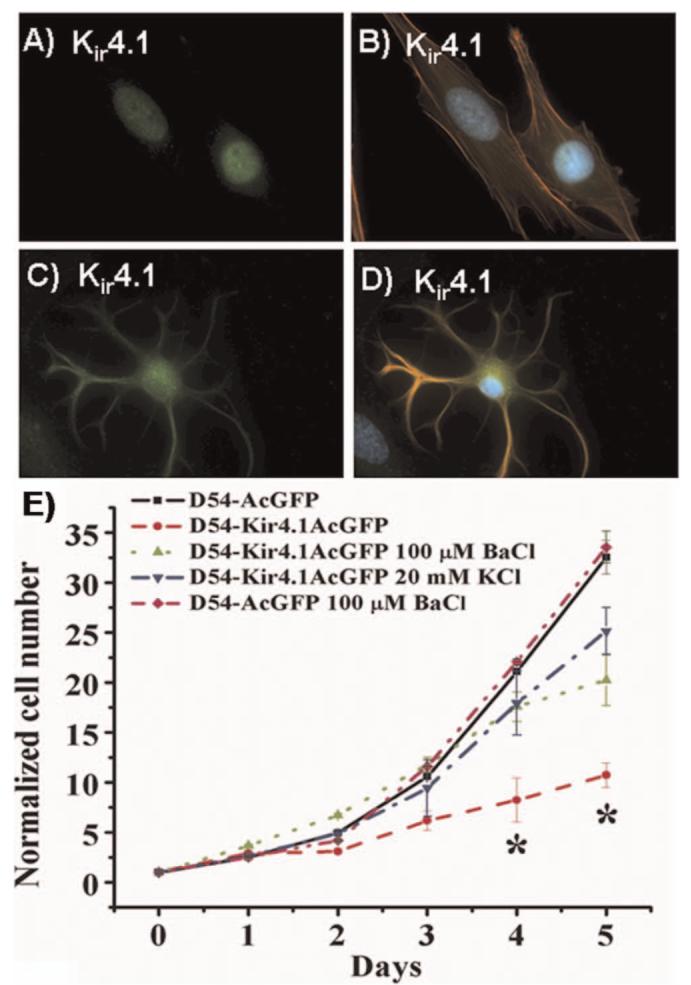
Glioma cells sequester Kir4.1 channels to the nucleus as they otherwise suppress cell growth. (A-B) Immunolabeling of cultured STTG1 glioma cells and spinal cord astrocytes (C-D) with Kir4.1 antibodies (green), phalloidin (red) and DAPI (blue). While both cell types show Kir4.1 channel expression, only astrocytes have them on the plasma membrane. (E) Cell growth over a 5-days period comparing wild type D54-MG glioma cells to cell expressing GFP tagged Kir4.1 and vector only D54-MG cells. Kir expression inhibited growth unless channel activity was blocked with Ba2+ or cells were depolarized by high K+. [(A-D) reproduced with permission from (17); (E) reproduced with permission from (22).] A color version of this figure is available in the online version of the journal.
