

Abstract
Cartilage extracellular matrix (ECM) contains large amounts of proteoglycans made of a protein core decorated by highly sulfated sugar chains, the glycosaminoglycans (GAGs). GAGs desulfation, a necessary step for their degradation, is exerted by sulfatases that are activated by another enzyme, Sulfatase-Modifying Factor 1 (SUMF1), whose inactivation in humans leads to severe skeletal abnormalities. We show here that despite being expressed in both osteoblasts and chondrocytes Sumf1 does not affect osteoblast differentiation. Conversely, in chondrocytes it favors ECM production and autophagy and promotes proliferation and differentiation by limiting FGF signaling. Thus, proteoglycan desulfation is a critical regulator of chondrogenesis.
Keywords: Proteoglycan, desulfation, FGF, chondrocytes
Endochondral ossification, a multistep process responsible for embryonic bone formation and postnatal longitudinal bone growth (Kronenberg 2003), begins once mesenchymal cells condense, forming the blueprint of the future skeleton. Then, these mesenchymal cells differentiate into resting and proliferating chondrocytes that express Aggrecan and α1(II) Collagen while cells at the periphery express α1(I) Collagen and form the perichondrium. Subsequently, chondrocytes in the center of this structure further differentiate into hypertrophic chondrocytes expressing α1(X) Collagen but not Aggrecan and α1(II) Collagen. The extracellular matrix (ECM) secreted by hypertrophic chondrocytes allows vascular invasion, degradation of the calcified ECM, and initiation of osteogenesis. Thus, the ECM plays important functions during skeletal development (Olsen et al. 2000).
Alongside collagen fibrils the cartilagineous ECM contains large amounts of proteoglycans that protect these fibrils and provide resistance against compression. Proteoglycans are composed of a protein core to which one or more highly sulfated polysaccharide chains (glycosaminoglycan [GAG]) bind. The GAGs moiety of proteoglycans can regulate the distribution and binding ability of several signaling molecules, thereby influencing developmental processes. GAGs fulfill these functions according to their degree of sulfation (Esko and Selleck 2002; Perrimon and Hacker 2004) a process determined by two classes of intracellular enzymes: the sulfotransferases, that catalyze incorporation of sulfated groups into a nascent molecule, and the sulfatases, that remove them (Diez-Roux and Ballabio 2005; Bulow and Hobert 2006).
There are at least 17 different sulfatases in vertebrates, whose function is determined by their subcellular localization in the lysosomes, cell surface, or ER/Golgi, and by substrates specificity (Diez-Roux and Ballabio 2005). In addition, sulfatase becomes active following a post-translational formylglycination catalyzed by the enzyme Sulfatase-Modifying Factor 1 (Sumf1) (Cosma et al. 2003). To date, no other substrates besides the sulfatases have been identified for Sumf1 (Diez-Roux and Ballabio 2005); hence, it can be viewed as the master regulator of proteoglycan desulfation. Further underscoring the biological importance of Sumf1 and of proteoglycan desulfation a broad spectrum of diseases, most of them including skeletal abnormalities, results from loss of function mutations in various sulfatases or in Sumf1 itself (Diez-Roux and Ballabio 2005). In most of these diseases, called mucopolysaccharidoses, the lack of lysosomal sulfatases causes a block in GAG degradation and an intralysosomal GAG accumulation (Neufeld and Muenzer 2001).
In order to define the importance of proteoglycan desulfation during skeletogenesis we studied mice lacking Sumf1 (Settembre et al. 2007). We show here that although Sumf1 is expressed in osteoblasts during skeletogenesis, it has no overt function in osteoblasts. In contrast, Sumf1 and, as a result, proteoglycan desulfation, favors ECM production, chondrocyte autophagy, and promotes chondrocyte proliferation and differentiation. Genetic and biochemical evidence show that Sumf1 regulation of chondrocyte proliferation and differentiation occurs by inhibiting FGF signaling.
Results and Discussion
Sumf1 expression during skeletogenesis
To determine whether Sumf1 is an appropriate tool to study the role(s) of proteoglycan desulfation during skeletogenesis we examined its pattern of expression using a LacZ gene inserted in the Sumf1 locus (Settembre et al. 2007). β-Galactosidase staining of whole embryos showed that Sumf1 starts to be expressed in skeletal elements as early as embryonic day 14.5 (E14.5) (Fig. 1A–C). Histological analysis revealed that this staining was equally strong in proliferating chondrocytes of the growth plate and in osteoblasts of the developing bones, while staining in hypertrophic chondrocytes was weak (Fig. 1D); in situ hybridization analysis of Sumf1 expression while confirming that Sumf1 was expressed in proliferating chondrocytes and osteoblasts also revealed a strong expression in hypertrophic chondrocytes (Fig. 1E). Thus, Sumf1 is a molecular marker of all cell types in the developing skeleton.
