

Summary
The tumor necrosis factor-alpha (TNF)-induced inflammatory response in human lung microvascular endothelial cells (MVECs) is an early event in acute lung injury. Studies have shown that p38 mitogen-activated protein kinase (MAPK), MAPK-activated protein kinase 2 (MK2) and heat shock protein 27 (HSP27) are involved in the expression of pro-inflammatory mediators in other cell types. However, their role in the TNF-induced inflammatory response in lung MVECs has not been determined. We evaluated the role of p38 MAPK, MK2 and HSP27 in regulating the TNF-induced expression of ICAM-1 and IL-8 in human lung MVECs. Inhibition of p38 MAPK reduced ICAM-1 and IL-8 expression without influencing NF-κB activation or ICAM-1 and IL-8 mRNA levels. TNF stimulation induced p38 MAPK-dependent phosphorylation of MK2 and HSP27. MK2 silencing reduced ICAM-1 and IL-8 expression without influencing NF-κB activation or ICAM-1 and IL-8 mRNA levels. HSP27 silencing reduced cellular HSP27 levels and HSP27 phosphorylation following TNF stimulation but had no effect on ICAM-1 and IL-8 expression. Our study demonstrates for the first time that MK2 mediates post-transcriptional regulation by p38 MAPK of the TNF-induced expression of ICAM-1 and IL-8 in human lung MVECs, and that this regulation by the p38 MAPK/MK2 pathway is dissociated from HSP27 phosphorylation.
Keywords: human lung microvascular endothelial cells, MK2, HSP27, siRNA, ICAM-1, IL-8
1. Introduction
One of the earliest events in the pathophysiology of acute lung injury and its more severe form, acute respiratory distress syndrome (ARDS), is an increase in circulating levels of pro-inflammatory cytokines, particularly tumor necrosis factor-α (TNF) [1, 2]. Circulating TNF interacts with lung microvascular endothelial cells to induce the expression of chemokines and adhesion molecules, including IL-8 and ICAM-1 [3]. Both IL-8 and ICAM-1 are necessary for neutrophil accumulation and infiltration in the lung during acute lung injury and ARDS [1, 2, 4, 5].
Although it is well known that TNF induces the expression of ICAM-1 and IL-8 in endothelial cells [3], the mechanisms involved are not fully understood. Several studies on human umbilical vein endothelial cells (HUVECs) found that inhibition of the p38 mitogen-activated protein kinase (MAPK) pathway reduces TNF-stimulated expression of IL-8 without influencing ICAM-1 expression [6, 7]. However, how endothelial cells in other vascular beds utilize the p38 MAPK pathway in the inflammatory response to TNF is unclear, and the role of the p38 MAPK pathway in ICAM-1 and IL-8 expression in human lung microvascular endothelial cells (MVECs) remains to be determined.
The transcription factor NF-κB plays a major role in controlling the TNF-induced expression of ICAM-1, IL-8 and other pro-inflammatory factors in HUVECs [6, 8, 9]. An association has been found between p38 MAPK and NF-κB activation in several cell types, including HeLa cells [10], and a recent study linked the p38 MAPK/MK2/HSP27 pathway to NF-κB intranuclear retention, a mechanism for controlling the level and duration of NF-κB activation [11]. However, the influence of the p38 MAPK pathway on TNF-induced NF-κB activation in lung MVECs has not been determined.
MAPK-activated protein kinase 2 (MK2) is a downstream kinase of p38 MAPK [12, 13]. It is phosphorylated by p38 MAPK and it, in turn, phosphorylates heat shock protein 27 (HSP27) to induce smooth muscle cell migration [14]. In addition, the p38 MAPK/MK2/HSP27 pathway has been shown to mediate NF-κB activation in HUVECs [11] and cytoskeletal changes in rat pulmonary endothelial cells [15]. Recently, HSP27 was found to be required for the IL-1-induced expression of the pro-inflammatory mediators cyclooxygenase-2, IL-6, and IL-8 in HeLa cells and fibroblasts [16]. However, it is unknown whether MK2 and HSP27 are involved in regulating the TNF-induced pro-inflammatory response in lung MVECs. Evaluating the role of the p38 MAPK/MK2/HSP27 pathway in the pro-inflammatory response to TNF in human lung MVECs may provide insight into the mechanism(s) by which TNF contributes to early events in acute lung injury and ARDS. Therefore in this study we examined: 1) the relative role of the p38 MAPK and ERK 1/2 pathways in the TNF-induced expression of ICAM-1 and IL-8 in human lung MVECs, 2) whether p38 MAPK regulates the TNF-induced expression of ICAM-1 and IL-8 at the level of transcriptional control by NF-κB, and 3) whether MK2 and/or HSP27 is involved in this regulation.
2. Materials and Methods
2.1. Materials
Human lung MVECs and cell culture reagents were purchased from Lonza (Walkersville, MD). TNF and Accutase solution were purchased from Sigma-Aldrich (St. Louis, MO). SB203580 (p38 MAPK inhibitor) and PD98059 (MAPK kinase inhibitor that inhibits ERK1/2 activation) were purchased from Calbiochem (San Diego, CA). Protein assay reagent and ECL immunoblotting substrate were purchased from Pierce (Rockford, IL). Antibodies against phosphorylated p38 (Thr180/Tyr182, #9211), phosphorylated ERK1/2 (Thr202/Tyr204, #9102), total MK2 (#3042), phosphorylated MK2 (Thr-334, #3041), phosphorylated HSP27 (Ser 78, #2405), phosphorylated NF-κB p65 (Ser536, #3033) and β-actin (#4967) were purchased from Cell Signaling (Danvers, MA). Total HSP27 antibody (spa-803) was purchased from Stressgen (Ann Arbor, MI). HSP27 siRNA (sc-29350), scrambled siRNA (sc-37007), transfection reagents (sc-29528), transfection medium (sc-36868) and ICAM-1 antibody (H108, sc-7891) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Film was bought from Bioexpress (Kaysville, UT). ON-TARGET plus SMART pool human MK2 siRNA (L-003516-00-0005), scrambled siRNA (D-001700-01-20), DharmaFECT1 transfection reagent (T-2001-02) and other transfection-related reagents for MK2 knockdown experiments were purchased from Dharmacon (Lafayette, CO). Opti-MEM I reduced serum medium was purchased from Invitrogen (Carlsbad, CA). PE-conjugated anti-human ICAM-1 antibody and PE-conjugated mouse IgG1 isotype control antibody were purchased from eBioscience (San Diego, CA). RNeasy micro kit was purchased from QiaGen (Valencia, CA). IL-8 and MCP-1 ELISA Kits were purchased from R&D (Minneapolis, MN).
2.2. Cell Culture and treatment
Human lung MVECs were cultured in complete medium (EGM-2 medium with 5% fetal bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin) at 37 °C in an incubator supplied with 5% CO2. Cells from passages 5 to 8 were used for all experiments. Unless otherwise specified, cells were at 90% confluence. For certain experiments, cells were stimulated with TNF (10 ng/ml). MAPK inhibitors (SB203580 and PD98059, various concentrations) were added to culture media 30 min before TNF stimulation and were present during TNF stimulation.
2.3. Immunoblotting
Immunoblotting was used to detect ICAM-1, phosphorylated p38 and ERK1/2, total and phosphorylated MK2 (Thr-334), total and phosphorylated HSP27 (Ser78 and Ser82), and phosphorylated NF-κB p65, with β-actin as a control. After treatment, human lung MVECs were lysed with lysis buffer (PBS containing a protease inhibitor cocktail and 1 % Triton X100, pH 7.4). Cell samples were separated on 4–20 % SDS-polyacrylamide gels (Bio-Rad, Hercules, CA), then transferred to nitrocellulose membranes. Membranes were rinsed in PBS and blocked for 1 h at room temperature with 5 % dry milk in TPBS (PBS containing 0.1% Tween 20), then incubated with the appropriate primary antibodies (ICAM-1 antibody was diluted 1:200, phosphorylated ERK1/2 antibody 1:500, and all others 1:1000) overnight at 4°C. After washing 4×5 min with TPBS, membranes were incubated with Horseradish peroxidase (HRP)-linked secondary antibodies (1:5000 dilution with TPBS containing 5% dry milk) at room temperature for 1 h. After washing 4×5 min with TPBS, target bands were developed using ECL and exposed on film. Band density was measured using NIH software ImageJ.
2.4. Gene knockdown
SiRNA transfection was performed to silence MK2 and HSP27. Human lung MVECs were cultured in antibiotic-free growth medium in 12-well plates (for immunoblotting) or 6-well plates (for PCR) until 60% confluent. For HSP27 siRNA transfection, cells were incubated with a mixture of HSP27 siRNA (60 nM) and transfection reagent (sc-29528, 6 µl per ml transfection medium) in antibiotic- and serum-free medium for 6 h, which we found to be the optimal time. The 70–80% of cells still alive after transfection were allowed to recover in growth medium for 48 h, then stimulated with TNF. Controls were treated with scrambled siRNA (sc-37007) and transfection reagent (sc-29528). For MK2 siRNA transfection, a transfection mixture of MK2 siRNA (30 nM) and DharmaFECT1 transfection reagent (0.5 µl per ml) was made in Opti-MEM I reduced serum medium. Cells were incubated with the transfection mixture in complete medium for 48 h, which we found to be the optimal transfection time. The more than 95% of cells still alive after transfection were incubated in growth medium for 24 h, then stimulated with TNF. Controls were treated with scrambled siRNA (D-001700-01-20) and DharmaFECT1 transfection reagent (T-2001-02). The sense and antisense siRNA oligonucleotides used to generate siRNA are detailed in Table 1.