Although glioma cells do not express functional Kir4.1 channels, they do express prominent K+ currents that are outwardly rectifying, i.e. predominantly allow K+ efflux, and these are mediated by channels from a different gene family. Specifically, glioma cells highly express Ca2+-activated K+ channels that represent a new splice variant of the human slo gene that gives rise to so-called big conductance K+ (BK) channels. The glioma variant of this channels has been termed glioma BK (gBK) and contains a 34 amino acid insert near the Ca2+ sensor of the channel (23). This makes glioma BK channels much more sensitive to small and physiologically relevant changes in intracellular Ca2+ (24). As a consequence, gBK channels are activated by many physiological stimuli such as exposure to neurotransmitters or growth factors (24). Figure 3A shows example recordings from two glioma cells, one recorded within a glioma section in situ, the other from an isolated glioma cell. Both show characteristic outwardly rectifying K+ currents that are inhibited by the application of paxilline, a BK channel specific inhibitor. In Figure 3B, recordings from an isolated glioma cell demonstrate that the application of 50μM Ach caused an increase in intracellular Ca2+ as determined by Fura-II imaging that causes the activation of BK channels resulting in a large hyperpolarization of the cell membrane. The fact that channels can be activated by a physiological increase in Ca2+ alone is significant because it is difficult to envision conditions under which the cells would experience large voltage changes that would otherwise be required for channel activation. Of note, gBK channels are expressed at very high density in gliomas, comparable to Kir4.1 channels in non-malignant glia. Moreover, protein expression increases with the malignancy of the tumor. As illustrated in Figure 3C, the protein concentration increases significantly when one compares Western blots obtained from 2 patients each diagnosed with tumor ranging from a grade I-IV with highest levels in a grade IV tumor. Despite the enhanced Ca2+ sensitivity, activation of BK channels still requires intracellular Ca2+ concentrations on the order of 500 nM for activation. While global increase in Ca2+ of such magnitude is rare, it has been hypothesized that BK channels may localize in the vicinity of a physiological Ca2+ source, i.e. a Ca2+ entry channels or release site from intracellular stores. This has indeed been demonstrated in a recent study that shows the co-localization of gBK channels with IP3 receptors on the invadipodia of glioma cells (25). While the channels do not interact directly with IP3 receptors, they jointly localize to specialized lipid domains called lipid rafts. These rafts can be isolated biochemically and contain BK and IP3-Rs in a lipid raft fraction that is also positive for caveolin-1, a structural protein that defines the subset of caveolar lipid rafts. Functionally, this clustering into defined lipid domains creates a privileged microenvironment in which BK channels can experience significant increases in Ca2+ sufficient for channels activation. Given that BK channels are the only significant K+ conductance in gliomas and the fact that they are activated by physiological ligands that are important in glioma invasion, we suggest that gBK is an important diffusional pathway for K+ release required for the shrinkage of invading cells or their processes.
Figure 3.

Glioma cells express Ca2+-activated K+ channels. (A) Representative recordings from glioma cells in a brain section or isolated cell. Outward currents were sensitive to paxilline, a specific inhibitor of Ca2+-activated K+ channels. Block was incomplete in brain slices because drug perfusion was limited in tissue. (B) Application of Acetylcholin (Ach) to a cultured glioma cell recorded using amphotericin perforated patch, hence without disturbing intracellular Ca2+ caused a rapid increase in Ca2+ determined by Fura-II recording (top) associated with a large hyperpolarizing voltage shift indicating activation of BK channels. (C) Two biopsies each from patients with gliomas ranging in malignancy grade from WHO-I-IV show an increase in BK channel protein with increasing malignancy on Western blot. [(C) reproduced with permission from (23); (B) reproduced with permission from (25).]
Glioma Cl- Channels