Figure 1.
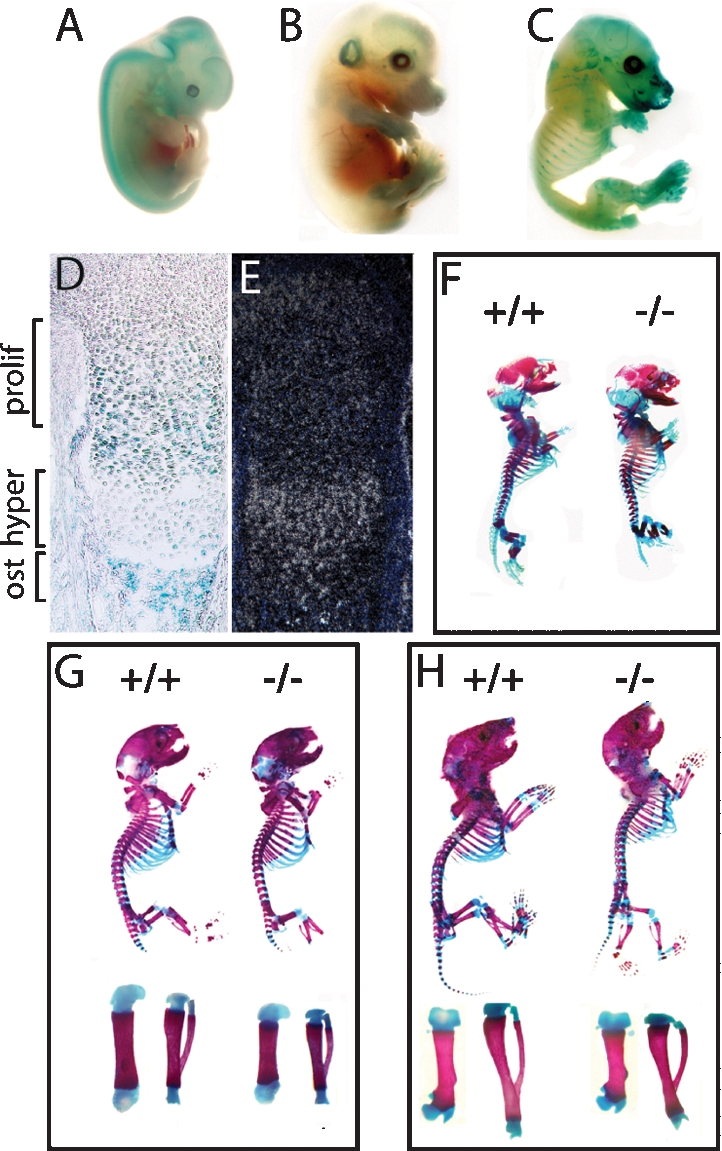
Skeletal development in Sumf1−/− embryos and mice. (A–C) X-gal-stained Sumf1+/− E12.5, E14.5, and E16.5 embryos. LacZ was expressed in all cartilagineous elements starting at E14.5. (D) Femoral growth plate section showing LacZ staining in proliferating chondrocytes and osteoblasts. (E) In situ hybridization of Sumf1 in E16.5 growth plate. (F–H) Alcian blue/alizarin red staining of E16.5 (F), newborn (G), and P4 (H) wild-type and Sumf1−/− embryos and mice. (G,H) Femur and tibia magnification of newborn and P4 Sumf1−/− and wild-type mice.
