

Abstract
Background
Sudden death syndrome (SDS) of soybean (Glycine max L. Merr.) is an economically important disease, caused by the semi-biotrophic fungus Fusarium solani f. sp. glycines, recently renamed Fusarium virguliforme (Fv). Due to the complexity and length of the soybean-Fusarium interaction, the molecular mechanisms underlying plant resistance and susceptibility to the pathogen are not fully understood. F. virguliforme has a very wide host range for the ability to cause root rot and a very narrow host range for the ability to cause a leaf scorch. Arabidopsis thaliana is a host for many types of phytopathogens including bacteria, fungi, viruses and nematodes. Deciphering the variations among transcript abundances (TAs) of functional orthologous genes of soybean and A. thaliana involved in the interaction will provide insights into plant resistance to F. viguliforme.
Results
In this study, we reported the analyses of microarrays measuring TA in whole plants after A. thaliana cv 'Columbia' was challenged with fungal pathogen F. virguliforme. Infection caused significant variations in TAs. The total number of increased transcripts was nearly four times more than that of decreased transcripts in abundance. A putative resistance pathway involved in responding to the pathogen infection in A. thaliana was identified and compared to that reported in soybean.
Conclusion
Microarray experiments allow the interrogation of tens of thousands of transcripts simultaneously and thus, the identification of plant pathways is likely to be involved in plant resistance to Fusarial pathogens. Dissection of the set functional orthologous genes between soybean and A. thaliana enabled a broad view of the functional relationships and molecular interactions among plant genes involved in F. virguliforme resistance.
Background
Transcriptional changes play a major role in many plant defense processes [1]. Investigation of alterations in transcript abundance in functional genomics has provided unique opportunities to delve into gene functions by the comparison of species, tissue and time specific transcript accumulation for thousands of genes simultaneously [2-4]. The transcript abundances of the annotated genes of Arabidopsis, soybean and many other crops can be evaluated in parallel using high-density microarrays of sequenced cDNAs (AGI, 2000) or oligomers [5]. Microarray experiments have enabled the detection of significant variation in mRNA abundance and improved the understanding of the molecular mechanism of partial defense responses [6-9]. The host-pathogen interaction involved in incomplete, quantitative and partial resistance of soybean roots to F. virguliforme has been intensively investigated [9-11]. Transcription factors, chromatin remodeling proteins and transcript stabilizing factors are likely candidates to be involved. Regulated pathways are expected to include the synthesis of phytoalexins, signal molecules, cell wall deposition and carbon (C) and nitrogen partitioning.
Several studies suggested that disease resistance genes shared the same specificity across distantly related plant species [12-15]. The specificity of response was maintained, perhaps because of balancing selection, in lineages leading to multiple plant species [16]. However, it is difficult to conclude that a unified model of host-pathogen interactions has been determined because many of the genes underlying pathogen recognition were functional orthologs rather than the closest sequence homologous in different species.
Phytoalexins, phytoanticipins and signal molecules are three major natural products involved in plant defense with common precursors [17]. Phenylalanine ammonia-lyase (PAL; EC 4.3.1.5) expression has been associated with resistance to fungal pathogens in many plant species [18,19]. PAL catalyzes the deamination of phenylalanine to produce trans-cinnamic acid, the first step in the phenylpropanoid pathway leading to phytoalexins, lignins or coumarins. Multiple isoforms of the pal gene were identified in plants [20]. Manipulation of PAL, the first enzyme of the phenylpropanoid pathway together with the downstream enzymes such as cinnamate 4-hydroxylase (C4H; EC 1.14.13.11), diphenol oxidase (laccase; EC 1.10.3.2) and 4-hydroxycinnamoyl CoA ligase (4CL; EC 6.2.1.12), revealed an association with resistance to viral and fungal infection [21-23]. Reduction of phenylpropanoid biosynthesis in tobacco via down-regulation of PAL reduced local and systemic acquired resistance to fungal or viral infection [24,25]. Phenylpropanoid derived polymers like lignin also play an important role as a physical barrier against pathogen invasion [26]. Lignin, a complex racemic aromatic heteropolymer is the second most abundant cell wall polymer (after cellulose) and provides rigidity for the cell wall and a physical barrier against pathogens [27]. Lignin is synthesized from the phenylpropanoid metabolism reactions. These series of hydroxylation and O-methylation and conversion of side-chain carboxyl to an alcohol result in the building blocks of lignin, which is initiated by deamination of phenylalanine by the enzyme PAL where hydroxycinnamic acid esters play a central role [28,29]. 4CL is responsible for the CoA esterification of p-coumaric acid. Down-regulation of isozymes of 4CLs may alter metabolite concentrations other than those involved in lignin production, with a secondary effect on growth as a consequence [30]. Laccase was the first enzyme demonstrated to be able to perform lignin polymerization in vitro. Over-expressed laccase in the roots caused cell wall lignin deposition increases in the developing xylem [31,32,23]. Therefore lignin might be involved in the disease resistance mechanism of plant cells where wall fortification occurred in response to many pathogens and would be especially useful against root rots.
In plant, SnRKs possess a catalytic domain similar to that of sucrose non-fermenting-1 (SNF1) of yeast (Saccharomyces cerevisiae) and AMP-activated protein kinase (AMPK) of animals. SnRK1, SnRK2 and SnRK3 are the key members of SnRKs family but the SnRK1 subfamily appears to share direct structural and functional homology with the SNF1/AMPK family. The SnRK1 protein complex can be further divided into 3 subunits: the α subunit which is SNF1-like protein; the β subunit that is composed of SIP1-, SIP2- and GAL83-like proteins; and the γ subunit that is SNF4-like protein based on sequence structure and expression patterns [33,34]. Homologues of SnRKs occur in all kingdoms and they appear to be highly conserved among yeasts, animals and plants suggesting that they may play very similar roles across species [35]. SNF1 modulates the phosphorylation state of a number of metabolic enzymes whilst SnRK1 regulates several enzymes involved in sugar metabolism and cell energy metabolism [36,33]. In yeast, the function of SNF1 is to coordinate about 600 genes to respond to lower cellular glucose concentrations. Hong et al. [33] identified GAL83 as mediating carbon partitioning during the plant response to herbivore Manduca sexta attack. GAL83, a β-subunit of a heterotrimeric SnRK1, showed a decrease in source leaves whereas the abundance of the catalytic α-subunit of SNF remained unaltered. The herbivore-induced changes in sink-source relations in Nicotiana attenuata was regulated by the β-subunit of SnRK1 (SNF1-related kinase) protein kinase, GAL83. GAL83 silenced plants were unable to enhance root reserves, delay senescence or prolonged flowering following herbivore attack during early stages of development. In turn, SnRK1 can be used to alter resource allocation thereby plants may be equipped to better tolerate the pest attack [37].
Partial resistance can often be subcategorized as rate reducing resistance for fungal infections of roots [38,39]. Partial resistance may result from a reduced infection frequency, an extended latent period of infection, a reduced sporulation of the pathogen, or a combination of these [40-42]. However, the defense pathways that are induced during partial resistance do not share the same temporal and spatial patterns of gene expression observed in complete resistance [9,10,40,42,43]. Sudden death syndrome (SDS) of soybean caused by F. virguliforme [44] results from two distinct interactions. SDS has the root infection component, where the fungus exogenously penetrates root cell walls and infect specific cells and causes root rot whilst leaf scorch component, where some of the toxins produced in the roots are translocated to leaves and cause the leaf scorch. The leaf symptoms only occur in soybean [45,46] but the root rot occurs in all legumes, most dicots and some cereals. Both root rot and leaf scorch contributes to yield losses [38,47]. Arabidopsis thaliana is a host for many types of phytopathogens. We have observed that Arabidopsis is also an excellent model plant for F. virguliforme resistance (authors unpublished data). Arabidopsis was a host for F. virguliforme and the responses to the pathogen with nicely respect to the spore concentration (Figure 1). Our date showed that this ecotype demonstrated a rate reduction resistance to the fungal pathogen. Thus, A. thaliana should be useful for studying the interactions between plant and F. virguliforme.