Table 1.
Sequences of MK2 and HSP27 siRNA and corresponding scrambled siRNA.
| MK2 | HSP27 | |||
|---|---|---|---|---|
| Sense | 5’-CGAAUGGGCCAGUAUGAAUUU-3’ | Sense | 5’-GAGUGGUCGCAGUGGUUAGtt-3’ | |
| Antisense | 5’-PAUUCAUACUGGCCCAUUCGUU-3’ | Antisense | 5’-CUAACCACUGCGACCACUCtt-3’ | |
| Sense | 5’-GUUAUACACCGUACUAUGUUU-3’ | Sense | 5’-GACGAGCUGACGGUCAAGAtt-3’ | |
| Knock-down siRNA pool | Antisense | 5’-PACAUAGUACGGUGUAUAACUU-3’ | Antisense | 5’-UCUUGACCGUCAGCUCGUCtt-3’ |
| Sense | 5’-GGCAUCAACGGCAAAGUUUUU-3’ | Sense | 5’-CCACGCAGUCCAACGAGAUtt-3’ | |
| Antisense | 5’-PAAACUUUGCCGUUGAUGCCUU-3’ | Antisense | 5’-AUCUCGUUGGACUGCGUGGtt-3’ | |
| Sense | 5’-CCACCAGCCACAACUCUUUUU-3’ | |||
| Antisense | 5’-PAAAGAGUUGUGGCUGGUGGUU-3’ | |||
| Scrambled siRNA | Sense | 5’-UAGCGACUAAACACAUCAAUU-3’ | Sense | 5’- UUCUCCGAACGUGUCACGUTT -3’ |
| Antisense | 5’-PUUGAUGUGUUUAGUCGCUAUU-3’ | Antisense | 5’- ACGUGACACGUUCGGAGAATT-3’ | |
2.5. Flow cytometry
Flow cytometry was used to detect cell-surface expression of ICAM-1. Briefly, cells were detached using ACCUTASE (500 units/ml) and collected. After washing with PBS twice, cells were incubated with 20 µl PE-conjugated anti-human ICAM-1 antibodies for 30 min at 4 °C. PE-conjugated mouse IgG1 isotype antibody was used as the negative control. Cells were then washed with PBS twice, fixed with 2% paraformaldehyde and stored in the dark at 4°C. Samples were read on a flow cytometer (FACScan, Becton Dickinson, TX). MFI values were corrected for isotype-matched controls.
2.6. RNA isolation and real-time reverse transcriptase-polymerase chain reaction (RT-PCR) analysis
RNA was extracted using TRIZOL reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Five µg total RNA was treated with RNase-free DNase and purified with an RNeasy micro kit. Quantitative PCR confirmed that the RNA sample was not contaminated with DNA. RNA (1 µg) was then reverse transcribed using oligo(dT) primers and the SuperScript first strand synthesis kit (Invitrogen) according to the manufacturer’s protocol. We used GAPDH as normalization control. The sequences of the primers are detailed in Table 2. Gene expression was assessed by real-time PCR using SYBR Green Master Mix (Eurogentec, San Diego, CA). The real-time PCR thermal profiles are as follows: 30 to 35 amplification cycles of 10 sec at 95°C, 15 sec at 66°C and 20 sec at 72°C, followed by melting curve analysis. The Rotor-Gene 3000 (Corbett Research, Sydney, Australia) Real-Time DNA Detection System was used for these reactions. Specificity of amplicons was verified by melting curve analysis when using SYBR Green as a detector. Negative and positive controls and the standard curve were included in the experiments.
Table 2.
Sequences of ICAM-1, IL-8 and GAPDH primers.
| Primers | ||
|---|---|---|
| ICAM-1 | Sense | 5’- AGCTTCTCCTGCTCTGCAAC -3’ |
| Antisense | 5’- GTCTGCTGGGAATTTTCTGG -3’ | |
| IL-8 | Sense | 5’- CAGGAATTGAATGGGTTTGC -3’ |
| Antisense | 5’- AAACCAAGGCACAGTGGAAC -3’ | |
| GAPDH | Sense | 5’- CATGGCCTCCAAGGAGTAAG -3’ |
| Antisense | 5’- AGGGGTCTACATGGCAACTG -3’ |
2.7. ELISA
ELISA Kits (R&D, Minneapolis, MN) were utilized to analyze the release of IL-8 and MCP-1. Recombinant proteins were used to construct standard curves. Absorbance of standards and samples was determined spectrophotometrically at 450 nm using a microplate reader.
2.8. Immunofluorescent staining
Immunofluorescent staining was performed to examine NF-κB intranuclear translocation in human lung MVECs following TNF stimulation. Briefly, cells were cultured in 8-well chamber slides to 30–40% confluence. The P38 inhibitor SB203580 (10 µM) was added to cells 30 min prior to TNF stimulation. Cells were then stimulated with TNF for 1 or 2 h. After washing with cold PBS, cells were incubated with a mixture of 30% methanol and 70% acetone at room temperature for 5 min and then air dried. After fixing in PBS-buffered 3.5% paraformaldehyde at room temperature for 10 min and washing with PBS, cells were blocked with 10% donkey serum for 30 min. Then cells were incubated for 2 h with a rabbit polyclonal antibody against NF-κB p65. Control cells were incubated with non-immune rabbit IgG. After washing with PBS, cells were incubated with Cy3-conjugated donkey anti-rabbit IgG for 1h. Nuclei were counterstained with bis-benzimide and cell membranes with wheat germ agglutinin tagged with Alexa 488 (Molecular Probes, Eugene OR). Cells were then washed with PBS and mounted with aqueous mounting media. Photography was performed with a Leica DMRX microscope (Wetzlar, Germany).
2.9. NF-κB p65 DNA binding assay
This assay is based on the specific binding of activated NF-κB p65 to a consensus oligonucleotide and provides quantitative results [17]. Nuclear extracts were prepared using an extraction kit (Active Motif, Carlsbad, CA). NF-κB activity was assayed using an NF-κB p65 DNA-binding assay kit (Active Motif), following the manufacturer’s instructions.
2.10. Statistics
Statistical analysis was performed by ANOVA with a post hoc Bonferroni/Dunn test. Statistical significance was accepted within a 95% confidence limit.
3. Results
3.1. P38 MAPK, not ERK1/2, regulates TNF-induced ICAM-1 and IL-8 expression in human lung MVECs
Both p38 MAPK and ERK1/2 have been reported to play a role in the inflammatory response to TNF in HUVECs [9, 18]. We evaluated the roles of these two MAPK pathways in the TNF-induced expression of ICAM-1 and IL-8 in human lung MVECs. Levels of phosphorylated p38 and ERK1/2 increased 10 min after TNF stimulation and then declined (Fig. 1A). Total cellular ICAM-1 levels increased markedly 4 h after TNF stimulation and continued to increase to their highest observed levels after 24 h (Fig. 1A). Cell-surface levels of ICAM-1 were significantly elevated 24 h after TNF stimulation (Fig. 1E). TNF also stimulated the release of IL-8 (Fig. 1F). IL-8 release followed the same temporal pattern as ICAM-1 expression, increasing after 4 h and reaching its highest observed level by 24 h (Fig. 1G). After TNF stimulation for 24 h, IL-8 levels increased by 12 fold (P<0.05). Inhibiting ERK1/2 with the specific inhibitor PD98059 (12.5–50 µM) had no influence on total cellular ICAM-1 levels (Fig. 1B) or on IL-8 release (Fig. 1F). By contrast, inhibiting p38 MAPK with the specific inhibitor SB203580 reduced total cellular levels of ICAM-1 at all time points (Fig. 1C) when compared to cells treated with TNF alone (a representative immunoblot is shown in Fig. 1A). Further, the effect of the p38 MAPK inhibitor was dose-dependent (Fig. 1D). Inhibiting p38 MAPK also resulted in reduced cell-surface ICAM-1 levels (Fig. 1E). Moreover, inhibiting p38 MAPK reduced IL-8 release 4 to 24 h following TNF stimulation (Fig. 1G). These results demonstrate that in human lung MVECs, the p38 MAPK pathway regulates the TNF-induced expression of ICAM-1 and IL-8.
Fig. 1. p38 MAPK, not ERK1/2, mediates TNF-induced ICAM-1 and IL-8 expression in human lung MVECs.