Proteoglycan desulfation affects endochondral ossification
As a first approach to study the importance of proteoglycan desulfation during skeletogenesis we analyzed skeletal preparations of Sumf1−/− embryos and mice stained with alcian blue for unmineralized cartilagineous ECMs and alizarin red for mineralized cartilagineous and bony ECMs. E14.5 Sumf1−/− embryos were indistinguishable from wild-type littermates (data not shown), an observation consistent with the fact that Sumf1 is not express in the developing skeleton before this stage. In contrast, examination of E16.5 embryos revealed that femurs were significantly shorter in Sumf1−/− than in wild-type embryos (3.0 mm wild type vs. 2.8 mm Sumf1−/−; n = 3, P < 0.05). This dwarfism, that worsened in newborn Sumf1−/− mice (4.2 mm wild type vs. 3.8 mm Sumf1−/−; n = 5, P < 0.01) was generalized, as every bone analyzed (femurs, vertebrae, mandibles, components of the skull) were shorter in Sumf1−/− than in wild-type littermates (Fig. 1F,G; data not shown). These morphological abnormalities that were not caused by a decrease in insulin growth factor 1 production (Supplemental Fig. 1A), illustrate that the extent of proteoglycan desulfation is an important determinant of the growth of most skeletal elements during embryonic development.
Proteoglycan desulfation affects growth plate cellularity
Next, we performed histological and gene expression analyses. Consistent with the absence of morphological defects in E14.5 Sumf1−/− embryos there was no difference in the morphology of the various populations of chondrocytes between Sumf1−/− and wild-type embryos at that stage and expression of α1(II) Collagen, a marker of proliferating chondrocytes, and of α1(X) Collagen, a marker of hypertrophic chondrocytes, was similar in Sumf1−/− and wild-type embryos at E14.5 (Fig. 2A). At E16.5 the zone of proliferating chondrocytes, determined by cell morphology and the extent of α1(II) Collagen expression, was significantly shorter in Sumf1−/− than in wild-type embryos (Fig. 2B). The intensity of α1(X) Collagen expression was also weaker in E16.5 Sumf1−/− than in wild-type embryos (Fig. 2B). At birth, the classical columnar organization of the proliferating chondrocytes was lost in the Sumf1−/− growth plate and expression of α1(II) Collagen and of α1(X) Collagen was again noticeably weaker in mutant than in wild-type growth plate (Fig. 2C). Remarkably, despite the high level of Sumf1 expression in osteoblasts, bone formation defined by the presence of bone trabeculae and expression of α1(I) Collagen was not affected by Sumf1 inactivation (Fig. 2B,C,D). Taken together, these results suggest that inactivation of Sumf1—i.e., inhibition of proteoglycan desulfation—affects more severely chondrogenesis than osteogenesis during development.
Figure 2.

Sumf1−/− deficiency affects chondrogenesis but not osteogenesis. (A) Hematoxylin/eosin (H/E) staining and in situ hybridization analysis of femoral sections. No differences between Sumf1−/− and wild-type growth plates are observed in E14.5 embryos, while in E16.5 (B) and newborn (C) there is a progressive shortening of both proliferative and hypertrophic area in mutant mice. Expression of α1(II) and α1(X) Collagen was decreased in newborn Sumf1−/− compared with wild-type mice. α1(I) Collagen expression was similar in wild-type and Sumf1−/− samples at all stages analyzed. (D) Von Kossa-Van Gieson staining of femoral sections showed normal mineralization (black staining) in E16.5 and newborn Sumf1−/− mice. Magnification: A–C, 100×, D, 50×.
We next investigated whether the weaker expression of α1(II) and α1(X) Collagen in the Sumf1−/− growth plate was reflecting a decrease in chondrocyte number. We observed that there was a significant decrease in chondrocyte number in Sumf1−/− growth plates at E16.5 and at postnatal day 0 (P0) (Fig. 3A), indicating that the extent of proteoglycan desulfation regulates, in ways studied below, chondrocyte survival and/or proliferation during endochondral ossification.
Figure 3.

Defective ECM in Sumf1−/− growth plate. (A) Chondrocyte number in the growth plate proliferative zone. Values are the mean ± SD. Student’s test (*) P < 0.05. (B) Toluidin blue staining of chondrocostal cartilage of newborn Sumf1−/− and wild-type littermates. Note the presence of cytoplasmatic vacuolization in Sumf1−/− chondrocytes. (C) EM analysis of E14.5 Sumf1−/− and wild-type chondrocytes showing no evidence of lysosomal vacuolization in Sumf1−/− embryos. (D,E) Cytoplasmatic vacuoles in E16.5 (D) and newborn (E) Sumf1−/− chondrocytes filled with amorphous material (GAGs) and partially degraded collagen fiber (E, inset). (F) Sumf1−/− osteoblasts are less affected than chondrocytes by lysosomal vacuolization. (G) ECM proteoglycan produced by Sumf1−/− and wild-type primary chondrocytes labeled with 3H-glucosamine and Na35SO4. ECM amount was estimated by 3H and 35S incorporation and normalized for cells number. (H) Decreased alcian blue staining in P7 Sumf1−/− compared with wild-type growth plate.