Figure 1.

Arabidopsis thaliana responded to F. virguliforme with respect to the spore concentration. The plant exhibited a remarkable response to the concentration of the spore on the plant fresh weight (mean weight). The mean weights labeled with the same letter were not significantly different (P < 0.05).
Iqbal et al. [9] measured changes in TAs of 192 known plant defense and biotic/abiotic stress related genes in soybean roots at five time points over a period of 10 days after F. virguliforme inoculation. The cDNA arrays used were chosen from a soybean root cDNA library [48] and a subtractive hybridization experiment [9]. The temporal and spatial response differed among soybean genotypes with different numbers of SDS resistance genes. The responses were different in the partial resistance and susceptible genotypes among genes involved in the plant defense, signal recognition and transduction and metabolic processes. For most of the responding genes and all genotypes, there was an initial decrease in TAs in the inoculated roots. However, by later stages of post inoculation, the inoculated roots of the partially susceptible cultivars failed to increase abundance of any transcripts of known defense-related genes. In the most resistant cultivar, a set of 35 genes maintained at least a two-fold higher abundance at all time points. The increase in TA in RIL23 was in contrast to that observed in Essex, susceptible parent, where most of the ESTs showed either no change or a decreased TA [9].
Here, we analyzed the changes that occurred in the abundance of transcripts corresponding to 10,560 A. thaliana expressed sequence tags (ESTs) after A. thaliana cv 'Columbia' was treated with F. virguliforme. Reverse labeled slides were used. An ortholog analysis was exploited to understand the evolutionary roles of the regulated genes based on investigation of orthologous relationship between soybean and Arabidopsis. A set of resistance pathways involved in response to the pathogen infection in A. thaliana has been proposed. The comparison of the transcriptional activity in the resistance pathways between soybean and Arabidopsis after F. virguliforme pathogenesis allows for the examination of evolution of the disease response in both species.
Results
Transcript profile of Arabidopsis genes in response to F. virguliforme infection
Analysis of the microarray data demonstrated a significant variation within and between the slides after local and global normalization [49] and 6,109 genes correlated in both reverse labeled slides from the 10,560 EST arrayed. The position and label variations between two replications did not significantly alter the topography between slides. The fungal infestation caused 168 transcripts to increase in abundance more than 1 fold (>1 on Log2 scale). About 24 of the transcripts were increased in abundance more than 2 fold (Log2 scale). In contrast, only forty-two transcripts were observed to decrease in abundance by more than 1 fold (Log2 scale) and 14 of them went down more than 2 fold (Log2 scale) following F. virguliform infection. In addition, the number of the up-regulated genes was nearly four times more than that of down-regulated genes. Therefore, A. thaliana cv Columbia responses to F. virguliforme were more similar to resistant than susceptible soybean cultivars [9].
After the Arabidopsis EST-based microarray data were converted into Affymetrix 22 K array annotation, the 6,109 genes on the arrays were subjected to pathway construction using the MapMan platform [50]http://gabi.rzpd.de/projects/MapMan. The majority (3,541) of genes altered by the treatment (P < 0.05) could not be assigned to any of the known function (Table 1). In contrast, 2,568 genes were assembled into 14 major bins, each bin representing a set of related pathways (Table 1). Classification of the transcript abundances changed by F. virguliforme infestation showed several interesting features. There were 571 genes distributed among five protein metabolism related bins: synthesis, activation, posttranslational modification, degradation and folding. Bin16, secondary metabolism, bin29, protein metabolism and bin30, signal transduction were subjected further investigation. A large proportion of these protein metabolism related genes was assigned to bin29.2 (synthesis) and bin29.4 (posttranslational modification). Bin29.4 (protein post-translational modification) contained many genes involved in published plant defense schema [51]. The 186 genes encompassed in this bin category would allow various stages of plant response to pathogen challenge to be investigated. Of the 186 genes involved in protein posttranslational modification there were only 9 genes with significantly altered TAs. The synthesis of new proteins and the alteration of the activities of existing proteins by modification have been frequently reported to be important to plant pathogen resistance.
Table 1.
Description of bin distributions in the MapMan platform.
| Bin | Name | Elements | p-value |
| 1.1 | PS.lightreaction | 59 | 0.04 |
| 1.1.4 | PS.lightreaction.ATP synthase | 6 | 0.01 |
| TCA/org. transformation.other organic acid | |||
| 8.2.11 | transformaitons.atp-citrate lyase | 2 | 0.03 |
| 10.2 | cell wall.cellulose synthesis | 14 | 0.01 |
| lipid metabolism.FA synthesis and FA elongation.long | |||
| 11.1.9 | chain fatty acid CoA ligase | 5 | 0.004 |
| 12 | N-metabolism | 13 | 0.03 |
| 12.2 | N-metabolism.ammonia metabolism | 8 | 0.04 |
| amino acid metabolism.synthesis.aspartate | |||
| 13.1.3.4 | family.methionine | 9 | 0.046 |
| 16.1.4 | secondary metabolism.isoprenoids.carotenoids | 6 | 0.04 |
| secondary metabolism.phenylpropanoids.lignin | |||
| 16.2.1 | biosynthesis | 18 | 0.006 |
| 16.7 | secondary metabolism.wax | 4 | 0.027 |
| 16.8.2 | secondary metabolism.flavonoids.chalcones | 3 | 0.006 |
| hormone metabolism.brassinosteroid.synthesis- | |||
| 17.3.1.1.1 | degradation.BRs.DET2 | 3 | 0.02 |
| 19.1 | tetrapyrrole synthesis.magnesium chelatase | 2 | 0.03 |
| 26.3 | misc.gluco-, galacto- and mannosidases | 22 | 0.01 |
| 27.1 | RNA.processing | 71 | 0.01 |
| RNA.regulation of transcription.TCP transcription factor | |||
| 27.3.29 | family | 6 | 0.04 |
| 29.1 | protein.aa activation | 32 | 0.02 |
| 29.2 | protein.synthesis | 200 | 0.004 |
| protein.synthesis.chloroplast/mito – plastid ribosomal | |||
| 29.2.1 | protein | 36 | 0.007 |
| protein.synthesis.chloroplast/mito – plastid ribosomal | |||
| 29.2.1.1 | protein.plastid | 21 | 0.04 |
| 29.2.2 | protein.synthesis.misc ribososomal protein | 106 | 0.01 |
| 29.4 | protein.postranslational modification | 186 | 0.03 |
| protein.postranslational modification.kinase.receptor like | |||
| 29.4.1.51 | cytoplasmatic kinase I | 2 | 0.03 |
| 29.5.11.4.3.2 | protein.degradation.ubiquitin.E3.SCF.FBOX | 58 | 0.01 |
| 29.6 | protein.folding | 25 | 0.006 |
| 30 | signalling | 267 | 0.02 |
| 30.3 | signalling.calcium | 59 | 0.01 |
| 35 | not assigned | 2150 | 0.02 |
| 35.2 | not assigned.unknown | 1191 | 0.003 |
The bin number of the microarray was also denoted from 1–100 and it was only shown for the bins with significant changes in transcript abundance at P < 0.05.
Further in silico analysis, the platform of the Arabidopsis Interactions Viewer [52] was used to investigate the potential molecular protein-protein interaction based on the observed transcript changes. When the 186 genes were deployed in the Arabidopsis Interactions Viewer, there were more than 745 interlogs obtained. However, only two hubs (AMP kinase and phosphatase associated protein 46 and TAP46) were identified if these 9 genes were loaded into the Arabidopsis Interactions Viewer under higher stringency.
Secondary metabolism and lignin biosynthesis pathways
The 31 genes which had been classified as potentially being involved in secondary metabolism (bin 16; Table 2; Figure 2) were prominent. Several other genes that were reported to be involved in plant responses to pathogen attack were found within the schema. They were subdivided into pathways leading to isoprenoid, phenylpropanoid and lignin biosynthesis. The 18 genes were mapped into bin 16.1.4 (isoprenoids, carotenoids) included genes involved in signaling, signaling-calcium, cell wall and cellular synthesis and 13 genes were mapped into bin16.2.1 (phenylpropanoids) and bin16.7 (waxes).