For K+ efflux through BK channels to mediate cell shrinkage, the cation efflux must be balanced by the efflux of osmotically active anions, most likely Cl-. Indeed, glioma cells show an unusual resting conductance for Cl- which can be attributed to channels that are sensitive to NPPB, DIDS and tamoxifen (18). In cells voltage clamped at -40 mV using amphoterecin-perforated patch clamp so as not disturb intracellular Cl-, the Cl- channel blocker NPPB reversibly blocked a Cl- current and inhibited a resting membrane conductance (18). Similar experiments in non-malignant astrocytes show an almost complete absence of Cl- channels active at rest (26). This Cl- conductance contributes significantly to the depolarized resting potential of glioma cells as inhibition of these Cl- channels causes a-10.7 mV hyperpolarization of the membrane potential (Habela and Sontheimer, unpublished). Cells that extend processes show activation of an NPPB sensitive channel and process extension correlates directly with and requires channel activity (Fig. 4A). The identity of the underlying Cl- channels has been questioned for some time. Unfortunately, specific pharmacological inhibitors for Cl- channels are absent and hence one must rely on a combination of biochemical and biophysical approaches to identify the underlying channels. The examination of glioma biopsies from patients revealed prominent expression of ClC-2, -3 & -5 (Fig. 4B) and all glioma cell lines examined also express these proteins (Fig. 4B). ClC-2 is also abundantly expressed in normal brain yet ClC-3 and ClC-5 are believed to be expressed only on intracellular vesicles. In gliomas, however, ClC-3 is found on the cell membrane and immuno-gold conjugated antibodies to ClC-3 show clusters of ClC-3 channels on the cell membrane and in intracellular vesicles (Fig. 4C). Importantly, currents attributable to ClC-2 and ClC-3 channels can be readily identified using genetic knockdown approaches. The treatment of glioma cells with ClC-2 antisense causes the selective loss of inwardly rectifying Cd2+ sensitive Cl- currents with biophysical features characteristic of ClC-2 channels (27) (Fig. 5A). Similarly, knockdown of ClC-3 reduces outwardly rectifying, inactivating Cl- currents sensitive to NPPB and DIDS (27) (Fig. 5B). These data suggest that ClC-2 and ClC-3 channels are excellent candidates mediating the resting Cl- conductance and hence may be involved in Cl- efflux that mediates cell shrinkage of invading cells. To question this assumption more directly, glioma invasion was studied using Transwell migration chambers in which drug effects on cell invasion can be readily quantified. These studies suggest that replacement of Cl- with anions that cannot permeate ClC-2 or ClC-3 channels impairs cell migration (28), as does inhibition of either ClC-2 with Cd2+ (28) or ClC-3 with NPPB (18), the latter being most effective. Interestingly, currents with the biophysical signature of ClC-3 are also inhibited by chlorotoxin (Cltx) (Fig. 5C), a scorpion peptide reported to be a Cl- channel inhibitor (29). This peptide binds to essentially all glioma cells in vivo, as illustrated by an example staining using an OregonGreen conjugated peptide (Fig. 5C, right). Interestingly, ClC-3 channels localize to the same specialized lipid domains on the invadipodia that also contain BK K+ channels (Fig. 6A, B) and both are found in the same lipid raft fraction when biochemically isolated (Fig. 6C). This colocalization may facilitate the local regulation of water efflux and hence localized cell shrinkage. Moreover, these lipid domains may generate functional domains in which channels that work together are clustered into privileged domains, a concept illustrated in the cartoon in Figure 6D. In support of this, we recently discovered that these lipid raft domains are also enriched in aquaporin-4, the principle water channel expressed in gliomas (30).
Figure 4.
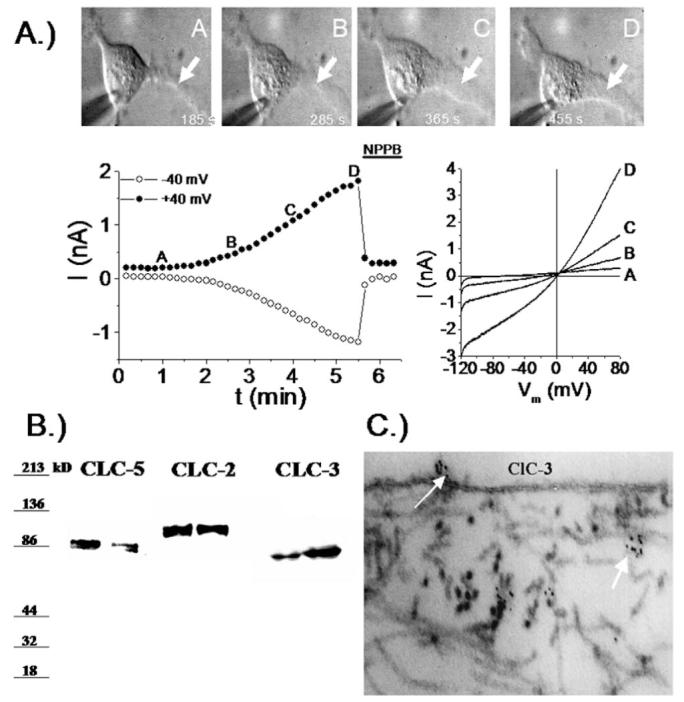
Expression and function of Cl- channels in glioma cells. (A) Extension of a process in a cultured glioma cells correlates with the progressive development of a NPPB sensitive outwardly rectifying Cl- current. (B) Western blots from 2 biopsies each probed for ClC-2, ClC-3 and ClC-5 show prominent expression of all 3 members of this channel family. (C) Using immuno-gold electron microscopy clusters of ClC-3 channels can be seen on the membrane surface of glioma cells. [(A) reproduced with permission from (18).]