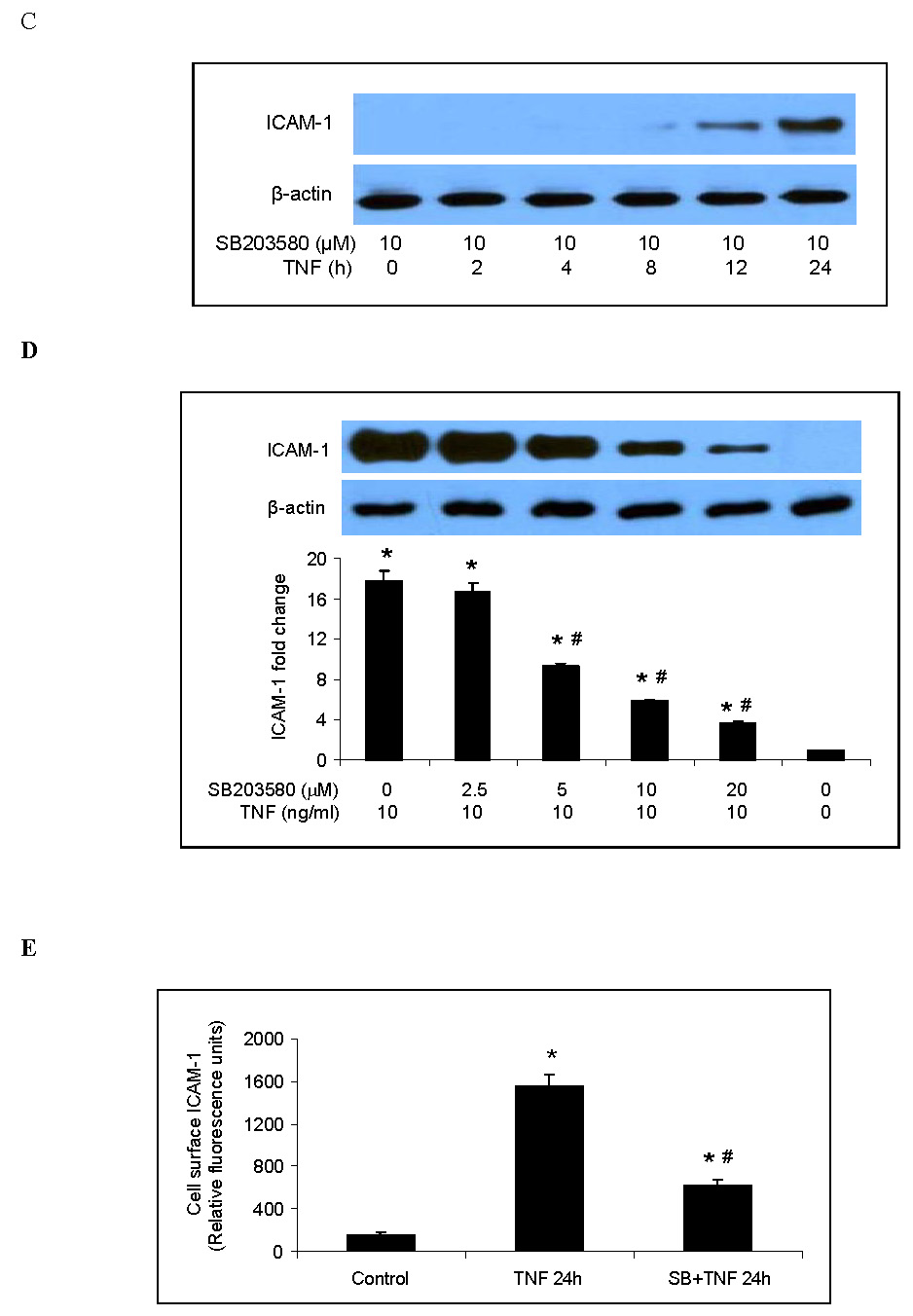
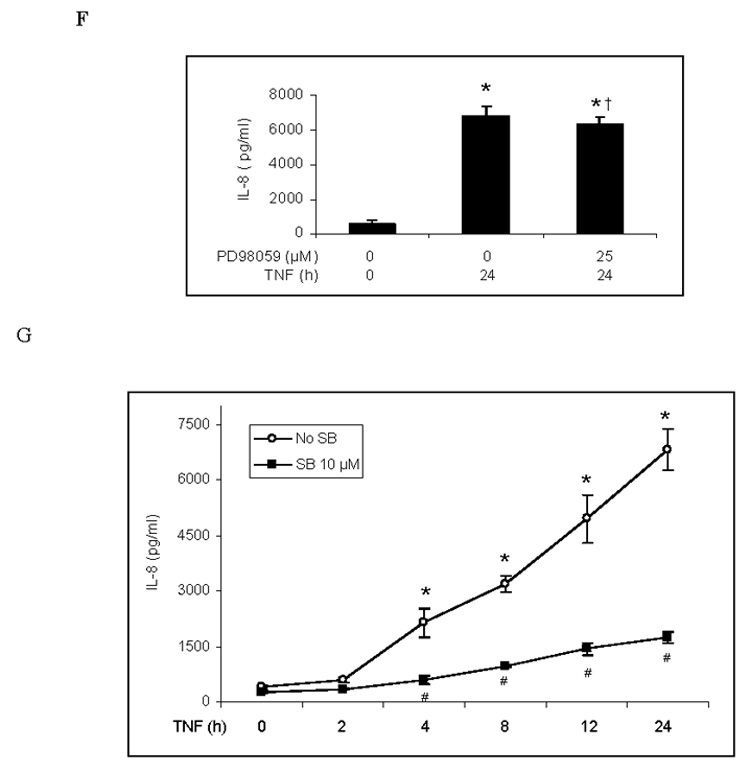
Representative immunoblots show that TNF rapidly induced phosphorylation of p38 MAPK and ERK1/2 in human lung MVECs and gradually increased cellular ICAM-1 levels (A). While inhibition of ERK1/2 activation with PD98059 did not influence total cellular ICAM-1 levels following TNF stimulation (B), inhibition of p38 MAPK with SB203580 reduced total cellular ICAM-1 levels at all time points we observed (C) and in a dose-dependent manner (D). Inhibition of p38 MAPK with SB203580 (10 µM) also reduced cell-surface ICAM-1 levels on cells stimulated with TNF for 24 h (E). While inhibition of ERK1/2 had no effect on IL-8 release (F), inhibition of p38 MAPK with SB203580 (10 µM) reduced IL-8 release at all time points we observed. Band density, flow cytometry and ELISA data are expressed as mean ± SEM. n = 4, *P<0.05 vs. untreated control, #P<0.05 vs. TNF alone (in D and E) or TNF alone at corresponding time point (in G), †non-significant vs. TNF alone. P-p38=phosphorylated p38, P-ERK1/2=phosphorylated ERK1/2, SB=SB203580.
3.2. P38 MAPK regulates TNF-induced ICAM-1 and IL-8 expression in human lung MVECs through a post-transcriptional mechanism
NF-κB is the primary transcription factor for the expression of ICAM-1 and IL-8. To determine whether p38 mediates the TNF-induced expression of ICAM-1 and IL-8 by influencing NF-κB activation, we examined the effect of SB203580 on NF-κB p65 phosphorylation, intranuclear translocation and DNA binding activity. Inhibiting p38 MAPK with SB203580 did not influence NF-κB p65 phosphorylation (Fig. 2A), intranuclear translocation (Fig. 2B) or DNA-binding activity (Fig. 2C) in human lung MVECs stimulated with TNF.
Fig. 2. Inhibition of p38 MAPK has no influence on NF-κB activation or ICAM-1 and IL-8 mRNA levels in human lung MVECs following TNF stimulation.

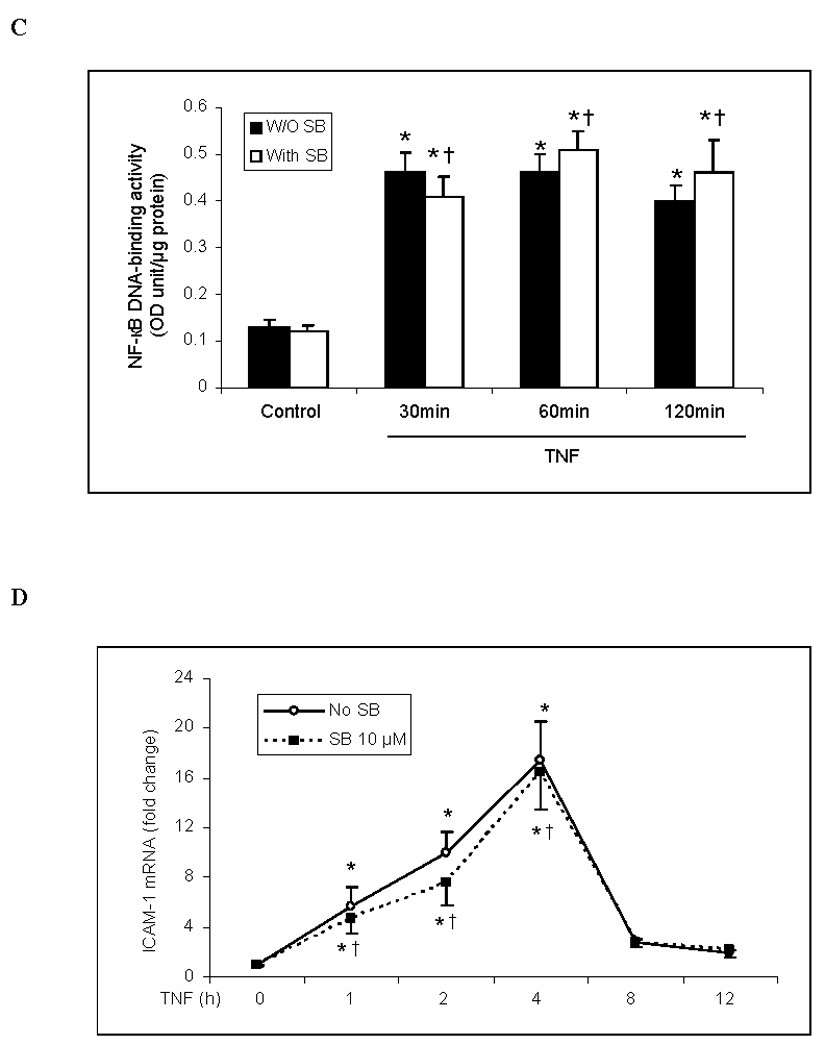
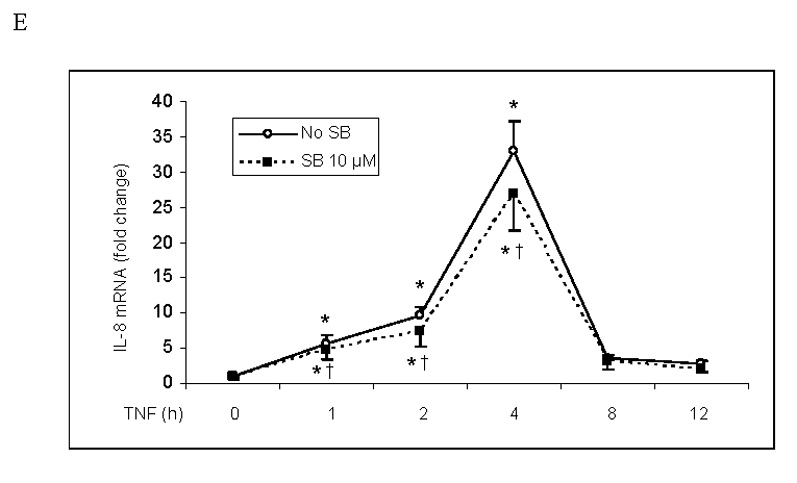
Inhibition of p38 MAPK with SB203580 did not influence NF-κB p65 phosphorylation (A), intranuclear translocation (B) or DNA-binding activity (C) in human lung MVECs stimulated with TNF. (The concentration of SB203580 in (B) and (C) is 10 µM.) ICAM-1 and IL-8 mRNA levels (normalized with GAPDH) increased 1 h after TNF stimulation, peaked at 4 h and returned to baseline after 8 h. Inhibition of p38 MAPK did not influence ICAM-1 or IL-8 mRNA levels (D, E). Band density, NF-κB DNA-binding activity and mRNA data are expressed as mean ± SEM, n = 3, *P<0.05 vs. corresponding control, †non-significant vs. TNF alone at the corresponding time point. P-NF-κB=phosphorylated NF-κB, SB=SB203580.