Proteoglycan desulfation influences ECM production and turnover
Since abnormalities in proteoglycan desulfation result in lysosomal defects in several tissues in vertebrates (Neufeld and Muenzer 2001), we asked if lysosomal function was affected in Sumf1−/− chondrocytes and/or osteoblasts.
Electron microscopic (EM) analysis of wild-type and mutant growth plates did not reveal any morphological difference between Sumf1−/− and wild-type chondrocytes at E14.5 (Fig. 3C), while in E16.5 Sumf1−/− embryos and newborn mice the cytoplasm of chondrocytes was filled with vacuolar structures that had characteristic appearance of lysosomes filled with GAGs as seen in cells from patients affected by mucopolysaccharidoses (Neufeld and Muenzer 2001) (Fig. 3D,E, inset; Supplemental Fig. 1B). Lysosomal vacuolization in osteoblasts was not nearly as dramatic, further suggesting that proteoglycan desulfation is a more important process during chondrogenesis than osteogenesis (Fig. 3F).
This impaired lysosomal degradation of GAGs in chondrocytes led us to ask whether the amount of GAGs present in the ECM was affected by the absence of Sumf1 in chondrocytes by measuring the amount of GAGs in wild-type and in Sumf1−/− chondrocytes. As hypothesized, mutant chondrocytes produced 30% fewer membrane-bound GAGs than wild-type chondrocytes; GAG secretion in the medium was also reduced by 50% (Fig. 3G). This explained why staining with alcian blue, a dye marking proteoglycans, was significantly weaker in Sumf1−/− than in wild-type growth plates (Fig. 3H). EM analysis of ER and Golgi did not shown any morphological differences between wild-type and Sumf1−/− chondrocytes at E16.5 or P0, indicating that the reduced amount of ECM is not caused by a block in the GAGs biosynthetic pathway (Supplemental Fig. 1C).
Chondrocyte autophagy is hampered in the absence of proteoglycan desulfation
We next asked what were the mechanisms leading the decrease in chondrocyte number in Sumf1−/− growth plate. There was no overt increase in chondrocyte apoptosis in Sumf1−/− mice before or after birth (data not shown). BrdU incorporation did not reveal any significant difference in chondrocyte proliferation between Sumf1−/− and wild-type embryos at E14.5 and E16.5 either, although chondrocyte proliferation was significantly decreased after birth in Sumf1−/− mice (Fig. 4A). Thus, at least two mechanisms explain the paucity of chondrocytes in the Sumf1−/− growth plate: a decrease in chondrocyte proliferation beyond birth, and another one, yet to be determined, during embryonic development.
Figure 4.

Abnormal autophagy in Sumf1−/− chondrocytes. (A) Number of BrdU positive cells present in the proliferative zone of the growth plate. Values are the mean ± SD. Student’s test (*) P < 0.05. (B) EM analysis of newborn chondrochostal cartilage revealed the presence of autophagosomes in wild-type chondrocytes. (Boxed inset) Note the double membrane vesicles surrounding a portion of cytoplasm (arrows). (C) Chondrocyte from newborn Sumf1−/− showing more autophagosomes (AV) surrounded by enlarged lysosomes (L). (D) Confocal microscopy analysis of Sumf1−/−;GFP-LC3 and wt;GFP-LC3 growth plate. In GFP-LC3 chondrocytes (left) the GFP fluorescence was more diffused throughout the cytoplasm while in Sumf1−/−;GFP-LC3 it was aggregated in cytoplasmatic dots (right). (E) Western blot analysis showing a 2.5-fold increase in LC3II level in newborn Sumf1−/− chondrocytes. No difference was observed in osteoblasts. Values shown are means of triplicate experiments. (F) Abnormal autophagy in Sumf1−/− chondrocytes during serum and nutrient starvation. Wild-type and Sumf1−/− chondrocytes were starved for the indicated period of time, harvested, and subjected to LC3 immunoblotting. Sumf1−/− chondrocytes and wild type stimulated with Baf presented an increased amount of LC3II compared with wild-type chondrocytes at all time points analyzed. (G) ATP amount is decreased in wild-type chondrocytes when autophagy is inhibited with Baf. Sumf1−/− chondrocytes displayed a lower level of ATP compared with wild-type and Baf treatment did not affected significantly ATP concentration. (H) Wild-type and Sumf1−/− chondrocytes were cultured in serum and glucose-free medium for 2 d. Wild-type chondrocytes were also treated with baf and 3-methyladenine, another inhibitor of autophagy. Cell viability was monitored after 12 h, 24 h, and 48 h. Error bars represent SEM. Student’s test (*) P < 0.05; (**)P < 0.01.