Figure 5.

Glioma cells express functional ClC-2 and ClC-3 channels. Using specific antisense oligonucleotides to ClC-2 (A) and ClC-3 (B) it was possible to identify currents attributable to these channels respectively. The Western blots illustrate effective reduction in protein after antisense treatment and show specificity. (C) Currents with a biophysical signature of ClC-3 are inhibited by 1 μM Chlorotoxin. OregonGreen-conjugated Chlorotoxin was used to label an acute biopsy section from a glioblastoma patient (right). [(A) reproduced with permission from (27); (C) reproduced with permission from (34).] A color version of this figure is available in the online version of the journal.
Figure 6.

ClC-3 and BK channels colocalize to lipid rafts. D54-MG glioma cells were immunostained with the beta subunit of Cholera toxin (CTxB) to identify lipid rafts. (A) ClC-3 antibodies labeled the same domains as indicated in the merged image. (B) Similarly, BK antibodies localize to lipid rafts suggesting a co-localization of ClC-3 and BK channels to lipid rafts. (C) Biochemical isolation of lipid rafts indeed demonstrates that BK and ClC-3 channels are enriched in the lipid raft domain that also contains the structural protein caveolin-1 found in caveolar rafts. (D) We hypothesize that Cl- and K+ channels colocalize to invadipodia to facilitate cell shrinkage and that lipid rafts localize them in close proximity. [Reproduced in modified form with permission from (34).] A color version of this figure is available in the online version of the journal.
Clinical Application
Following the initial discovery that glioma cells express Cl- channels that may aid the invasion of these cells, and electrophysiological studies showing that chlorotoxin acted as relatively specific inhibitor for glioma Cl- channels (31) it was demonstrated that this drug is an effective inhibitor of glioma cell invasion in vitro and in vivo (32), ultimately paving the way for its clinical use. Figure 7A & B show examples of Transwell migration assays in which natural and recombinant Cltx were directly compared regarding their ability to inhibit glioma invasion. Both drugs showed a dose-dependent inhibition of migration with an IC50 around 100 μM (A). Surprisingly, however, further studies on the interaction of Cltx with Cl- channels suggested that these effects were indirect. The actual receptor for Cltx appears to be a protein complex that contains MMP-2 (33) and ClC-3 and binding of Cltx induces the endocytosis of this complex and hence the ClC-3 channels (34). This is a surprising action for a scorpion peptide but explains the irreversible action of this peptide and its relatively slow time course of Cl- channel block (35). As illustrated in Figure 7C, surface expression of ClC-3 channels is almost completely eliminated due to channel endocytosis into caveolar vesicles when glioma cells were exposed to Cltx. The drug effect could be overcome if the lipid rafts were stabilized, hence preventing endocytosis using the drug Filipin. In the presence of Filipin, Cltx also lost its ability to inhibit cell invasion (Fig. 7D). Even preceding the identification of this mechanism of action, the relative glioma specificity of this peptide spurred research toward a clinical application of Cltx as a drug to target gliomas clinically. This work demonstrated for example that Cltx binds exclusively to glial derived tumors and some cancers that are embryologically related (36), melanoma being one example. No binding was observed to normal human tissues in numerous autopsies and biopsies evaluated. In line with these findings, a recent study used a Cy5-labelled Cltx to demarcate tumor boundaries of medulloblastomas in vivo (37). A synthetic version of Cltx showed equal biological activity and tumor specificity to the native toxin and was submitted to the FDA for clinical evaluation. Beginning in 2004, a phase 1 clinical study began administering Cltx labeled with 10 mCu 131I to patients with late stage, high-grade malignant glioma. These patients had received prior radiation and chemotherapy and have shown recurrence. A single injection of the drug through a micro-release catheter into the tumor cavity allowed for subsequent imaging (38). Examples of whole body scans are shown in Figure 8 (top), which illustrates a series of images taken from a patient over a 5-day period. As predicted from prior animal studies, Cltx showed remarkable tumor specificity and did not show any non-specific labeling. Early scans showed radiolabel only in those organs involved with the excretion of excessive material such as the liver kidney and urinary bladder. Importantly, the peptide remained bound to the tumor tissue for over 5 days, and this is more clearly visible on the MRI scans in Figure 8 (bottom). Coregistration of the SPECT image, identifying I-Cltx with T1-MRI as a structural image showed tumor specific retention 8 days post injection of the drug (39). This finding is consistent with the above-discussed discovery that Cltx is being endocytosed into caveolar vesicles and hence trapped inside cells. Typically, radio labeled compounds clear the body in less than 24 h. From a therapeutic standpoint, the endocytosis enhances the effectiveness of 131I conjugated to the Cltx.