ICAM-1 and IL-8 mRNA levels increased 1 h after TNF stimulation, reached a peak after 4 h and returned to baseline after 8 h (Figs. 2D and 2E). Inhibiting p38 MAPK with SB203580 did not reduce ICAM-1 and IL-8 mRNA levels, nor did it change the temporal pattern of ICAM-1 and IL-8 mRNA expression. Therefore the p38 MAPK pathway appears to regulate the expression of ICAM-1 and IL-8 in human lung MVECs at a post-transcriptional level.
3.3. MK2 mediates the effect of p38 MAPK on TNF-induced ICAM-1 and IL-8 expression
Studies of other endothelial cells have shown that MK2 is a downstream factor in the p38 MAPK pathway and that inhibition of p38 MAPK reduces the phosphorylation of MK2 in response to pro-inflammatory stimuli. To determine the role of MK2 in the p38 MAPK pathway in human lung MVECs, we applied SB203580 (5 or 10 µM) 30 min before TNF stimulation and examined MK2 phosphorylation. As shown in Fig. 3A, phosphorylated MK2 was detected 10 and 30 min after TNF stimulation, but was reduced when p38 MAPK was inhibited with SB203580. Therefore MK2 is downstream of p38 MAPK in human lung MVECs.
Fig. 3. MK2 is involved in regulating the TNF-induced expression of ICAM-1 and IL-8 in human lung MVECs.
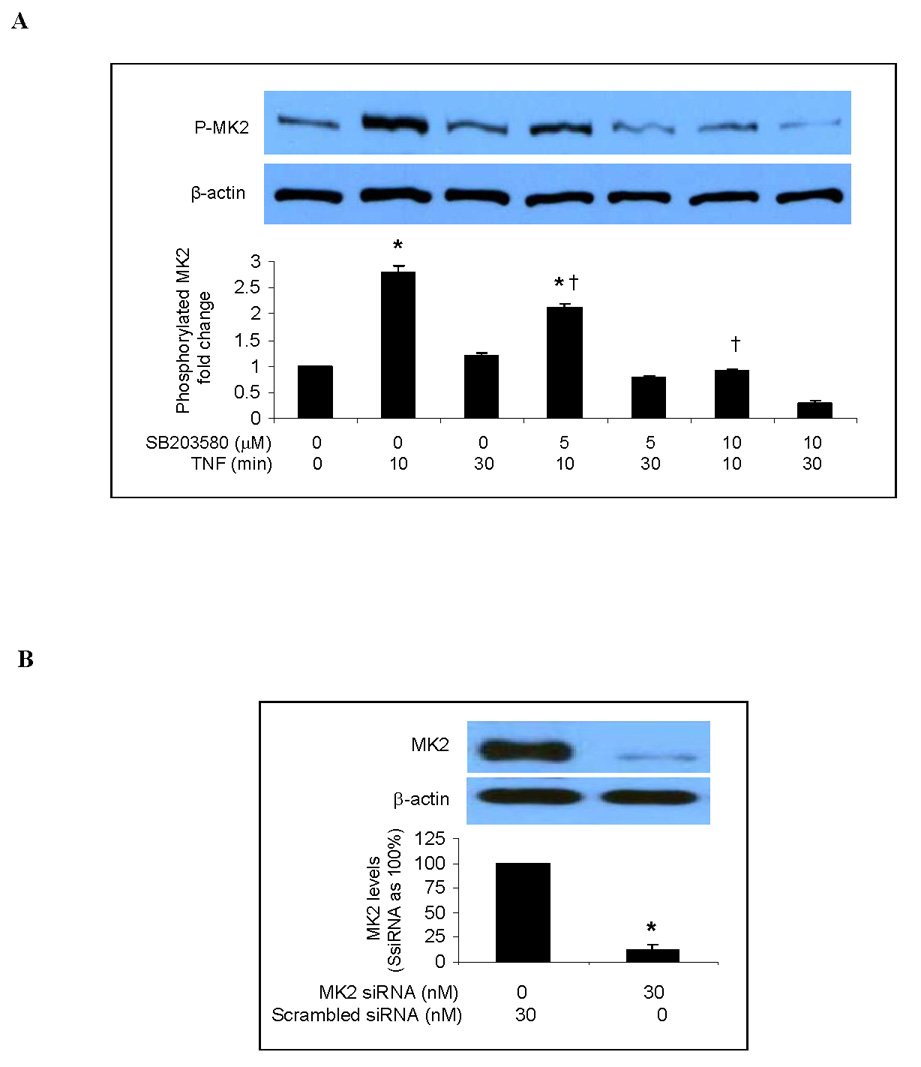
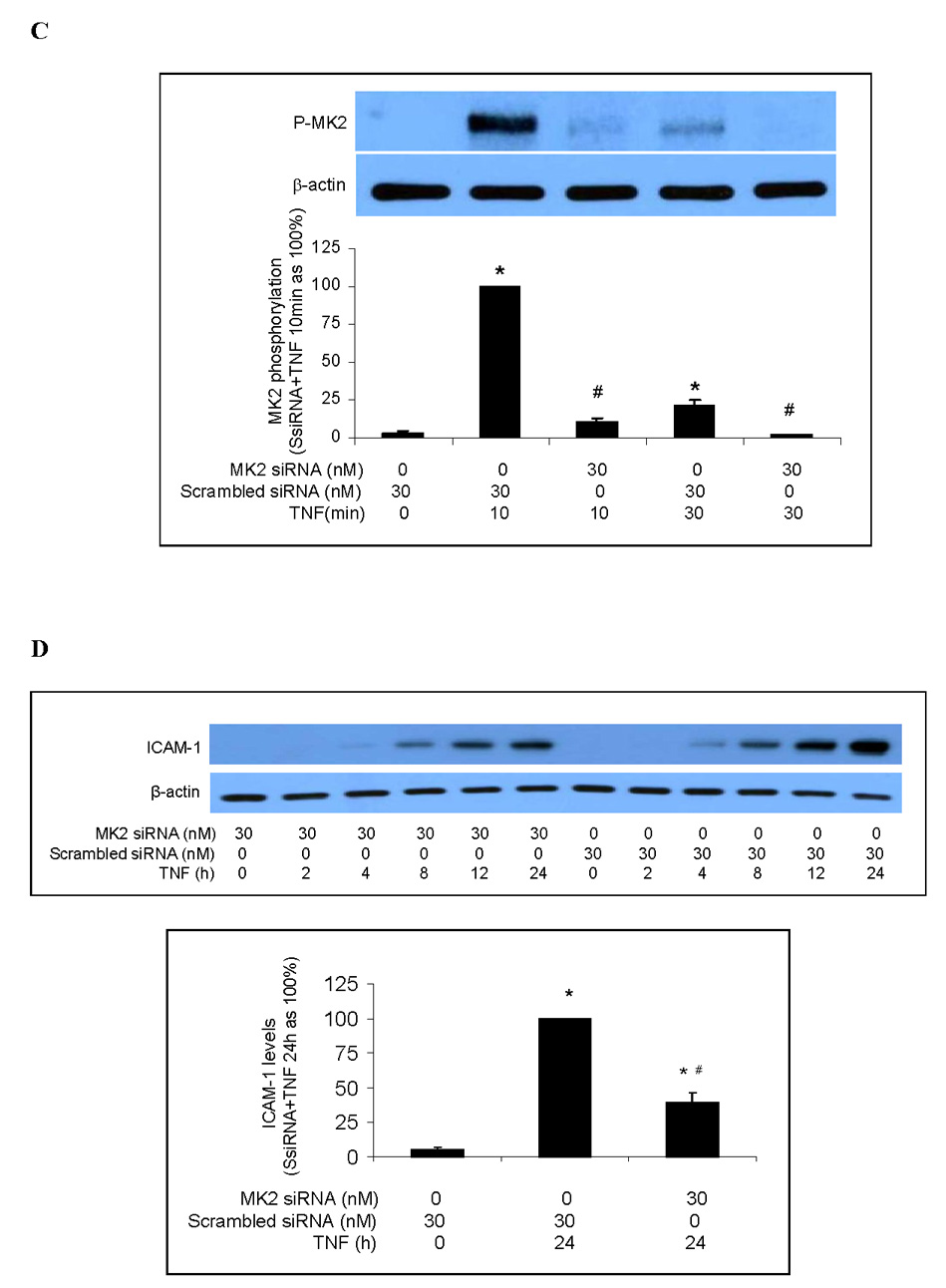
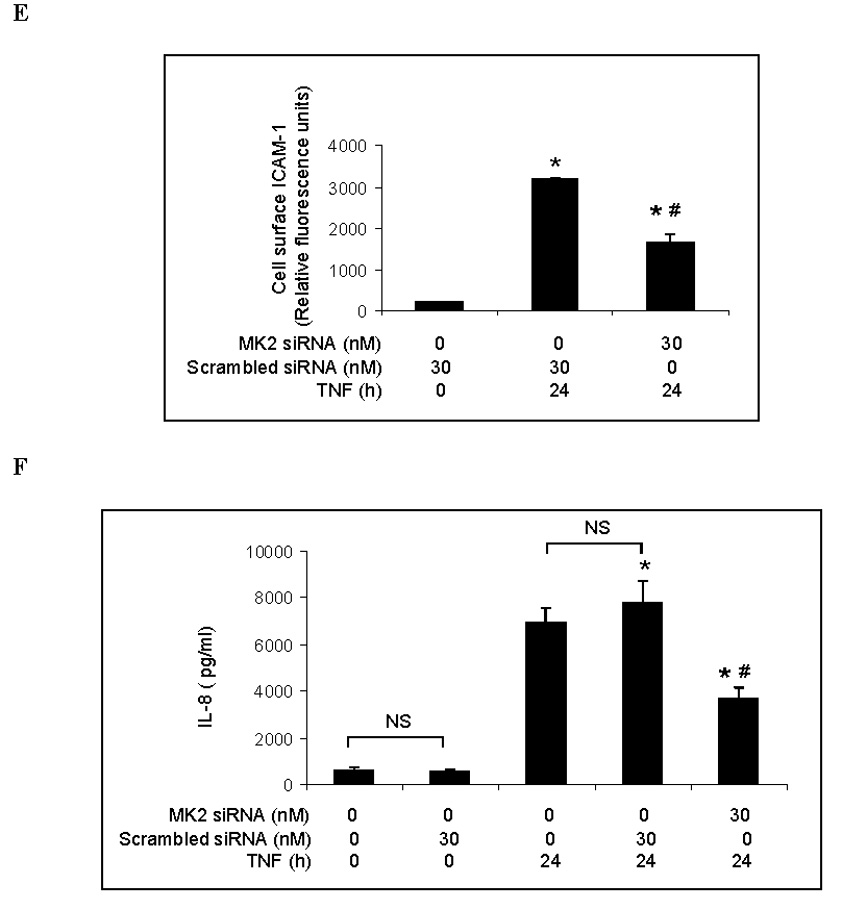
Representative immunoblots with band density data show that MK2 phosphorylation is stimulated by TNF but inhibited by the p38 MAPK inhibitor SB203580 (A). MK2 silencing markedly reduced levels of MK2 (B) and phosphorylated MK2 (C) following TNF stimulation. MK2 silencing attenuated the TNF-induced increase in total cellular ICAM-1 levels (D) and cell-surface ICAM-1 levels (E). MK2 silencing also significantly reduced IL-8 release following TNF stimulation (F). Band density, flow cytometry and ELISA data are expressed as mean ± SEM. n = 4, *P<0.05 vs. untreated controls (in A) or scrambled siRNA-treated controls (in B, C, D, E, F), †P<0.05 vs. TNF alone, #P<0.05 vs. scrambled siRNA plus TNF. NS = non-significant, P-MK2 = phosphorylated MK2, SsiRNA=scrambled siRNA.