Table 2.
The protein-protein interaction carried out by the Arabidopsis Interaction Viewer on Bin 16.
| Locus | Description | Fold change |
| At3g10340 | phenylalanine ammonia-lyase | 3.395 |
| At2g23910 | cinnamoyl-CoA reductase | 2.688 |
| At4g09500 | glycosyltransferase family protein | 1.131 |
| At5g12210 | geranylgeranyl transferase type II beta subunit | 1.117 |
| At4g17190 | farnesyl pyrophosphate synthetase 2 | 1.032 |
| At5g62790 | 1-deoxy-D-xylulose 5-phosphate reductoisomerase | 1.013 |
| At2g40230 | transferase family protein | 0.924 |
| At1g74020 | atss-2 strictosidine synthase | 0.899 |
| At1g26410 | FAD-binding domain-containing protein | 0.889 |
| At3g10230 | lycopnene beta-cyclase | 0.887 |
| At1g62570 | flavin-containing monooxygenase family protein | 0.826 |
| At5g57840 | transferase family protein | 0.796 |
| At1g08550 | violaxanthin de-epoxidase precursor | 0.665 |
| At4g34540 | isoflavone reductase family protein | 0.665 |
| At1g35190 | oxidoreductase | 0.633 |
| At2g29330 | tropinone reductase, putative | 0.603 |
| At4g16330 | oxidoreductase | 0.559 |
| At1g06570 | 1;4-hydroxyphenylpyruvate dioxygenase | 0.552 |
| At1g58180 | carbonic anhydrase family protein | 0.541 |
| At4g33360 | terpene cyclase | 0.523 |
| At3g21240 | 4-coumaroyl-CoA synthase 2 | -0.057 |
| At2g30490 | cinnamic acid 4-hydroxylase | -0.274 |
| At5g13930 | chalcone synthase | -0.479 |
| At3g55120 | chalcone-flavanone isomerase | -0.51 |
| At3g51240 | naringenin 3-dioxygenase/flavanone 3-hydroxylase | -0.52 |
| At4g34230 | cinnamyl-alcohol dehydrogenase | -0.556 |
| At4g39330 | mannitol dehydrogenase | -0.645 |
| At1g17050 | geranyl diphosphate synthase | -0.821 |
Key genes were in bold. The value was on Log 2 scale.
Figure 2.
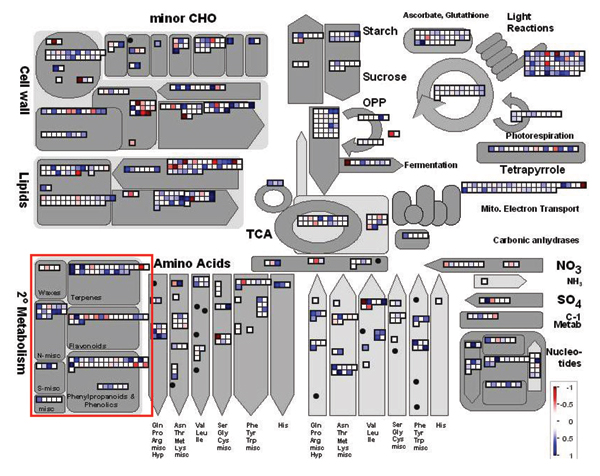
TA changes in responses of Arabidopsis plant to F. virguliforme. The secondary metabolism in the MapMan program [50] was marked in red square. Blue and Red squares were denoted increased (positive) and decreased (negative) transcript abundances (TAs), respectively. The bar was shown on log2 scale.
The transcriptional analysis of genes from the multiple branches of the phenylpropanoid pathway showed that 6 of the 31 genes were significantly altered in TA (Table 2). Further, TA changes inferred that synthesis of cinnamic acid in Arabidopsis was one of the early responses to F. viguliforme infection. Phytoalexins and phytoanticipins serve both structural and metabolic functions in disease resistance. Our microarray data showed that the resistance response of the phenylpropanoid pathway was different in Arabidopsis and soybean [9]. The microarray data showed that the phenylpropanoid pathway was activated during the resistance response in the Arabidopsis. In contrast to soybean [9], despite the fact that the PAL was induced in this metabolic pathway, other major genes such as cinnamic acid 4-hydroxylase (C4L), chalcone synthase (CHS) and cinnamyl-alcohol dehydrogenase (CAD) in the phenylpropanoid pathway were all suppressed (Table 2), indicating that soybean and Arabidopsis did not share similar strategies in the specific pathway to resistance F. virguliforme. Based on the up-regulation of ESTs, total six genes in the phenylpropanoid pathway had more than 1-fold increase in abundances (Log2 scale). However, the rest of genes involved in major branches in the pathway were either down-regulated or no significant increase in TAs (Table 2). The suppression of these branches of the phenylpropanoid pathway seemed somehow different from the soybean response to the pathogen [9]. Since transcripts of those enzymes leading to the synthesis of flavonols, terpenes and proanthocyanidins, decrease in abundances of down-stream genes in the procedure of the resistance response in the pathway but increase in PAL and cinnamoyl-CoA reductase transcripts during this same time suggesting the existence of a potential bypass to synthesize secondary metabolites in the defense response. After the 192 soybean genes have been converted into 158 functional orthologs of Arabidopsis genes, comparison of the protein-protein interaction network in both Arabidopsis and soybean demonstrated a high specificity trend in gene regulation of the two species (Figure 3). There were 12 hubs identified in the 158 functional orthologous soybean genes whilst more than 15 network hubs were observed to mediate the resistance in the up-regulated Arabidopsis genes. Unfortunately, no resistance pathway was generated from our Arabidopsis microarray data by the Arabidopsis Interactions Viewer program in the bin16 and no significant interlog among genes in bin16 of the data obtained.
Figure 3.

Network of protein-protein interaction of A. thaliana and soybean infested with F. virguliforme. The networks were generated by Cytoscape [67] and visual displays were saved as Cytoscape graphs. Interaction hubs were marked by red.
SNF1 (sucrose non-fermenting-1)-related protein kinase (SnRK1, AKIN10) gene
When all 50 genes altered in TA from in bin30 (signaling) were subjected to Arabidopsis Interactions Viewer platform, more than 300 interlogs were identified. The genes clustered into three major categories based on both interlog confidence value and Pearson correlation coefficient. The first group of the interlogs was mediated around MAP (mitogen-activated protein) kinases, the second group was centered on SNF1-related protein kinase 1 (SnRK1; Figure 4), and the third group was formed around ATMPK (Table 3). The second group contained 32 genes altered in TAs and this group was composed of three putative major nodes with a total 111 hits and 443 interlog confidence values (Table 3). A putative signal transduction pathway was derived from the interaction network (Figure 5). The results suggested that the SnRK1 gene could be important for coordinating the signal assembly of a cellular apparatus associated with the "endogenous fuel gauge" [53], since of the 31 genes in the reaction center may be differentially regulated by the endogenous AMP and sugar content in order to maintain cell defense. As shown in Figure 3, SnRK1 was centered among 31 interacting genes. The group was composed of 3 clades and most genes corresponded to the signal transduction cascade and cellular responses (data not shown).
Figure 4.

Interlogs of Bin30 based on Arabidopsis Interactions Viewer [52] SNF1 (sucrose non-fermenting-1)-related protein kinase (SnRK1) gene was positioned in lower part of the figure by the computer program.
Table 3.