Macroautophagy (hereafter referred as autophagy) is a lysosomal process of cellular self-digestion required for turnover of cytoplasmic structure and for producing energy in conditions of cellular starvation (Mizushima 2007). Since chondrocytes reside in a hypoxic and avascular environment (Schipani et al. 2001) we hypothesized that autophagy may be a physiological means allowing them to survive, and that impairment in this process could cause chondrocyte death in Sumf1−/− embryos. Indeed, EM analysis of E16.5 embryos and newborn wild-type mice identified vacuoles containing a portion of cytoplasm and degraded organelles, a feature characteristic of autophagy (Mizushima 2007) (Fig. 4B, arrows). Remarkably, the number of autophagosome vacuoles (autophagosomes) was greatly increased in mutant chondrocytes (Fig. 4C).
To establish rigorously that the number of autophagosomes was increased in Sumf1−/− chondrocytes we used in vivo and biochemical approaches. First, we generated Sumf1−/− mice harboring in all cells a transgene expressing a GFP-tagged LC3 protein (Mizushima et al. 2004). During autophagy the free cytoplasmatic LC3I isoform of the LC3 protein is converted into LC3II, a specific marker of autophagosomes (Kabeya et al. 2000). Consistent with an increase in the number of autophagosomes GFP immunoreactivity was noticeably stronger in Sumf1−/−;GFP-LC3 than in GFP-LC3 chondrocytes (Supplemental Fig. 1D). Moreover, while the GFP fluorescence was distributed throughout the cytoplasm in wild-type chondrocytes, it was clustered in autophagosomes in Sumf1−/−;GFP-LC3 chondrocytes (Fig. 4D, arrows). Western blot quantification of LC3II proteins showed also a 2.5-fold increase in Sumf1−/− compared with wild-type growth plates (Fig. 4E). This was not observed in Sumf1−/− osteoblasts (Fig. 4E), further illustrating that these cells are not overtly influenced by the absence of Sumf1.
Since GAGs accumulation can inhibit lysosomal function (Li et al. 2004), we tested whether this increase in the number of autophagosomes in Sumf1−/− chondrocytes was reflecting a failure of lysosome to digest autophagosomes by treating wild-type and Sumf1−/− chondrocytes with bafilomycin (Baf), an inhibitor of the autophagosome–lysosome fusion (Yamamoto et al. 1998), a necessary step during autophagy (Mizushima 2007). Baf treatment of wild-type chondrocytes triggered an increase in autophagosome accumulation as measured by LC3-II level and a significant decrease of energy (ATP) production (Fig. 4F,G). In contrast, in Sumf1−/− chondrocyte the level of LC3II was higher than in wild-type chondrocytes, and Baf did not decrease energy production significantly (Fig. 4F,G). These results support the notion that autophagy is impaired in Sumf1−/− chondrocytes. To determine if this impairment of autophagy could lead to chondrocyte death, wild-type and mutant chondrocytes were cultured in glucose-free and serum-free medium, a condition in which autophagy is required for energy production and cell survival (Lum et al. 2005). LC3-II immunoreactivity decreased rapidly upon nutrient starvation, suggesting that autophagosomes were efficiently digested by lysosomes in wild-type but not in Sumf1−/− chondrocytes (Fig. 4F). Moreover, when measured with colorimetric assay, cell viability was decreased in Sumf1−/− chondrocytes compared with wild-type cells (Fig. 4H).
In summary, in vivo and cell-based assays establish that autophagy is used by wild-type chondrocytes to produce energy and suggest that disruption of intralysosomal GAG digestion impairs autophagy in Sumf1−/− chondrocytes. This impairment in turn leads to cell death.