Figure 7.

Inhibition of Cl- channels with Chlorotoxin retards cell invasion. (A) Dose-dependent inhibition of cell invasion using Transwell migration/invasion assays shows effective inhibition with natural (Cltx) and recombinant (His-Cltx) chlorotoxin. (B) Underside of Transwell filters shows a significant decrease in migrated cells in the presence of 5 μM Cltx. (C) Cltx interferes with trafficking of ClC-3 channels. Isolation of membrane associated ClC-3 channels was achieved after cell surface biotinylation. 30 min incubation with Cltx causes a significant loss of ClC-3 from the plasma membrane presumably by internalization of channels into caveolar vesicles. Disruption of caveoli with the sterol binding drug Filipin eliminated the effect of Cltx and retained all ClC-3 protein on the cell surface. (D) The inhibitory effects of Cltx on cell migration/invasion were also overcome by treatment of cells with Filipin which would prevent internalization of ClC-3 channels. [(B) reproduced with permission from (32); (A, C, D) reproduced with permission from (33).] A color version of this figure is available in the online version of the journal.
Figure 8.
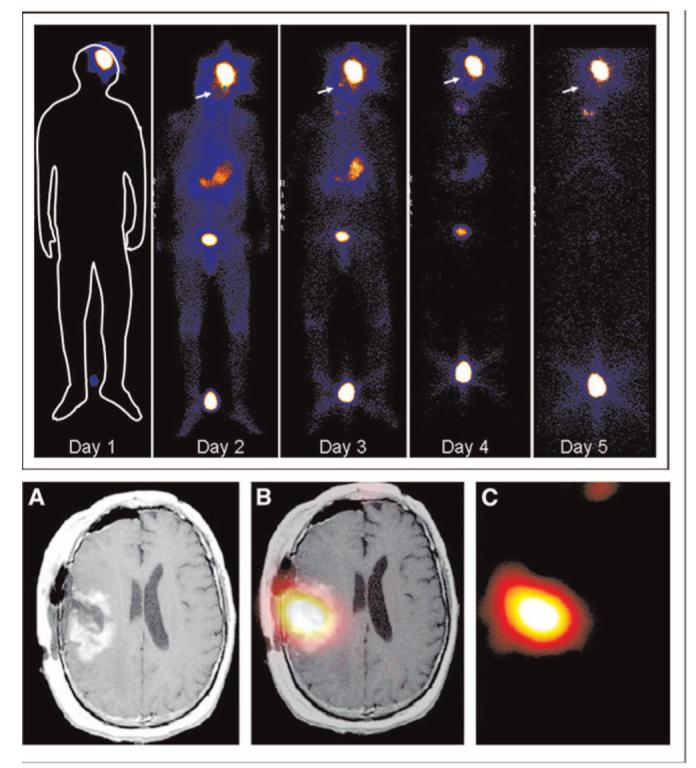
Chlorotoxin in clinical trial: (top) Whole body gamma scan shows specific localization and retention of 131I-ClTx to a glioblastoma in a patient from a recent Phase I study [reproduced with permission from (38)]. Hot spot between the subject’s feet is a radiation standard. (Bottom) Axial view of T1-Wc (A), coregistered (B), and SPECT (day 8) images of intrathecal-delivered peptide. [Reproduced with permission from (39).] A color version of this figure is available in the online version of the journal.