Overview of protein-protein interaction of bin30 based on Arabidopsis Interactions Viewer program.
| Protein 1 | Protein 2 | Interolog Confidence Value | Interolog Confidence | Pearson Correlation Coefficient | Protein2 Subcellular Localisation | Protein1 Annotation | Protein2 Annotation | Total hits | Protein2 fold change |
| At3g45640 | At4g26070 | 27 | High | 0.65 | N/A | MAP kinase 3 | MAP kinase kinase1 | 9 | |
| At3g45640 | At4g29810 | 9 | Medium | 0.633 | cytosol | MAP kinase 3 | MAP kinase kinase2 | 3 | 1.02 |
| At3g45640 | At5g50260 | 1 | Low | -0.053 | N/A | MAP kinase 3 | cysteine proteinase | 1 | |
| At3g01090 | At1g09020 | 287 | High | 0.822 | N/A | SnRK1 | Sucrose NonFermenting 4 | 41 | |
| At3g01090 | At3g45240 | 27 | High | 0.829 | N/A | SnRK1 | GRIK kinase 1 | 9 | 1.17 |
| At3g01090 | At5g48630 | 24 | High | 0.661 | N/A | SnRK1 | cyclin family protein | 6 | 2.93 |
| At3g01090 | At4g16360 | 20 | High | 0.732 | N/A | SnRK1 | AMP-activated protein kinase | 10 | 1.16 |
| At3g01090 | At5g10270 | 16 | High | 0.892 | N/A | SnRK1 | cyclin dependent kinase C 1 | 4 | 1.2 |
| At3g01090 | At5g63610 | 16 | High | 0.726 | N/A | SnRK1 | HUA ENHANCER 3 | 4 | |
| At3g01090 | At1g15780 | 6 | Medium | 0.878 | N/A | SnRK1 | unknown protein | 3 | 1.16 |
| At3g01090 | At4g05320 | 6 | Medium | 0.74 | N/A | SnRK1 | polyubiquitin 10 | 3 | 1.12 |
| At3g01090 | At1g01960 | 4 | Medium | 0.829 | N/A | SnRK1 | G-nucleotide exchange factor | 2 | |
| At3g01090 | At1g09200 | 4 | Medium | 0.565 | N/A | SnRK1 | histone H3 | 2 | 1.4 |
| At3g01090 | At1g24290 | 4 | Medium | 0.694 | N/A | SnRK1 | AAA-type ATPase protein | 2 | |
| At3g01090 | At3g54610 | 4 | Medium | 0.766 | nucleus | SnRK1 | Histon acetyltransferase 1 | 2 | |
| At3g01090 | At4g18880 | 4 | Medium | 0.199 | nucleus | SnRK1 | HS transcription factor A4A | 2 | |
| At3g01090 | At5g21170 | 3 | Medium | 0.571 | N/A | SnRK1 | AMP-activated protein kinase | 3 | 2.17 |
| At3g01090 | At1g05570 | 2 | Medium | 0.586 | PM | SnRK1 | callose synthase | 2 | |
| At3g01090 | At1g14400 | 1 | Low | 0.831 | N/A | SnRK1 | ubiquitin carrier protein 1 | 1 | 1.15 |
| At3g01090 | At1g16030 | 1 | Low | -0.04 | plastid | SnRK1 | HS protein 70B | 1 | |
| At3g01090 | At1g20970 | 1 | Low | 0.744 | N/A | SnRK1 | adhesin-related | 1 | |
| At3g01090 | At1g35160 | 1 | Low | 0.807 | NC, cytosol | SnRK1 | general regulatory factor 4 | 1 | |
| At3g01090 | At1g36160 | 1 | Low | 0.747 | PM, cytosol | SnRK1 | acetyl-CoA carboxylase | 1 | 0.33 |
| At3g01090 | At1g50370 | 1 | Low | 0.854 | N/A | SnRK1 | ser/thr protein phosphatase | 1 | |
| At3g01090 | At1g75560 | 1 | Low | 0.741 | nucleus | SnRK1 | zinc knuckle family protein | 1 | |
| At3g01090 | At2g32850 | 1 | Low | 0.837 | N/A | SnRK1 | protein kinase family protein | 1 | 1.24 |
| At3g01090 | At2g37840 | 1 | Low | 0.71 | N/A | SnRK1 | protein kinase family protein | 1 | |
| At3g01090 | At2g43790 | 1 | Low | 0.665 | N/A | SnRK1 | ATMPK6 | 1 | 0.86 |
| At3g01090 | At3g56640 | 1 | Low | 0.777 | N/A | SnRK1 | exocyst complex subunit | 1 | |
| At3g01090 | At4g10310 | 1 | Low | 0.385 | N/A | SnRK1 | sodium ion transporter | 1 | |
| At3g01090 | At4g26070 | 1 | Low | 0.297 | N/A | SnRK1 | MAP kinase kinase 1 | 1 | |
| At3g01090 | At4g26840 | 1 | Low | 0.72 | nucleus | SnRK1 | small ubiquitin-like modifier 1 | 1 | 0.79 |
| At3g01090 | At5g44740 | 1 | Low | 0.686 | N/A | SnRK1 | UMUC-like DNA repair protein | 1 | |
| At3g01090 | At5g53770 | 1 | Low | 0.668 | N/A | SnRK1 | nucleotidyltransferase | 1 | |
| At1g59580 | At4g29810 | 12 | High | 0.621 | cytosol | MAP kinase 2 | ATMKK2 | 12 | 1.02 |
| At1g59580 | At2g31660 | 8 | Medium | 0.521 | nucleus | MAP kinase 2 | SAD2 | 8 | |
| At1g59580 | At2g17700 | 6 | Medium | 0.358 | N/A | MAP kinase 2 | protein kinase | 6 | 0.88 |
| At1g59580 | At1g58230 | 4 | Medium | 0.57 | mitochondria | MAP kinase 2 | WD-40 repeat protein | 4 | |
| At1g59580 | At2g35320 | 4 | Medium | 0.601 | N/A | MAP kinase 2 | protein tyrosine phosphatase | 4 | |
| At1g59580 | At4g21820 | 4 | Medium | 0.099 | N/A | MAP kinase 2 | calmodulin-binding protein | 4 | |
| At1g59580 | At5g17690 | 4 | Medium | 0.447 | nucleus | MAP kinase 2 | terminal flower 2 | 4 | |
| At1g59580 | At3g58040 | 2 | Medium | 0.278 | N/A | MAP kinase 2 | seven in absentia protein | 2 | |
| At1g59580 | At4g24440 | 2 | Medium | 0.345 | nucleus | MAP kinase 2 | TFIIA-gamma | 2 | |
| At1g59580 | At4g35780 | 2 | Medium | 0.657 | N/A | MAP kinase 2 | protein kinase protein | 2 | |
| At1g59580 | At5g03415 | 2 | Medium | 0.269 | nucleus | MAP kinase 2 | DPB__DPB | 2 | |
| At1g59580 | At1g53570 | 1 | Low | 0.685 | N/A | MAP kinase 2 | MAP kinase kinase kinase 3 | 1 | 1.15 |
| At1g59580 | At1g59580 | 1 | Low | 1 | N/A | MAP kinase 2 | ATMPK2 | 1 | 1.47 |
Fold change was on Log2 scale.
Figure 5.

Simplified diagram of putative SnRK1 pathway. SnRK1 hub was marked by a green ring.
Upstream of the SnRK1 signal cascade, the ATMPK cascade may be connected to cellular calcium (through calmodulin binding protein) and protein kinases (Figure 4, 5). In animals, LKB1 (also called STK11, a recently identified tumor suppressor gene) is required for the activation of AMPK in response to cellular concentrations of AMP induced by cellular stresses. The increase in AMP promotes phosphorylation by LKB1 [54]. The SnRK1 shared 33% identify with one of the mouse LKB1 paralogs (BAA76749) and there was a significant interlog predicted between the SnRK1 and LKB1. Pathogen elicitor or biotic and abiotic stresses may activate MAPK pathway through an unknown protein. On the other hand, cellular C and AMP concentrations may also trigger the AMPK to induce SnRK1via SNF4. However, the role of the AMPK of Arabidopsis and its molecular significance and molecular fragments located immediately in the upstream of SnRK1 are still unknown. The TAs of the AMPK (At5g21170) and cyclin family protein (At5g48630) were significantly increased by F. virguliform but ACC1 (acetyl-CoA carboxylase 1) was suppressed (Table 3). Analysis of functional orthologs of the soybean genes in Arabidopsis indicated that significant variations existed between two species facing the same pathogen. Interestingly, SnRK1 and SNF4 were also found in the soybean resistance network (Figure 3).