We took the knockdown approach to determine the role of MK2 in the regulation of TNF-induced ICAM-1 and IL-8 expression by p38 MAPK. As shown in Fig. 3B, MK2 levels in cells transfected with MK2 siRNA (30 nM) were 10% of controls (cells transfected with scrambled siRNA). Similarly, MK2 silencing reduced levels of phosphorylated MK2 to approximately 10% of controls (Fig. 3C). We determined the effect of MK2 silencing on TNF-induced ICAM-1 and IL-8 expression. As shown in Figs. 3D and 3E, both total cellular and cell-surface ICAM-1 levels were greatly reduced in MK2-silenced cells compared to controls. Further, MK2 silencing reduced IL-8 release to 48% of controls treated with scrambled siRNA and TNF (Fig 3F). These results indicate that MK2 is involved in regulating the TNF-induced expression of ICAM-1 and IL-8 in human lung MVECs by the p38 MAPK pathway.
To determine whether MK2 regulates ICAM-1 and IL-8 gene expression, we examined the effect of MK2 silencing on TNF-induced NF-κB activation and ICAM-1 and IL-8 mRNA expression. Consistent with our results on p38 MAPK inhibition, MK2 silencing had no effect on NF-κB p65 phosphorylation (Fig. 4A) or NF-κB DNA-binding activity (Fig. 4B). Similarly, MK2 silencing had no influence on ICAM-1 and IL-8 mRNA levels or on the temporal pattern of ICAM-1 and IL-8 mRNA expression (Fig. 4C). These results further indicate that the p38 MAPK/MK2 pathway regulates TNF-induced ICAM-1 and IL-8 expression in human lung MVECs at a post-transcriptional level.
Fig. 4. MK2 silencing has no influence on NF-κB activation or ICAM-1 and IL-8 mRNA levels in human lung MVECs following TNF stimulation.

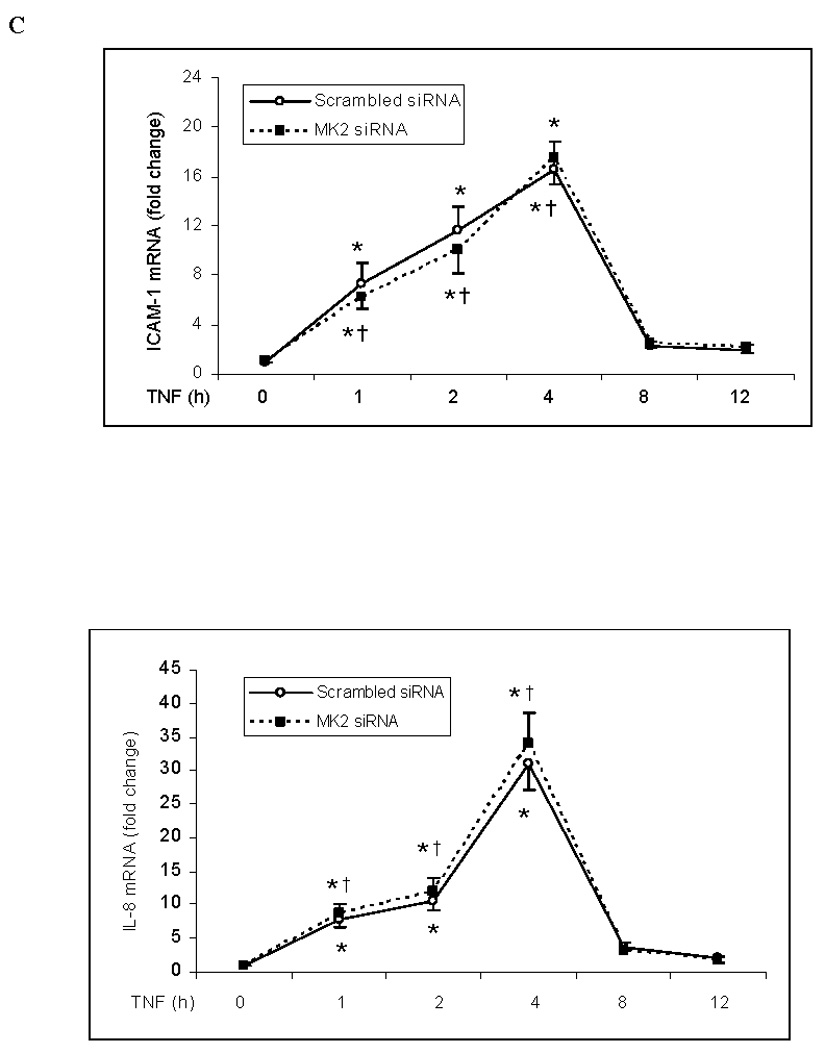
MK2 silencing (30 nM) did not influence NF-κB p65 phosphorylation (A), NF-κB DNA-binding activity (B) or ICAM-1 and IL-8 mRNA levels (normalized with GAPDH) (C) in human lung MVECs stimulated with TNF. Band density, NF-κB DNA-binding activity and mRNA data are expressed as mean ± SEM. n = 3, *P<0.05 vs. controls treated with scrambled siRNA alone, †non-significant vs. scrambled siRNA plus TNF at the corresponding time point. P-NF-κB=phosphorylated NF-κB, SsiRNA=scrambled siRNA.
