

Abstract
Motivation
BLAST programs are very efficient in finding similarities for sequences. However for large datasets such as ESTs, manual extraction of the information from the batch BLAST output is needed. This can be time consuming, insufficient, and inaccurate. Therefore implementation of a parser application would be extremely useful in extracting information from BLAST outputs.
Results
We have developed a java application, Batch Blast Extractor, with a user friendly graphical interface to extract information from BLAST output. The application generates a tab delimited text file that can be easily imported into any statistical package such as Excel or SPSS for further analysis. For each BLAST hit, the program obtains and saves the essential features from the BLAST output file that would allow further analysis. The program was written in Java and therefore is OS independent. It works on both Windows and Linux OS with java 1.4 and higher. It is freely available from: http://mcbc.usm.edu/BatchBlastExtractor/
Background
The NCBI BLAST database search tool is one of the most popular programs designed to solve single query problems. BLAST (Basic Local Alignment Search Tool) is the heuristic search algorithm employed by the programs blastp, blastn, blastx, tblastn, and tblastx. The BLAST programs were tailored for sequence similarity searching for example to identify homologs of a given query sequence [1].
The five common BLAST programs perform the following tasks: 1) blastp compares an amino acid query sequence against a protein sequence database; 2) blastn compares a nucleotide query sequence against a nucleotide sequence database; 3) blastx compares the six-frame conceptual translation products of a nucleotide query sequence (both strands) against a protein sequence database 4) tblastn compares a protein query sequence against a nucleotide sequence database dynamically translated in all six reading frames (both strands), and 5) tblastx compares the six-frame translations of a nucleotide query sequence against the six-frame translations of a nucleotide sequence database.
The BLAST programs all provide information in roughly the same format. First comes (A) an introduction to the program; (B) a histogram of expectations if one was requested; (C) a series of one-line descriptions of matching database sequences; (D) the actual sequence alignments, and finally the parameters and other statistics gathered during the search. However, for genome-wide comparisons involving multiple queries (batch query), the search is a challenge. For instance, EST collections are currently produced for many species as an efficient strategy for gene identification. Analysis of the ESTs involves clustering, contig formation and annotation of thousands of fragments, interpretation of which may involve thousands of individual BLAST searches [2-5]. An automated post processing of the output (Figure 1) can simplify the analysis in such cases. The blast parser (BlastLikeSaxParser) in BioJava [6] and BPlite from BioPerl [7] are frequently being used to parse a variety of different blast outputs, but neither are user friendly and therefore programming skills are needed to use these applications.
Figure 1.
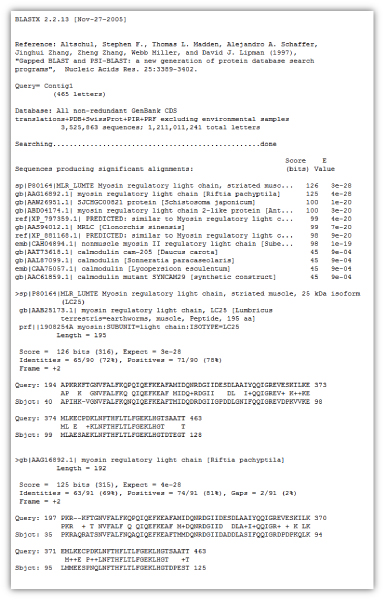
Screenshot of a Blastx Output.
We developed the "Batch Blast Extractor" program (Figure 2 and 3) for use in this regard. It serves as a parser storing only the essential features of BLAST hits in a tabular form. The user can then apply a number of selection criteria to filter out hits with particular attributes. "Batch Blast Extractor" thus serves as a powerful annotation tool for large sets of query sequences.
Figure 2.
Screenshot of the Batch Blast Extractor Web site.
Figure 3.
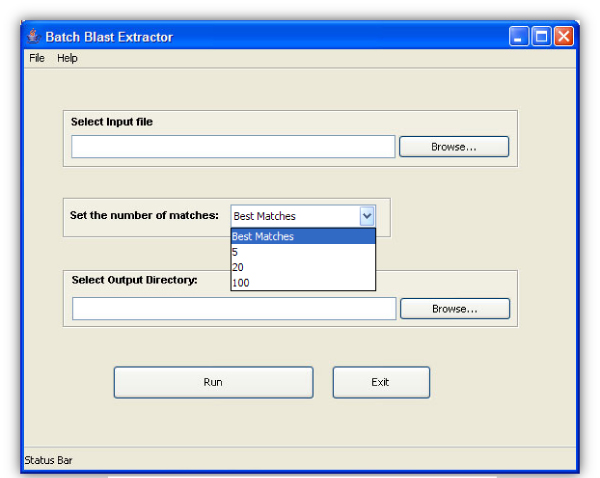
The Bach Blast Extractor Graphical User Interface.