Comparison of the fungal resistance genes between Arabidopsis and soybean
168 genes with altered TAs in the Arabidopsis microarray data shared homology to the 192 soybean genes involved in resistance, signal transduction, plant defense and transport of metabolites. Those TAs from both species were functionally clustered into three major groups (data not shown) using CLUSTAL X [55]. The results of multiple alignments were subjected to phylogenetic analysis with algorithm of the MEGA4 software [56] using the Maximum Parsimony analysis with Kimura two parameter distances. The reliabilities of each branch point were assessed by the analysis of 500 bootstrap replicates. The Maximum Parsimony (MP) analysis showed that these genes possessed homology by evolutionary descent. Maximum Parsimony phylogenetic analyses partitioned these resistance genes in Arabidopsis and soybean [9] jointly into 7 clades (denoted I-VII, data not shown). Each clade contained various members. Some clades could be further subdivided into subclasses. Clade III contained members that were the most divergent compared with those encoded by the other clades. Interestingly, there were several ESTs that were only found in one species. Majority families or subfamilies in the higher nodes tended to give low bootstrap values (data not shown).
Discussion
It is well-known that over-expression PAL in tobacco plant can effectively be used against a virulent fungal pathogen Cercospora nicotiana infection and also altered the lesion phenotype inoculated with TMV [57]. Reduction of phenylpropanoid biosynthesis in tobacco via down-regulation of PAL reduces local and systemic acquired resistance to fungal or viral infection [27,28]. The major function of PAL is to catalyze phytoalexins and phytoanticipins production and thereby reduce disease severity [58]. Constitutive over-expression of the PAL gene from the tropical pasture legume Stylosanthes humilis in tobacco plants provided resistance to Cercospora nicotianae and to pathogen Phytophthora parasitica pv. Nicotianae [59]. The key enzymes of phenylpropanoid pathway, PAL, C4L and CHS were all induced in soybean resistance to F. virguliforme [9]. The global expression analysis of the Arabidopsis plant challenged by F. virguliforme appeared to be different. The fungal pathogen affected the secondary metabolism not only in the phenylpropanoid pathway but also via pathways leading to other cellular functions (terpenes, phenolics, special N compounds metabolism). Transcripts of the PAL gene encode the key enzyme feeding all these branches. Therefore, the decreases in most key transcripts at the times when PAL and cinnamoyl-CoA reductase transcripts increases in abundance is a novel finding showing that the synthesis of PAL and cinnamoyl-CoA reductase transcripts is with negative correlation to the activity of the other genes during the pathogen stress response. Perhaps in A. thaliana, the phenylpropanoid pathway did not directly participate in the cascade of reactions elicited during the defense and probably, the biosynthesis of phytoalexins and phytoanticipins was regulated in different fashions in soybean and Arabidopsis. Transcripts of genes involved in the synthesis of lignin, flavonols, anthocyanins and proanthocyanidins (data not shown) seemed to be down-regulated during the periods of time when the genes involved in the synthesis of cinnamic acid was up-regulated. Arabidopsis lacks isoflavonoid and phytoalexins and produces mainly camalexin rather than a cocktail of isoflavonoids [60]. Therefore, in Arabidopsis the flavonoids (anthocyanins, proanthocyanidins, flavones and flavonols) may indeed not play a significant role in defense. In contrast, there was evidence that cinnamoyl alcohol dehydrogenase (CAD) was induced rapidly in Arabidopsis infected with Xanthomonas [61] and CAD is in the pathway that leads to lignin biosynthesis. In the phenylpropanoid pathway, key genes like PAL are encoded by a group of genes showing a great degree of sequence diversity. Significantly up-regulated PAL transcripts support for the hypothesis that in Arabidopsis expression of the PAL is significantly associated with pathogen infection. However, the PAL over-expression plants caused a significant reduction in growth and delayed flowering. These phenotypes may also be related to energy status or amino acid pool availability being altered by PAL.
It has been shown that the addition of sugar activates genes related to disease resistance [62]. The metabolic change in the pathogenesis could negatively affect cellular C content based on the expression of sugar-repressive nuclear genes and may be involved in the regulation of these genes [63]. Therefore, there should be other mechanisms for maintaining the cellular C concentrations. Moreover, metabolic movement should provide some explanation. Our results infer that the metabolite profiles may affect the status of disease resistance in the Arabidopsis plant. The response to C stresses is probably the key role of SnRK1 in eukaryotes [64]. SnRK1 may represent a primordial protein that in plants plays an important role in all resistances. It should be noted that SnRK1 interacts with 31 proteins, and each is located up- and down-stream of the gene in the interactome. Perhaps, the SnRK1 sequence has shared motifs with the rest of other proteins in the network. Interestedly, the presence of proteins in Arabidopsis related to three subunits of the SnRK1 protein might indicate an early duplication of these subunits in the process of evolution. To date, it is still not clear how, during the course of evolution of multicellular organisms, the SnRK1 system acquired the ability to be a signal system. One important implication is that SnRK1 serves as one of the hubs for the signal cascades in the plant. Although the relative specificity of SnRK1 in plants is yet unknown, it is possible that further investigation of such role may allow us to draw new resistance pathways based on energy status. Indeed, plant SnRK1 associated with herbivore tolerance was recently identified [53]. Our results suggest that interaction between SnRK1 with its network partners may be induced by the fungal pathogen. Further studies of the gene and the interacting partners should help to uncover important specific aspects of SNF1-mediated signaling during pathogen infection.
Orthologous genes are defined by direct evolutionary descent and should play similar developmental or physiological roles. Several gene groups were identified according to the outcome of functional orthologous analysis in this study, showing same functional orthologous relationships in these two species. In our primary results, the 1 to 1 ratio of the orthologous relationship was found among those several paired groups, indicating these gene groups were descended from a common ancestor and corresponded to well-conserved functions. These 1 to 1 ortholog classes are presumed to represent conserved functions in Arabidopsis and soybean, but they shared diverse bootstrapping value. Diversification following gene duplication may have occurred but the degree is still unknown. Twenty-eight pairs of the genes from both species were taxoned together and each pair possessed unique bootstrapping value and Kimura phylogenetic distance (data not shown). Based on phylogenetic analysis, the organization of soybean phenylalanine ammonia-lyase (PAL) proteins were very similar to that of the Arabidopsis PAL proteins, implying that the soybean and Arabidopsis PAL proteins analyzed here were all derived from a common ancestor. The fact that these clades were formed by both Arabidopsis and soybean PAL genes gave a suggestion that these PAL genes existed before the divergence of monocots and dicots. Interestingly, fair numbers of EST sequences were not found in the other organism, even in closely related gene families that were associated with the fungal resistance. However, detailed screening phylogenetic relationship among those resistance genes is necessary for detection of the gene evolution.
Plants possess an ability of resistance to most potentially pathogenic microbes. Gene transcriptional changes are critical for many plant defense processes [1]. Soybean SDS consists of root infection and leaf scorch. With the limitations in the study of solely predicted gene interactions, definite conclusions about the nature of the resistant response to F. virguliforme infection cannot yet be made. Investigation on differentially expressed genes of Arabidopsis and soybean in response to F. virguliforme can lead to better understanding of the mechanisms of resistant crops for certain disease. Microarray experiment allows interrogation of tens of thousands of genes simultaneously. To understand the molecular interactions involved in F. virguliforme resistance, we have integrated results from the soybean SDS resistance with Arabidopsis DNA microarray studies by in silico analyses. The results of this study can be used as a model system to facilitate the understanding of plant resistance to F. virguliforme.