Results from the completed phase 1 study have recently been published (38) as have images form patients like those shown in Figure 8. Overall the drug was deemed safe and tumor specific and FDA approved the use of this compound in a multi-center phase II efficacy study in which multiple doses of Cltx are administered. This study is currently enrolling patients at over 10 trial sites (see www.clinicaltrials.gov). An additional clinical trial has just released preliminary findings from 6 patients indicating that 131I-Cltx administered intravenously crosses the blood-brain barrier and binds to malignant gliomas (Dr. John Fiveash, UAB unpublished). Hence, it is possible that even inoperable gliomas may soon be treated with this peptide that targets Cl- channels, and, importantly these studies may suggest the administration of this drug much sooner in the progression of the disease.
Following the cloning of glioma specific BK channels (23), described above, and the discovery that a functional loss of Kir4.1 channels is associated with the malignant growth of these tumors (22), both classes of K+ channels appeared to also be logical targets for the development of novel therapeutic. Indeed, K+ channels have been demonstrated to govern growth control in many other cancers (40) and K+ channel inhibitors can generally inhibit cell proliferation in culture. Moreover, overexpression of the gene encoding the “ether a go-go” (EAG) channel in fibroblasts is sufficient to induce solid cancers in mice (41) suggesting that these channels act as oncogenes. Surprisingly, these channels may act as regulators for mitogen-activated protein kinase stimulating growth without acting as ion conducting pores (42). While these studies are providing fascinating insight into the interplay of biophysics and cancer biology, they have unfortunately not yet yielded any clinical application. Despite much research, K+ channels remain elusive therapeutic targets. This is largely due to the inability to develop drugs that only target cancer-associated K+ channels. In the case of glioma cells, for example, BK channels would represent an excellent glioma specific target. However drugs that inhibit glioma BK channels also inhibit BK channels that are vital for the function of vascular smooth muscle cells and many interneurons. With regards to Kir4.1 channels, an opportunity would exist to arrest growth of gliomas if one could identify signals that enhance their expression and proper targeting to the cell membrane (22), neither of which is currently available.
Outlook and Implications
The notion that ion channels may have important functions in cancer has been around for some time and certain K+ channels have received most of the attention (43). In most instances it has been difficult to assign a specific, mechanistic role for these channels as it pertains to the cancerous growth that they are supporting. Gliomas may chart the way in this regard as the proposed model is intuitive and the clinical data compelling. It is possible, even likely, that other cancers use Cl- and K+ channels, albeit from different molecular families in similar ways. Cancer cells must undergo profound shape changes as they extravasate into blood vessels or as they penetrate into organ sites. Consequently the findings discussed here may have much broader applicability for other cancers. In addition, the author subscribes to the notion that cancers recapitulate many biological phenomena of early development. In this vein, gliomas may be a useful model system to understand cell migration in normal brain development where most cells are on the move. It is conceivable that migratory glia and neurons may transiently utilize the here-described cell volume changes as a means to support their journey through the brain placing ion channels into an important biological context.
Acknowledgments
The author is grateful for the continued supported by grants from the National Institutes of Health RO1 NS-31234, RO1 NS-52634, and NS-36692, and P50-CA97247.