The p38MAPK/MK2 pathway has been shown to play an important role in the post-transcriptional regulation of LPS-induced TNF expression. This regulation is dependent on the presence of adenine/uridine-rich elements (AREs) in the 3' untranslated region (UTR) of TNF mRNA [19, 20]. A typical ARE sequence is AUUUA. According to the Nucleotide database, ICAM-1 and IL-8 have two and eight copies, respectively, of the AUUUA motif at the 3’UTR of their mRNA sequences. By contrast, MCP-1 does not have an ARE in its mRNA. To determine whether p38 MAPK/MK2 signaling is ARE-dependent in human lung MVECs, we examined the influence of p38 MAPK inhibition and MK2 silencing on MCP-1 expression. TNF stimulation significantly increased MCP-1 mRNA levels in cells (peak MCP-1 mRNA/GAPDH mRNA ratio compared to untreated controls: 41.3±3.5 in TNF 4 h) and MCP-1 protein levels in culture medium (5.80±0.38 ng/ml vs. 0.18±0.33 ng/ml in untreated controls, P<0.01). However, neither p38 MAPK inhibition nor MK2 silencing had any effect on TNF-induced MCP-1 mRNA expression (peak MCP-1 mRNA/GAPDH mRNA ratio compared to untreated controls: 37.1±4.7 and 39.3±3.6 in SB+TNF and MK2 siRNA+TNF, respectively, vs. 41.3±3.5 and 43.6±3.9 in TNF alone and scrambled siRNA+TNF, respectively) or MCP-1 protein production (6.26±0.73 ng/ml in cells pretreated with SB203580 and 5.12±0.57 ng/ml in cells pretreated with MK2 siRNA, both P>0.05). These results suggest that the post-transcriptional regulation of TNF-induced ICAM-1 and IL-8 expression by the p38 MAPK/MK2 pathway may be at the level of ARE-dependent translation.
3.4. HSP27 is not involved in the regulation of TNF-induced ICAM-1 and IL-8 expression by the p38 MAPK/MK2 pathway
Another downstream component of the p38 MAPK signaling pathway is HSP27 [21, 22]. Hsp27 has been found to play a role in the post-transcriptional expression of pro-inflammatory factors in HeLa cells and fibroblasts [16] and to regulate the release of pro-inflammatory mediators in keratinocytes by modulating NF-κB activation [11]. To determine whether HSP27 is involved in the p38 MAPK/MK2-mediated post-transcriptional regulation of TNF-stimulated ICAM-1 and IL-8 expression in human lung MVECs, we first determined the role of the p38 MAPK/MK2 pathway in HSP27 phosphorylation. TNF stimulation increased levels of phosphorylated HSP27, but this effect was absent in cells pretreated with the p38 MAPK inhibitor SB203580 (Fig. 5A). MK2 silencing reduced HSP27 phosphorylation by 87% after 10 min of TNF stimulation and approximately 50% after 30 min of TNF stimulation (Figure 5B). Therefore the TNF-induced phosphorylation of HSP27 in human lung MVECs is primarily mediated by the p38 MAPK/MK2 pathway.
Fig. 5. TNF induces HSP27 phosphorylation in human lung MVECs through the p38 MAPK/MK2 pathway.
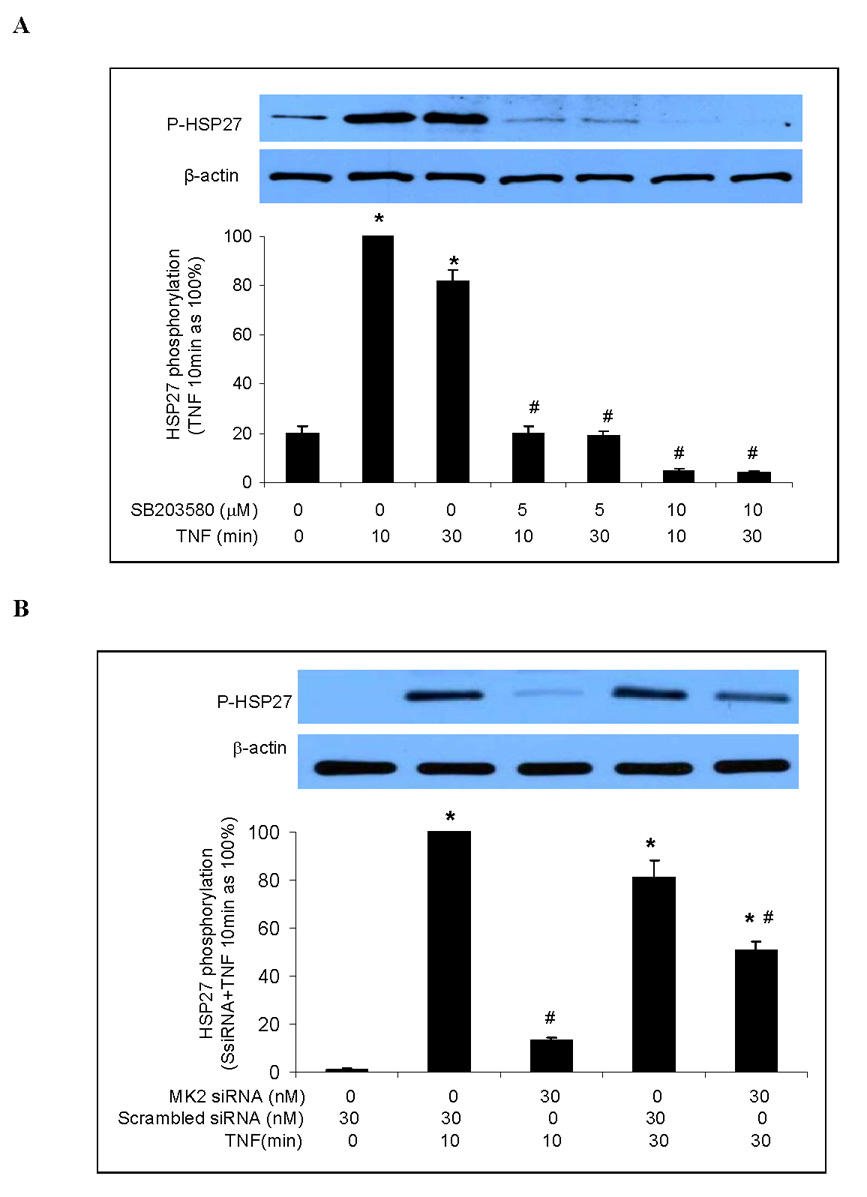
Representative immunoblots with band density data show that TNF-induced HSP27 phosphorylation was abolished by p38 MAPK inhibition (A) and markedly attenuated by MK2 silencing (B). Band density data are expressed as mean ± SEM. n = 3, *P<0.05 vs. untreated controls (in A) or scrambled siRNA-treated controls (in B) controls, #P<0.05 vs. TNF (in A) or scrambled siRNA plus TNF (in B) at the corresponding time point. P-HSP27=phosphorylated HSP27, SsiRNA=scrambled siRNA.
Next we examined the effect of HSP27 silencing on TNF-induced ICAM-1 and IL-8 expression. We transfected human lung MVECs with HSP27 siRNA for 6 h, allowed them to recover for 48 h, and then stimulated them with TNF. Immunoblotting confirmed that HSP27 siRNA (60 nM) effectively reduced levels of both HSP27 (Fig. 6A) and phosphorylated HSP27 (Fig. 6B). However, neither total cellular nor cell-surface ICAM-1 levels were affected by HSP27 silencing (Figs. 6C and 6D). Similarly, HSP27 silencing did not affect the amount of IL-8 released into the culture medium in response to TNF (Fig. 6E). These results show that, although HSP27 phosphorylation is mediated by the p38 MAPK/MK2 pathway, reducing total and phosphorylated HSP27 levels does not influence ICAM-1 and IL-8 expression in human lung MVECs.
Fig. 6. HSP27 silencing fails to influence TNF-induced ICAM-1 and IL-8 expression in human lung MVECs.