Methods and materials
Plant materials
Arabidopsis thaliana cv. Columbia plants were germinated from seeds under conditions of 16 h photoperiod (500 μE/M2/sec) with temperatures at 22°C day/18°C night and 80% (v/v) relative humidity in a growth chamber. Plants were grown on rafts floating on liquid MS medium [65]. There were 30 plants per treatment arranged in a randomized complete block with three plants per treatment per block. Twenty-one days after planting, synchronously growing plants were selected and collected.
Inoculation of roots with F. virguliforme spores
The F. virguliforme isolate 'Mont-1' was obtained from Dr. Shiuxian Li at the National Soybean Research Laboratory (Urbana, IL). F. virguliforme was cultured on potato dextrose agar medium (PDA, Difco, Detroit, MI) supplemented with 80 mg ml-1 tetracycline and a few drops of Tween 20. A spore suspension of F. virguliforme isolate 'Mont-1' was prepared as described [66]. The spore suspension, at 5 × 104 spore ml-1 with sterile distilled water, was made by adding F. virguliforme spores from several F. virguliforme culture plates and was continuously stirred on a stir flask to keep a uniform suspension. The spore suspension was poured on the growth medium for the infested plants and the same volume of sterile distilled water was added to non-infested plants.
RNA isolation and microarray procedure
RNA was isolated separately from both inoculated and non-inoculated roots of 30 A. thaliana plants that were bulked and ground to a fine powder in liquid nitrogen. RNA was extracted with a RNeasy Plant Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. RNA samples were treated with DNase in order to remove any residual DNA using the RNase-free DNase kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instructions. After DNase treatment, RNA was purified on RNeasy mini spin columns (Qiagen, Valencia, CA). The quantity and quality of the RNA recovered was determined by spectrophotometry at 260–280 nm and electrophoresis on a 1.2% (w/v) agarose, 20% (v/v) formaldehyde gel. The microarray hybridization and slide scanning were carried out by the facility at AFGC http://afgc.stanford.edu. Microarrays (16561.xls and 27314.xls at ftp://smd-ftp.stanford.edu/smd/organisms/AT/ were used in the experiment. The mRNA samples corresponding to treatment (infested) and control (non-infested) was labeled during the cDNA synthesis with Cy3- or Cy5- labeled dUTP and with one technical replicate labeled by reversed dye compared to the first hybridization.
Data analysis
A visualization software, MapMan was used to perform a gene ontology where the set of Arabidopsis genes of the microarray was assigned based on the non-redundant and hierarchically categorized assignment of BINs and sub-BINs at TIGR (The Institute for Genomic Research). The ontology was derived from the Affymetrix 22 K array corresponding to similar or sub-modal biological functions [50]http://gabi.rzpd.de/projects/MapMan/. The classification of Image Annotator in the software was also used to diagram the data display. The changes were expressed relative to those in pathogen challenged roots. Transcripts that increased in abundance were denoted in blue, and transcripts were decreased in abundance were denoted in red. In the scale used for the visualized data, a 1-fold change (Log2 scale) was required to produce a visible coloration, and the scale saturates at a 3-fold (log2 scale) change. Blue and Red squares were denoted increased (positive) and decreased (negative) transcript abundances (TAs), respectively. The bar was shown on log2 scale. The Arabidopsis Interaction Viewer queries a database of 19,979 predicted and 1,499 confirmed interacting proteins. The predicted interactions (interologs) were generated by Geisler-Lee et al. [52]. Output of two interlogs was transferred to the Cytoscape software environment [67] for network visualization and modeling against each other in order to catalog all of their conserved pathways and gene interaction networks. The program was equipped with a plug-in architecture for customizing applications. The visual displays were saved as a Cytoscape graphs.
Stringent quality control measures were applied to all stages of data analysis. The Microarray data were normalized by local (local background value was subtracted from the intensity value of each spot) and global metrics. The procedures described by Pevsner [49] were followed to adjust for differences in the intensity of the two labels. Coefficients of means and variances on the signal intensities in each channel and ratio of signals from two replicates were calculated by our C++ program (available on request), which was also used to handle the missing and extra data values. The average ratio for a signal microarray from two replicates was computed by the equation of [Ratio1st+ (1/Ratio2nd)]/2. The Student's t-test was used to determine the statistical significance for genes considered between and within Microarrays slides and the plant mean weight differences (P < 0.05).
Phylogenetic analyses
Functional sequence analysis was performed on amino acid sequences using Clustal X [55] with the default settings. Nucleotide sequences were aligned with Clustal W [68]. The results of multiple alignments were subjected to phylogenetic analysis using the algorithm of the MEGA package version 4.0 using the Maximum Parsimony analysis with Kimura two parameter distances [56]. The Maximum-Parsimony was assessed by 500 bootstrap replicates. Only nucleotide sequences were employed in phylogenetic analysis based on higher stringy consideration for the phylogenetic tree construction.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
DAL and JI envisioned the research. JY and JI grew plants, infested them and made RNA. AFGC carried out the microarrays. JY and MZ did data analysis. All authors helped in manuscript preparation. All authors read and approved the final manuscript. DAL, KM and JI envisioned the research.
Acknowledgments
Acknowledgements
We thank Dr. Matt Geisler for the guidance on data analysis. This research was funded in part by the grant from the National Science Foundation, the Illinois Soybean Association and the United Soy Board.
This article has been published as part of BMC Genomics Volume 9 Supplement 2, 2008: IEEE 7th International Conference on Bioinformatics and Bioengineering at Harvard Medical School. The full contents of the supplement are available online at http://www.biomedcentral.com/1471-2164/9?issue=S2
Contributor Information
Jiazheng Yuan, Email: jyuan@siu.edu.
Mengxia Zhu, Email: mzhu@cs.siu.edu.
David A Lightfoot, Email: ga4082@siu.edu.
M Javed Iqbal, Email: mjiqbal@siu.edu.
Jack Y Yang, Email: jyang@bwh.harvard.edu.
Khalid Meksem, Email: meksemk@siu.edu.