References
- 1.Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 2.Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2:494–503. doi: 10.1038/ncpneuro0289. [DOI] [PubMed] [Google Scholar]
- 3.Kleihues P, Soylemezoglu F, Schaueble B, Scheithauer BW, Burger PC. Histopathology, classification and grading of gliomas. Glia. 1995;15:211–221. doi: 10.1002/glia.440150303. [DOI] [PubMed] [Google Scholar]
- 4.Kleihues P, Burger PC, Scheithauer BW. The new who classification of brain tumours. Brain Pathol. 1993;3:255–268. doi: 10.1111/j.1750-3639.1993.tb00752.x. [DOI] [PubMed] [Google Scholar]
- 5.Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 6.Huncharek M, Muscat J. Treatment of recurrent high grade astrocytoma; results of a systematic review of 1,415 patients. Anticancer Res. 1998;18:1303–1311. [PubMed] [Google Scholar]
- 7.Zalutsky MR. Current status of therapy of solid tumors: brain tumor therapy. J Nucl Med. 2005;46(Suppl 1):151S–156S. [PubMed] [Google Scholar]
- 8.Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383–4391. [PubMed] [Google Scholar]
- 9.Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor. TINS. 1987;10:299–302. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- 10.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. New Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 11.Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat Med. 2001;7:1010–1015. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- 12.Lyons SA, Chung WJ, Weaver AK, Ogunrinu T, Sontheimer H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer Res. 2007;67:9463–9471. doi: 10.1158/0008-5472.CAN-07-2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sontheimer H. Ion channels and amino acid transporters support the growth and invasion of primary brain tumors. Mol Neurobiol. 2004;29:61–71. doi: 10.1385/MN:29:1:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Owens DF, Boyce LH, Davis MB, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J Neurosci. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamada J, Okabe A, Toyoda H, Kilb W, Luhmann HJ, Fukuda A. Cl-uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J Physiol. 2004;557:3–41. doi: 10.1113/jphysiol.2004.062471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernest NJ, Weaver AK, Van Duyn LB, Sontheimer HW. Relative contribution of chloride channels and transporters to regulatory volume decrease in human glioma cells. Am J Physiol Cell Physiol. 2005;288:C1451–C1460. doi: 10.1152/ajpcell.00503.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olsen ML, Sontheimer H. Mislocalization of Kir channels in malignant glia. Glia. 2004;46:63–73. doi: 10.1002/glia.10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ransom CB, O’Neal JT, Sontheimer H. Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J Neurosci. 2001;21:7674–7683. doi: 10.1523/JNEUROSCI.21-19-07674.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J Neurophysiol. 1995;73:333–345. doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- 20.Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neurosci. 2004;129:1043–1054. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacFarlane SN, Sontheimer H. Changes in ion channel expression accompany cell cycle progression of spinal cord astrocytes. Glia. 2000;30:39–48. doi: 10.1002/(sici)1098-1136(200003)30:1<39::aid-glia5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 22.Higashimori H, Sontheimer H. Role of Kir4.1 channels in growth control of glia. Glia. 2007;55:1668–1679. doi: 10.1002/glia.20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X, Chang Y, Reinhart PH, Sontheimer H, Chang Y. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J Neurosci. 2002;22:1840–1849. doi: 10.1523/JNEUROSCI.22-05-01840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ransom CB, Liu X, Sontheimer H. BK channels in human glioma cells have enhanced calcium sensitivity. Glia. 2002;38:281–291. doi: 10.1002/glia.10064. [DOI] [PubMed] [Google Scholar]