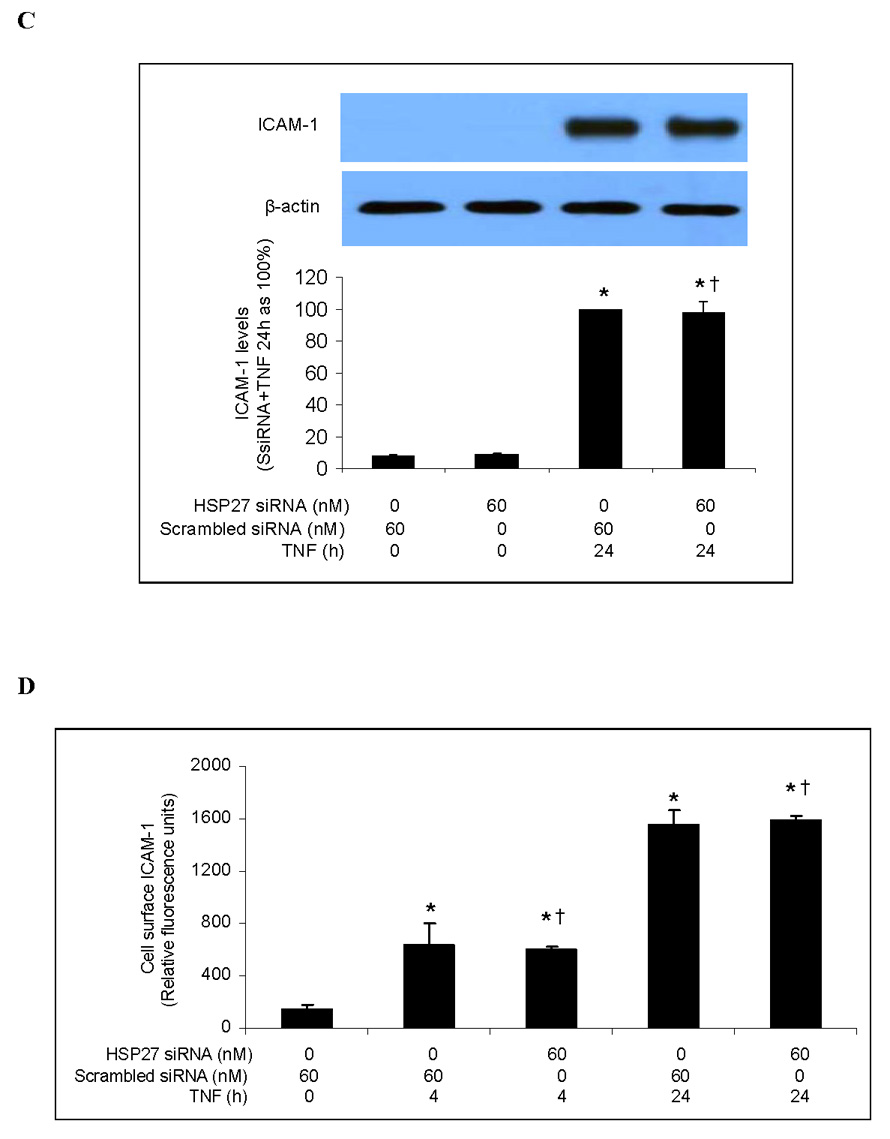
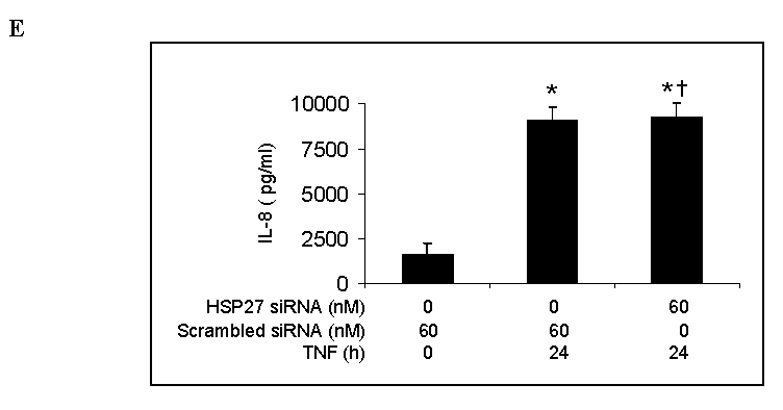
HSP27 silencing markedly reduced levels of cellular HSP27 (A). TNF-stimulated levels of phosphorylated HSP27 were reduced in cells subjected to HSP27 silencing (B). HSP27 silencing failed to influence either total (C) or cell-surface (D) levels of ICAM-1. HSP27 silencing also failed to influence IL-8 expression (E). Band density, flow cytometry and ELISA data are expressed as mean ± SEM. n = 4, *P<0.05 vs. controls (untreated or treated with scrambled siRNA alone), #P<0.05 vs. scrambled siRNA plus TNF at the corresponding time point, †non-significant vs. scrambled siRNA plus TNF at the corresponding time point. P-HSP27=phosphorylated HSP27, SsiRNA=scrambled siRNA.
4. Discussion
Multiple MAPK pathways regulate the cellular inflammatory response. The ERK1/2 and p38 MAPK pathways have been shown to be involved in regulating the expression of ICAM-1 and IL-8 in several cell types [9, 18, 23]. In HUVECs, the p38 MAPK pathway is critically important in regulating IL-8 expression [6], but both p38 MAPK and ERK1/2 appear to be minimally involved in regulating ICAM-1 expression [6, 8]. In the present study on human lung MVECs, we found that the pro-inflammatory stimulus TNF induced rapid phosphorylation of both ERK1/2 and p38 MAPK and increased protein levels of ICAM-1 and IL-8 up to 24 hours afterwards. Treatment with the ERK1/2 inhibitor PD98059 at several concentrations did not affect ICAM-1 and IL-8 levels following TNF stimulation. By contrast, inhibition of p38 MAPK with SB203580 significantly reduced TNF-induced IL-8 release and both total cellular and cell-surface ICAM-1 expression. Thus p38 MAPK, but not ERK1/2, regulates TNF-induced expression of ICAM-1 and IL-8 in human lung MVECs.
A previous study has examined the influence of p38 MAPK inhibition on TNF-induced ICAM-1 expression in human lung MVECs [24]. That study found reduced cell-surface distribution of ICAM-1 6 h after TNF stimulation in cells treated with SB203580, so the authors concluded that the p38 MAPK pathway regulates ICAM-1 redistribution in response to TNF stimulation. In our study, we analyzed both cell-surface and total cellular ICAM-1 levels following 24-h TNF stimulation and found that inhibition of p38 MAPK reduces both total cellular and cell-surface ICAM-1 levels. Moreover, we found that the reduction in cell-surface levels of ICAM-1 is proportional to the decrease in total cellular ICAM-1 levels. Therefore p38 MAPK inhibition seems to influence cell-surface ICAM-1 distribution primarily by down-regulating ICAM-1 expression.
Although the p38 MAPK pathway plays a critical role in regulating the inflammatory response to TNF in a variety of cell types, the mechanism by which it regulates that response appears to differ in endothelial cells from different vascular beds. The p38 MAPK pathway was found to be involved in regulating the TNF-induced expression of IL-8 but not ICAM-1 in HUVECs [6, 8], whereas we found that the p38 MAPK pathway is involved in regulating the TNF-induced expression of both IL-8 and ICAM-1 in human lung MVECs. Apparently, endothelial cells from different microenvironments use different signaling networks.
NF-κB is the primary transcription factor involved in regulating the inflammatory response to TNF in endothelial cells [8, 11]. Indeed, NF-κB inhibition drastically reduced ICAM-1 expression in HUVECs [8]. To evaluate the role of NF-κB in the p38 MAPK-mediated expression of ICAM-1 and IL-8 in human lung MVECs, we examined the effect of p38 inhibition on NF-κB p65 phosphorylation, intranuclear translocation and DNA-binding activity. Surprisingly, inhibition of p38 MAPK with SB203580 had no influence on any of these indices of NF-κB activation, indicating that the p38 MAPK pathway regulates TNF-induced expression of ICAM-1 and IL-8 in human lung MVECs independent of NF-κB. Consistent with these findings, inhibition of p38 MAPK did not influence the TNF-stimulated increase in ICAM-1 and IL-8 mRNA levels nor change the kinetics of ICAM-1 and IL-8 mRNA expression. Although inhibition of p38 MAPK did not affect ICAM-1 and IL-8 mRNA expression, it significantly reduced the subsequent production and accumulation/release of ICAM-1 and IL-8 proteins up to 24 h after TNF stimulation. Thus the p38 MAPK pathway appears to regulate ICAM-1 and IL-8 expression in human lung MVECs at a post-transcriptional level. This finding is consistent with an earlier report that inhibition of p38 MAPK in rheumatoid synovial fibroblasts reduces TNF-induced IL-8 peptide expression without affecting IL-8 mRNA levels [25].
MK2 and HSP27 are downstream factors in the p38 MAPK pathway. The p38 MAPK/MK2/HSP27 pathway has been found to mediate TNF-induced cytoskeletal changes in rat pulmonary endothelial cells [15]. In addition, HSP27 was recently found to play a critical role in the IL-1-induced expression of pro-inflammatory mediators such as cyclooxygenase-2, IL-6, and IL-8 in HeLa cells and fibroblasts [16]. Our study showed that TNF induced the phosphorylation of both MK2 and HSP27 in human lung MVECs in a p38 MAPK-dependent fashion, suggesting that MK2 and HSP27 play a role in regulating the pro-inflammatory response to TNF in lung MVECs by p38 MAPK. MK2 silencing did indeed significantly reduce levels of both IL-8 and ICAM-1 following TNF stimulation, but HSP27 silencing had no effect on TNF-stimulated IL-8 and ICAM-1 expression. Therefore, the TNF-induced expression of ICAM-1 and IL-8 in human lung MVECs seems to involve MK2 but is independent of HSP27.