References
- Journot-Catalino N, Somssich IE, Roby D, Kroj T. The transcription factors WRKY11 and WRKY17 Act as negative regulators of basal resistance in Arabidopsis thaliana. Plant Cell. 2006;18:3289–3302. doi: 10.1105/tpc.106.044149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk PM, Kazan K, Wilson I, Anderson JP, Richmond T, Somerville SC, Manners JM. Coordinated plant defense responses in Arabidopsis revealed by microarray analysis. Proc Natl Acad Sci USA. 2000;97:11655–11660. doi: 10.1073/pnas.97.21.11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Guegler K, LaBrie ST, Crawford NM. Genomic analysis of a nutrient response in Arabidopsis reveals diverse expression patterns and novel metabolic and potential regulatory genes induced by nitrate. Plant Cell. 2000;12:1491–1510. doi: 10.1105/tpc.12.8.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Tischner R, Gutierrez RA, Hoffman M, Xing X, Chen M, Coruzzi G, Crawford NM. Genomic analysis of the nitrate response using a nitrate reductase null mutant of Arabidopsis. Plant Physiol. 2004;136:2512–2522. doi: 10.1104/pp.104.044610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature. 2000;405:827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- Gutierrez RA, Ewing RM, Cherry JM, Green PJ. Identification of unstable transcripts in Arabidopsis by cDNA microarray analysis: rapid decay is associated with a group of touch- and specific clock-controlled genes. Proc Natl Acad Sci USA. 2002;99:11513–11518. doi: 10.1073/pnas.152204099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd JC, Zakhleniuk OV. Responses of primary and secondary metabolism to sugar accumulation revealed by microarray expression analysis of the Arabidopsis mutant, pho3. J Exp Bot. 2004;55:1221–1230. doi: 10.1093/jxb/erh143. [DOI] [PubMed] [Google Scholar]
- Zhao B, Ardales E, Raymundo A, Bai J, Trick H, Leach J, Hulbert S. The avrRxo1 gene from the rice pathogen Xanthomonas oryzae pv. oryzicola confers a nonhost defense reaction on maize with resistance gene Rxo1. Mol Plant-Microbe Interact. 2004;17:771–779. doi: 10.1094/MPMI.2004.17.7.771. [DOI] [PubMed] [Google Scholar]
- Iqbal MJ, Yaegashi S, Ahsan R, Shopinski K, Lightfoot DA. Root response to F. solani f. sp. glycines: Temporal accumulation of transcripts in partially resistant and susceptible soybean. Theor and Appl Genet. 2005;110:1429–1438. doi: 10.1007/s00122-005-1969-9. [DOI] [PubMed] [Google Scholar]
- Iqbal MJ, Yaegashi S, Njiti V, Ahsan R, Cryder KL, Lightfoot DA. Resistance locus pyramids alter transcript abundance in soybean roots inoculated by Fusarium solani f. sp. glycines. Mol Genet and Genom. 2002;268:407–417. doi: 10.1007/s00438-002-0762-6. [DOI] [PubMed] [Google Scholar]
- Iqbal MJ, Afzal AJ, Yaegashi S, Ruben E, Triwitayakorn K, Njiti V, Ahsan R, Wood AJ, Lightfoot DA. A pyramid of loci for partial resistance to Fusarium solani fsp. Theor and Appl Genet. 2002;105:1115–1123. doi: 10.1007/s00122-002-0987-0. [DOI] [PubMed] [Google Scholar]
- Dangl JL, Ritter C, Gibbon MJ, Muir LA, Wood JR, Goss S, Mansfield I, Taylor JD, Vivian A. Functional homologs of the Arabidopsis RPM7 disease resistance gene in bean and pea. Plant Cell. 1992;4:1359–1369. doi: 10.1105/tpc.4.11.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innes RW, Bisgrove SR, Smith NM, Bent AF, Staskawicz BJ, Liu YC. ldentification of a disease resistance locus in Arabidopsis that is functionally homologous to the Rpgl locus of soybean. Plant J. 1993;4:813–820. doi: 10.1046/j.1365-313X.1993.04050813.x. [DOI] [PubMed] [Google Scholar]
- Simonich MT, Innes RW. A disease resistance gene in Arabidopsis with specificity for the avrPph3 gene of Pseudomonas syringae pv. phaseolicola. Mol Plant Microbe lnteract. 1995;8:637–640. doi: 10.1094/mpmi-8-0637. [DOI] [PubMed] [Google Scholar]
- Meyers BC, Kaushik S, Nandety RS. Evolving disease resistance genes. Curr Opin Plant Biol. 2005;8:129–134. doi: 10.1016/j.pbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Labrador M, Corces VG. Setting the boundaries of chromatin domains and nuclear organization. Cell. 2002;111:151–154. doi: 10.1016/S0092-8674(02)01004-8. [DOI] [PubMed] [Google Scholar]
- Dixon RA, Achnine L, Kota P, Liu CJ, Reddy MSS, Wang L. The phenylpropanoid pathway and plant defense – a genomics perspective. Mol Plant Pathol. 2002;3:371–390. doi: 10.1046/j.1364-3703.2002.00131.x. [DOI] [PubMed] [Google Scholar]
- Durner J, Wendehenne D, Klessig DF. Defense gene induction in tobacco by nitric oxide, cyclic GMP, and cyclic ADP-ribose. Proc Natl Acad Sci. 1998;95:10328–10333. doi: 10.1073/pnas.95.17.10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell JM, Woffenden BJ. Plant disease resistance genes: recent insights and potential applications. Trends in Biotechnology. 2003;21:178–183. doi: 10.1016/S0167-7799(03)00053-2. [DOI] [PubMed] [Google Scholar]
- Cochrane FC, Davin LB, Lewis NG. The Arabidopsis phenylalanine ammonia lyase gene family: Kinetic characterization of the four PAL isoforms. Phytochemistry. 2004;65:1557–1564. doi: 10.1016/j.phytochem.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Pellegrini L, Geoffroy P, Fritig B, Legrand M. Molecular cloning and expression of a new class of ortho-diphenol-O-methyltransferases induced in tobacco (Nicotiana tabacum L.) leaves by infection or elicitor treatment. Plant Physiol. 1993;103:509–517. doi: 10.1104/pp.103.2.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrin L, Rohfritsch O, Fritig B, Legrand M. Phenylalanine ammonia-lyase in tobacco. Molecular cloning and gene expression during the hypersensitive reaction to tobacco mosaic virus and the response to a fungal elicitor. Plant Physiol. 1994;106:877–886. doi: 10.1104/pp.106.3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozovaya V, Lygin A, Zernova O, Li S, Hartman G, Widholm J. Isoflavonoid accumulation in soybean hairy roots upon treatment with Fusarium solani. Plant Physiol Biochem. 2004;42:671–679. doi: 10.1016/j.plaphy.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Maher EA, Bate NJ, Ni W, Elkind Y, Dixon RA, Lamb CJ. Increased disease susceptibility of transgenic tobacco plants with suppressed levels of preformed phenylpropanoid products. Proc Natl Acad Sci. 1994;91:7802–7806. doi: 10.1073/pnas.91.16.7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallas JA, Paiva NL, Lamb CJ, Dixon RA. Tobacco plants epigenetically suppressed in phenylalanine ammonia-lyase expression do not develop systemic acquired resistance in response to infection by tobacco mosaic virus. Plant J. 1996;10:281–293. doi: 10.1046/j.1365-313X.1996.10020281.x. [DOI] [Google Scholar]
- Mitchell HJ, Hall SA, Stratford R, Hall JL, Barber MS. Differential induction of cinnamyl alcohol dehydrogenase during defensive lignification in wheat (Triticum aestivum L.): Characterisation of the major inducible form. Planta. 1999;208:31–37. doi: 10.1007/s004250050531. [DOI] [Google Scholar]
- Buchanan BB, Gruissem W, Jones RL. Biochemistry and molecular biology of plants. American Society of Plant Physiologists; 2000. [Google Scholar]
- Humphreys JM, Chapple C. Rewriting the lignin roadmap. Curr Opin Plant Biol. 2002;5:224–229. doi: 10.1016/S1369-5266(02)00257-1. [DOI] [PubMed] [Google Scholar]
- Boerjan W, Ralph J, Baucher M. Lignin biosynthesis. Annu Rev Plant Biol. 2003;54:519–546. doi: 10.1146/annurev.arplant.54.031902.134938. [DOI] [PubMed] [Google Scholar]
- Baucher M, Halpin C, Petit-Conil M, Boerjan W. Lignin: Genetic engineering and impact on pulping. Crit Rev Biochem Mol Biol. 2003;38:305–350. doi: 10.1080/10409230391036757. [DOI] [PubMed] [Google Scholar]
- Davin LB, Bedgar DL, Katayama T, Lewis NG. On the stereospecific synthesis of (+) pinoresinol in Forsythia suspensa from its achiral precursor, coniferyl alcohol. Phytochemistry. 1992;31:3869–3874. doi: 10.1016/S0031-9422(00)97544-7. [DOI] [PubMed] [Google Scholar]
- Dean JFD, LaFayette PR, Rugh C, Tristram AH, Hoopes JT, Eriksson K-EL, Merkle SA. Laccases associated with lignifying vascular tissue. In: Lewis NG, Sarkanen S, editor. Lignin and Lignan Biosynthesis. Washington, DC: American Chemical Society; 1998. pp. 96–108. [Google Scholar]
- Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci. 2003;100:8839–8843. doi: 10.1073/pnas.1533136100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Simon A, Hawley SA, Scott JW. AMP-activated protein kinase – development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford NG, Hardie DG. SNF1-related protein kinases: Global regulators of carbon metabolism in plants? Plant Mol Biol. 1999;37:735–748. doi: 10.1023/A:1006024231305. [DOI] [PubMed] [Google Scholar]
- Woods A, Munday MR, Scott J, Yang X, Carlson M, Carling D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J Biol Chem. 1994;269:19509–19515. [PubMed] [Google Scholar]
- Schwachtje J, Minchin EHP, Jahnke S, van Dongen TJ, Schittko U, Baldwin TI. SNF1-related kinases allow plants to tolerate herbivory by allocating carbon to roots. Proc Natl Acad Sci USA. 2006;103:12935–12940. doi: 10.1073/pnas.0602316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooley PW, Grau CR. Identification and quantitative characterization of rate-reducing resistance to Phytophthora megasperma f.sp. glycinea in soybean seedlings. Phytopathology. 1982;72:727–733. [Google Scholar]
- Njiti VN, Gray LE, Lightfoot DA. Rate-reducing resistance to Fusarium solani f.sp. phaseoli underlies field resistance to soybean sudden death syndrome (SDS) Crop Sci. 1997;37:1–12. [Google Scholar]
- Yu I, Fengler KA, Clough SJ, Bent AF. Identification of Arabidopsis mutants exhibiting an altered hypersensitive response in gene-for-gene disease resistance. Mol Plant Microbe Interact. 2000;13:277–286. doi: 10.1094/MPMI.2000.13.3.277. [DOI] [PubMed] [Google Scholar]
- Gebhardt C, Valkonen JP. Organization of genes controlling disease resistance in the potato genome. Annu Rev Phytopathol. 2001;39:79–102. doi: 10.1146/annurev.phyto.39.1.79. [DOI] [PubMed] [Google Scholar]
- Piffanelli P, Zhou F, Casais C, Orme J, Jarosch B, Schaffrath U, Collins NC, Panstruga R, Schulze-Lefert P. The barley MLO modulator of defense and cell death is responsive to biotic and abiotic stress stimuli. Plant Physiol. 2002;129:1076–1085. doi: 10.1104/pp.010954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultheiss H, Dechert C, Kogel KH, Huckelhoven Functional analysis of barley RAC/ROP G-protein family members in susceptibility to the powdery mildew fungus. Plant J. 2003;36:589–601. doi: 10.1046/j.1365-313X.2003.01905.x. [DOI] [PubMed] [Google Scholar]
- Aoki T, O'Donnell K, Homma Y, Lattanzi AR. Sudden death syndrome of soybean is caused by two morphologically and phylogenetically distinct species within the Fusarium solani species complex – F. virguliforme in North America and F. tucumaniae in South America. Mycologia. 2003;95:660–684. doi: 10.2307/3761942. [DOI] [PubMed] [Google Scholar]
- Jin H, Hartman GL, Nickell CD, Widholm JM. Characterization and purification of a phytotoxin produced by Fusarium solani, the causal agent of soybean sudden death syndrome. Phytopathology. 1996;86:277–282. doi: 10.1094/Phyto-86-277. [DOI] [Google Scholar]
- Achenbach LA, Patrick J, Gary L. Use of RAPD markers as a diagnostic tool for the identification of Fusarium solani isolates that cause soybean sudden death syndrome. Plant Disease. 1999;80:1228–1232. [Google Scholar]
- Wrather JA, Sleper DA, Steven WE, Shannon JG, Wilson RF. Planting date and cultivar effects on soybean yield, seed quality, and Phomopsis sp. seed infection. Plant Dis. 2003;87:529–532. doi: 10.1094/PDIS.2003.87.5.529. [DOI] [PubMed] [Google Scholar]
- Shoemaker R, Keim P, Vodkin L, Retzel E, Clifton SW, Waterson R, Smoller D, Coryell V, Khanna A, Erpelding J, Gai X, Brendel V, Raph-Schmidt C, Shoop EG, Vielweber CJ, Schmatz M, Pape D, Bowers Y, Theising B, Martin J, Dante M, Wylie T, Granger C. A compilation of soybean ESTs: generation and analysis. Genome. 2002;45:329–338. doi: 10.1139/g01-150. [DOI] [PubMed] [Google Scholar]
- Pevsner J. Bioinformatics and Functional Genomics. New York: John Wiley & Sons Inc; 2003. [Google Scholar]
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metaboli pathways and other biological processes. Plant J. 2004;37:914–939. doi: 10.1111/j.1365-313X.2004.02016.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yun B, Kwon Y, Hong J, Yoon J, Loake G. S-Nitrosylation: an emerging redox-based post-translational modification in plants. J Exp Bot. 2006;57:1777–1784. doi: 10.1093/jxb/erj211. [DOI] [PubMed] [Google Scholar]
- Geisler-Lee J, O'Toole N, Ammar R, Provart NJ, Millar AH, Geisler M. A Predicted Interactome for Arabidopsis. Plant Physiology. 2007;145:317–329. doi: 10.1104/pp.107.103465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwachtje J, Minchin PEH, Jahnke S, van Dongen JT, Schittko U, Balswin IT. SNF1-related kinases allow plants to tolerate herbivory by allocating carbon to roots. Proc Natl Acad Sci USA. 2006;103:12935–12940. doi: 10.1073/pnas.0602316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Tamura K, Nei M. MEGA 3: Integrated software for Molecular Evolutionary Genetics Analysis sequence alignment. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- Shadle GL, Wesley SV, Korth KL, Chen F, Lamb C, Dixon RA. Phenylpropanoid compounds and disease resistance in transgenic tobacco with altered expression of L-phenylalanine ammonia-lyase. Phytochemistry. 2003;64:153–161. doi: 10.1016/S0031-9422(03)00151-1. [DOI] [PubMed] [Google Scholar]
- Chong J, Baltz R, Schmitt C, Beffa R, Fritig B, Saindrenan P. Downregulation of a pathogen-responsive tobacco UDP-Glc:phenylpropanoid glucosyltransferase reduces scopoletin glucoside accumulation, enhances oxidative stress, and weakens virus resistance. Plant Cell. 2002;14:1093–1107. doi: 10.1105/tpc.010436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way HM, Kazan K, Mitter N, Goulter KC, Birch RG, Manners JM. Constitutive expression of a phenylalanine ammonia-lyase gene from Stylosanthes humilis in transgenic tobacco leads to enhanced disease resistance but impaired plant growth. Physiol and Mol Plant Pathol. 2002;60:275–282. [Google Scholar]
- Tsuji J, Jackson EP, Gage DA, Hammerschmidt R, Somerville SC. Phytoalexin accumulation in Arabidopsis thaliana during the hypersensitive reaction to Pseudomonas syringae Pv syringa. Plant Physiol. 1992;98:1304–1309. doi: 10.1104/pp.98.4.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buell CR, Somerville SC. Expression of defense-related and putative signaling genes during tolerant and susceptible interactions of Arabidopsis with Xanthomonas campestris pv. campestri. Mol Plant-Microbe Interact. 1995;8:435–443. [Google Scholar]
- Thibaud MC, Gineste S, Nussaume L, Robaglia C. Sucrose increases athogenesis-related PR-2 gene expression in Arabidopsis thaliana through an SAdependent but NPR1-independent signaling pathway. Plant Physiol Biochem. 2004;42:81–88. doi: 10.1016/j.plaphy.2003.10.012. [DOI] [PubMed] [Google Scholar]
- Gaudet AD, Laroche A, Yoshida M. Low temperature-wheat-fungal interactions: A carbohydrate connection. Physiologia Plantarum. 1999;106:437–444. doi: 10.1034/j.1399-3054.1999.106412.x. [DOI] [Google Scholar]
- Polge C, Thomas M. SNF1/AMPK/SnRK1 kinases, global regulators at the heart of energy control? Trends Plant Sci. 2007;12:20–28. doi: 10.1016/j.tplants.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Meyer R, Yuan J, Afzal J, Iqbal J, Zhu M, Garvey G, Lightfoot DA. Identification of Gsr1: A locus inferred to regulate gene expression in response to exogenous glutamine. Euphytica. 2006;151:291–302. doi: 10.1007/s10681-006-9149-9. [DOI] [Google Scholar]
- Njiti VN, Johnson JE, Torto TA, Gray LE, Lightfoot DA. Inoculum rate influences selection for field resistance to soybean sudden death syndrome in the greenhouse. Crop Science. 2001;41:1726–1731. [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TG. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gp penalties and weight matrix choice. Nucl Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]