Results
The application generates a tab delimited text file that can be easily imported into any statistical package such as Excel or SPSS for further analysis. For each BLAST hit, the program derives and saves the following features: Query ID, Query Length, Accession version and GI number, Alignment Length, Score, bit, E-value, Identities, Positives, Gaps, Frame, Organism, and Description.
The extracted information includes the following:
▪ Query: headers of sequences to analyze
▪ Subject: headers of sequences found in the database
▪ Score: a number representation (e.g. 550)
▪ Score Text: full text representation plus BITS (e.g. 235 bits (450))
▪ Expect: the E-Value as number (e.g. 1e-166)
▪ Identities %: a number representation (e.g. 85)
▪ Identities Text: full text representation plus characters matching (e.g. 110/130 (90%))
▪ Positives %: a number representation (e.g. 92)
▪ Positives Text: full text representation (e.g. 110/130 (90%))
▪ Gaps %: a number representation (e.g. 11)
▪ Gaps Text: full text representation plus voids (e.g. 9/102 (9%))
▪ Frame: orientation of the translated ORF (e.g. +3)
▪ Length Query: the number of nucleotides or amino acids (e.g. 400)
▪ Length Subject: the number of nucleotides or amino acids (e.g. 500)
▪ Position Query: as text representation plus the length of the frame (e.g. 328–600 (360))
▪ Position Subject: as text representation plus the length of the frame (e.g. 1–110 (120))
The program was written in Java. It is OS independent and works on both Windows and Linux OS with java 1.4 and higher. It is freely available to noncommercial users from: http://mcbc.usm.edu/BatchBlastExtractor/ (Figure 2 and 3).
Currently the application works with blastx results. Efforts to extend functionality to other BLAST programs such as blastp and blastn are in progress.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MP and YD initiated the project. MP designed, programmed and implemented the application and drafted the manuscript. EJP and YP directed the project. All authors read and approved the final manuscript.
Acknowledgments
Acknowledgements
This work was supported by the Mississippi Functional Genomics Networks (MFGN), Mississippi Computational Biology Consortium (MCBC) (NSF Grant # EPS-0556308), and the Army Environmental Quality Program of the US Army Corps of Engineers under contract #W912HZ-05-P-0145. Permission was granted by the Chief of Engineers to publish this information.
This article has been published as part of BMC Genomics Volume 9 Supplement 2, 2008: IEEE 7th International Conference on Bioinformatics and Bioengineering at Harvard Medical School. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2164/9?issue=S2
Contributor Information
Mehdi Pirooznia, Email: Mehdi.Pirooznia@usm.edu.
Edward J Perkins, Email: Edward.J.Perkins@erdc.usace.army.mil.
Youping Deng, Email: youping.deng@usm.edu.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Wang G, Liu L, Ji G, Liu Y, Chen J, Webb JS, Reese G, Dean JF. WebTraceMiner: a web service for processing and mining EST sequence trace files. Nucleic Acids Res. 2007:W137–142. doi: 10.1093/nar/gkm299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj SH, Deshpande N, Gasser RB, Ranganathan S. ESTExplorer: an expressed sequence tag (EST) assembly and annotation platform. Nucleic Acids Res. 2007:W143–147. doi: 10.1093/nar/gkm378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia M, Gong P, Guan X, Inouye LS, Yang K, Perkins EJ, Deng Y. Cloning, analysis and functional annotation of expressed sequence tags from the Earthworm Eisenia fetida. BMC Bioinformatics. 2007;8:S7. doi: 10.1186/1471-2105-8-S7-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangalam H. The Bio* toolkits – a brief overview. Brief Bioinform. 2002;3:296–302. doi: 10.1093/bib/3.3.296. [DOI] [PubMed] [Google Scholar]
- Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JG, Korf I, Lapp H, et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002;12:1611–1618. doi: 10.1101/gr.361602. [DOI] [PMC free article] [PubMed] [Google Scholar]