We demonstrated that MK2 is involved in regulating the TNF-induced expression of ICAM-1 and IL-8 by p38 MAPK in human lung MVECs at a post-transcriptional level. MK2 has previously been shown to regulate ARE-dependent TNF translation without influencing either mRNA levels or mRNA stability [19, 20]. We compared the influence of p38 MAPK inhibition and MK2 silencing on the expression of ICAM-1 and IL-8, which have the AUUUA motif at the 3’UTR of their mRNA, and on the expression of MCP-1, which does not have an ARE in its mRNA. Since neither p38 MAPK inhibition nor MK2 silencing affected levels of MCP-1, it appears that the post-transcriptional regulation of TNF-induced ICAM-1 and IL-8 expression by the p38 MAPK/MK2 pathway is at the level of ARE-dependent translation. Further, our PCR results demonstrated no decrease in mRNA levels. Thus MK2 may regulate the expression of ICAM-1 and IL-8 by influencing the translational efficiency of ICAM-1 and IL-8 mRNA.
In this study, we examined the role of ERK1/2, p38 MAPK, MK2, and HSP27 in regulating the TNF-induced expression of ICAM-1 and IL-8 in human lung MVECs. Our results demonstrate for the first time that the p38 MAPK/MK2 pathway plays a critical role in regulating both ICAM-1 and IL-8 expression in human lung MVECs, and that this regulation is at the post-transcriptional level and does not involve HSP27. Since both IL-8 and ICAM-1 are necessary for neutrophil accumulation and infiltration in the lung [2], our findings indicate that the p38 MAPK pathway may play a critical role in the pulmonary inflammatory response. One limitation of our study is that it was performed on cultured human lung MVECs that may behave differently than cells in vivo. Further in vivo study is required to determine the role of the p38 MAPK/MK2 pathway in the pulmonary inflammatory response and acute lung injury.
Acknowledgments
This work was supported in part by NIH grant HL079051. The authors are grateful to Dr. Helen Kim for critically reading this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNFalpha in pulmonary pathophysiology. Respir Res. 2006;7:125–133. doi: 10.1186/1465-9921-7-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 3.Lucas R, Lou J, Morel DR, Ricou B, Suter PM, Grau GE. TNF receptors in the microvascular pathology of acute respiratory distress syndrome and cerebral malaria. J Leukoc Biol. 1997;61:551–558. doi: 10.1002/jlb.61.5.551. [DOI] [PubMed] [Google Scholar]
- 4.Strieter RM, Kunkel SL. Acute lung injury: the role of cytokines in the elicitation of neutrophils. J Investig Med. 1994;42:640–651. [PubMed] [Google Scholar]
- 5.Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol. 2001;70:317–325. doi: 10.1006/exmp.2001.2368. [DOI] [PubMed] [Google Scholar]
- 6.Kuldo JM, Westra J, Asgeirsdottir SA, Kok RJ, Oosterhuis K, Rots MG, Schouten JP, Limburg PC, Molema G. Differential effects of NF-{kappa}B and p38 MAPK inhibitors and combinations thereof on TNF-{alpha}- and IL-1{beta}-induced proinflammatory status of endothelial cells in vitro. Am J Physiol Cell Physiol. 2005;289:C1229–C1239. doi: 10.1152/ajpcell.00620.2004. [DOI] [PubMed] [Google Scholar]
- 7.Pietersma A, Tilly BC, Gaestel M, de Jong N, Lee JC, Koster JF, Sluiter W. p38 mitogen activated protein kinase regulates endothelial VCAM-1 expression at the post-transcriptional level. Biochem Biophys Res Commun. 1997;230:44–48. doi: 10.1006/bbrc.1996.5886. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Z, Connell MC, MacEwan DJ. TNFR1-induced NF-kappaB, but not ERK, p38MAPK or JNK activation, mediates TNF-induced ICAM-1 and VCAM-1 expression on endothelial cells. Cell Signal. 2007;19:1238–1248. doi: 10.1016/j.cellsig.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 9.Kim YS, Ahn Y, Hong MH, Joo SY, Kim KH, Sohn IS, Park HW, Hong YJ, Kim JH, Kim W, Jeong MH, Cho JG, Park JC, Kang JC. Curcumin attenuates inflammatory responses of TNF-alpha-stimulated human endothelial cells. J Cardiovasc Pharmacol. 2007;50:41–49. doi: 10.1097/FJC.0b013e31805559b9. [DOI] [PubMed] [Google Scholar]
- 10.Park KJ, Gaynor RB, Kwak YT. Heat shock protein 27 association with the I kappa B kinase complex regulates tumor necrosis factor alpha-induced NF-kappa B activation. J Biol Chem. 2003;278:35272–35278. doi: 10.1074/jbc.M305095200. [DOI] [PubMed] [Google Scholar]
- 11.Gorska MM, Liang Q, Stafford SJ, Goplen N, Dharajiya N, Guo L, Sur S, Gaestel M, Alam R. MK2 controls the level of negative feedback in the NF-kappaB pathway and is essential for vascular permeability and airway inflammation. J Exp Med. 2007;204:1637–1652. doi: 10.1084/jem.20062621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brook M, Tchen CR, Santalucia T, McIlrath J, Arthur JS, Saklatvala J, Clark AR. Posttranslational regulation of tristetraprolin subcellular localization and protein stability by p38 mitogen-activated protein kinase and extracellular signal-regulated kinase pathways. Mol Cell Biol. 2006;26:2408–2418. doi: 10.1128/MCB.26.6.2408-2418.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ronkina N, Kotlyarov A, Dittrich-Breiholz O, Kracht M, Hitti E, Milarski K, Askew R, Marusic S, Lin LL, Gaestel M, Telliez JB. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol Cell Biol. 2007;27:170–181. doi: 10.1128/MCB.01456-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hedges JC, Dechert MA, Yamboliev IA, Martin JL, Hickey E, Weber LA, Gerthoffer WT. A role for p38(MAPK)/HSP27 pathway in smooth muscle cell migration. J Biol Chem. 1999;274:24211–24219. doi: 10.1074/jbc.274.34.24211. [DOI] [PubMed] [Google Scholar]
- 15.Kayyali US, Pennella CM, Trujillo C, Villa O, Gaestel M, Hassoun PM. Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2. J Biol Chem. 2002;277:42596–42602. doi: 10.1074/jbc.M205863200. [DOI] [PubMed] [Google Scholar]
- 16.Alford KA, Glennie S, Turrell BR, Rawlinson L, Saklatvala J, Dean JL. Heat shock protein 27 functions in inflammatory gene expression and transforming growth factor-beta-activated kinase-1 (TAK1)-mediated signaling. J Biol Chem. 2007;282:6232–6241. doi: 10.1074/jbc.M610987200. [DOI] [PubMed] [Google Scholar]
- 17.Renard P, Ernest I, Houbion A, Art M, Le Calvez H, Raes M, Remacle J. Development of a sensitive multi-well colorimetric assay for active NFkappaB. Nucleic Acids Res. 2001;29:e21. doi: 10.1093/nar/29.4.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nizamutdinova IT, Oh HM, Min YN, Park SH, Lee MJ, Kim JS, Yean MH, Kang SS, Kim YS, Chang KC, Kim HJ. Paeonol suppresses intercellular adhesion molecule-1 expression in tumor necrosis factor-alpha-stimulated human umbilical vein endothelial cells by blocking p38, ERK and nuclear factor-kappaB signaling pathways. Int Immunopharmacol. 2007;7:343–350. doi: 10.1016/j.intimp.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 19.Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999;1:94–97. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- 20.Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M. MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem. 2002;277:3065–3068. doi: 10.1074/jbc.C100685200. [DOI] [PubMed] [Google Scholar]
- 21.Wu R, Kausar H, Johnson P, Montoya-Durango DE, Merchant M, Rane MJ. Hsp27 regulates Akt activation and polymorphonuclear leukocyte apoptosis by scaffolding MK2 to Akt signal complex. J Biol Chem. 2007;282:21598–21608. doi: 10.1074/jbc.M611316200. [DOI] [PubMed] [Google Scholar]
- 22.Zheng C, Lin Z, Zhao ZJ, Yang Y, Niu H, Shen X. MAPK-activated protein kinase-2 (MK2)-mediated formation and phosphorylation-regulated dissociation of the signal complex consisting of p38, MK2, Akt, and Hsp27. J Biol Chem. 2006;281:37215–37226. doi: 10.1074/jbc.M603622200. [DOI] [PubMed] [Google Scholar]
- 23.Chen XL, Grey JY, Thomas S, Qiu FH, Medford RM, Wasserman MA, Kunsch C. Sphingosine kinase-1 mediates TNF-alpha-induced MCP-1 gene expression in endothelial cells: upregulation by oscillatory flow. Am J Physiol Heart Circ Physiol. 2004;287:H1452–H1458. doi: 10.1152/ajpheart.01101.2003. [DOI] [PubMed] [Google Scholar]
- 24.Tamura DY, Moore EE, Johnson JL, Zallen G, Aiboshi J, Silliman CC. p38 mitogen-activated protein kinase inhibition attenuates intercellular adhesion molecule-1 up-regulation on human pulmonary microvascular endothelial cells. Surgery. 1998;124:403–407. discussion 408. [PubMed] [Google Scholar]
- 25.Suzuki M, Tetsuka T, Yoshida S, Watanabe N, Kobayashi M, Matsui N, Okamoto T. The role of p38 mitogen-activated protein kinase in IL-6 and IL-8 production from the TNF-alpha- or IL-1beta-stimulated rheumatoid synovial fibroblasts. FEBS Lett. 2000;465:23–27. doi: 10.1016/s0014-5793(99)01717-2. [DOI] [PubMed] [Google Scholar]