- 25.Weaver AK, Olsen ML, McFerrin MB, Sontheimer H. BK channels are linked to IP3-receptors via lipid rafts: a novel mechanism for coupling [CA2+]1 to ion channel activation. J Biol Chem. 2007;282:31558–31568. doi: 10.1074/jbc.M702866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parkerson KA, Sontheimer H. Contribution of chloride channels to volume regulation of cortical astrocytes. Am J Physiol Cell Physiol. 2003;284:C1460–C1467. doi: 10.1152/ajpcell.00603.2002. [DOI] [PubMed] [Google Scholar]
- 27.Olsen ML, Schade S, Lyons SA, Amarillo MD, Sontheimer H. Expresssion of voltage-gated chloride channels in human glioma cells. J Neurosci. 2003;23:5572–5582. doi: 10.1523/JNEUROSCI.23-13-05572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soroceanu L, Manning TJ, Jr, Sontheimer H. Modulation of glioma cell migration and invasion using Cl(-) and K(+) ion channel blockers. J Neurosci. 1999;19:5942–5954. doi: 10.1523/JNEUROSCI.19-14-05942.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeBin JA, Maggio JE, Strichartz GR. Purification and characterization of chlorotoxin, a chloride channel ligand from the venom of the scorpion. Am J Physiol. 1993;264:C361–369. doi: 10.1152/ajpcell.1993.264.2.C361. [DOI] [PubMed] [Google Scholar]
- 30.McCoy E, Sontheimer H. Expression and function of water channels (aquaporins) in migrating malignant astrocytes. Glia. 2007;55:1034–1043. doi: 10.1002/glia.20524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ullrich N, Bordey A, Gillespie GY, Sontheimer H. Expression of voltage-activated chloride currents in acute slices of human gliomas. Neuroscience. 1998;83:1161–1173. doi: 10.1016/s0306-4522(97)00456-9. [DOI] [PubMed] [Google Scholar]
- 32.Soroceanu L, Manning TJ, Jr, Sontheimer H. Modulation of glioma cell migration and invasion using Cl- and K+ ion channel blockers. J Neurosci. 1999;19:5942–5954. doi: 10.1523/JNEUROSCI.19-14-05942.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deshane J, Garner CC, Sontheimer H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J Biol Chem. 2003;278:4135–4144. doi: 10.1074/jbc.M205662200. [DOI] [PubMed] [Google Scholar]
- 34.McFerrin MB, Sontheimer H. A role for ion channels in glioma cell invasion. Neuron Glial Biol. 2006;2:39–49. doi: 10.1017/S17440925X06000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ullrich N, Gillespie GY, Sontheimer H. Human astrocytoma cells express a unique chloride current. Neuroreport. 1996;7:1020–1024. doi: 10.1097/00001756-199604100-00013. [DOI] [PubMed] [Google Scholar]
- 36.Lyons SA, O’Neal J, Sontheimer H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia. 2002;39:162–173. doi: 10.1002/glia.10083. [DOI] [PubMed] [Google Scholar]
- 37.Veiseh M, Gabikian P, Bahrami SB, Veiseh O, Zhang M, Hackman RC, Ravanpay AC, Stroud MR, Kusuma Y, Hansen SJ, Kwok D, Munoz NM, Sze RW, Grady WM, Greenberg NM, Ellenbogen RG, Olson JM. Tumor paint: a chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer Res. 2007;67:6882–6888. doi: 10.1158/0008-5472.CAN-06-3948. [DOI] [PubMed] [Google Scholar]
- 38.Mamelak AN, Rosenfeld S, Bucholz R, Raubitschek A, Nabors LB, Fiveash JB, Shen S, Khazaeli MB, Colcher D, Liu A, Osman M, Guthrie B, Schade-Bijur S, Hablitz DM, Alvarez VL, Gonda MA. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. J Clin Oncol. 2006;24:3644–3650. doi: 10.1200/JCO.2005.05.4569. [DOI] [PubMed] [Google Scholar]
- 39.Hockaday DC, Shen S, Fiveash J, Raubitschek A, Colcher D, Liu A, Alvarez V, Mamelak AN. Imaging glioma extent with 131I-TM-601. J Nucl Med. 2005;46:580–586. [PubMed] [Google Scholar]
- 40.Kunzelmann K. Ion channels and cancer. J Membr Biol. 2005;205:159–173. doi: 10.1007/s00232-005-0781-4. [DOI] [PubMed] [Google Scholar]
- 41.Pardo LA, del Camino D, Sanchez A, Alves F, Bruggemann A, Beckh S, Stuhmer W. Oncogenic potential of EAG K(+) channels. EMBO J. 1999;18:5540–5547. doi: 10.1093/emboj/18.20.5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hegle AP, Marble DD, Wilson GF. A voltage-driven switch for ion-independent signaling by ether-a-go-go K+ channels. Proc Natl Acad Sci U S A. 2006;103:2886–2891. doi: 10.1073/pnas.0505909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kunzelmann K. Ion channels and cancer. J Membr Biol. 2005;205:159–173. doi: 10.1007/s00232-005-0781-4. [DOI] [PubMed] [Google Scholar]