Proteoglycan desulfation regulates chondrocyte proliferation and differentiation
To explain the decrease in chondrocyte proliferation noted in Sumf1−/− mice we asked whether Sumf1 expression modulates growth factor signaling. We first looked at Indian Hedgehog (Ihh), since its signaling is influenced in vivo by GAGs (Koziel et al. 2004), but failed to detect any difference in the level of expression of Ihh or of its receptor Patched (Ptch) between Sumf1−/− and wild-type chondrocytes at E16.5, P0, or P4 (Supplemental Fig. 2; data not shown). These observations indicate that Sumf1 deletion does not affect overtly Ihh signaling during endochondral ossification.
The defect in chondrocyte proliferation and differentiation noticed in Sumf1−/− mice was reminiscent of what is observed in mice harboring an increase in FGF signaling (Ornitz and Marie 2002). To determine if FGF signaling was increased we generated Sumf1−/− mice lacking one copy of Fgf18, a known regulator of chondrocyte proliferation (Liu et al. 2002). Several lines of evidence indicate that proteoglycan desulfation indeed regulates, directly or indirectly, FGF signaling.
First, skeletal preparation showed that removing one allele of Fgf18 rescued Sumf1−/− mice short stature at P0 as determined by femur length (Fig. 5A [arrowhead], quantification in E). Second, chondrocyte proliferation measured by BrdU incorporation, which was reduced by 20% in the P0 Sumf1−/−, was indistinguishable from wild-type in Sumf1−/−;Fgf18+/− P0 mice (Fig. 5B, quantification in E). As a result, the zone of hypertrophic chondrocytes was larger, and there was a significant increase of growth plate cellularity in Sumf1−/−;Fgf18+/− compared with Sumf1−/− mice (Fig. 5E). Third α1(II) Collagen and α1(X) Collagen expression was restored almost to its normal intensity in Sumf1−/−;Fgf18+/− newborn mice (Fig. 5C,D). Together, these data support the notion that the absence of Sumf1 results in an increase in FGF signaling, explaining the decrease in chondrocyte proliferation observed in Sumf1−/− mice. Of note, removing one Fgf18 allele did not normalize the chondrocyte number (Fig. 5E), indicating that the abnormal autophagy seen in the Sumf1−/− embryos is not secondary to the increase in FGF signaling.
Figure 5.

Sumf1 regulates FGF18 activity during endochondral ossification. (A) H/E staining of femurs showing shortening of Sumf1−/− bone length (arrowheads) and its rescue in Sumf1−/−;fgf18+/− mice. Brdu staining (B) and in situ expression of α1(II) Collagen (C) and α1(X) Collagen (D) in wild-type (left), Sumf1−/− (middle), and Sumf1−/−;fgf18+/− mice. (E) Quantification of femoral length, BrdU index, and cell number in newborn wild-type, Sumf1−/−, and Sumf1−/−;fgf18+/− mice. At least three mice were analyzed per each genotype. Error bars represent SEM. Student’s test (*) P < 0.05; (**) P < 0.01. (F) Primary chondrocytes from wild-type and Sumf1−/− mice treated with Fgf18 (20 ng/mL) for the indicated period of time. Note the more sustained phosophorylation of ERK and 70S6k in Sumf1−/− than in wild-type chondrocytes following Fgf18 treatment.
To identify the cause(s) of this increased FGF signaling in Sumf1−/− chondrocytes we first compared Fgf18 expression in Sumf1−/− and wild-type newborn mice but failed to detect any significant difference (Supplemental Fig. 3), thus ruling out that Sumf1 is not a regulator of Fgf18 expression. It has been proposed that proteoglycans, through their degree of sulfation, modulate the affinity of FGFs for their cognate receptors (Bishop et al. 2007). To determine if this was the case in Sumf1−/− mice we stimulated wild-type and Sumf1−/− primary chondrocytes with FGF18 and measured activation of FGF signaling. Both the ERK kinase and the ribosomal protein S6, which are involved in FGF signaling (Murakami et al. 2004), were more phosphorylated in Sumf1−/− than in wild-type chondrocytes (Fig. 5F). These results support the hypothesis that Sumf1, and more generally proteoglycan desulfation, influence FGF signaling in chondrocytes during skeletal development.
Sulfatases catalyze desulfation of the GAGs moiety of proteoglycans in the intracellular and extracellular space. These enzymes are substrates of Sumf1 whose only known function is to activate sulfatases. By studying endochondral ossification in Sumf1−/− mice we show that proteoglycan desulfation regulates several aspects of chondrocyte biology. Indeed, the block in proteoglycan desulfation caused by Sumf1−/− deletion severely decreases chondrocytes viability by hampering their capacity to generate enough energy through autophagy to survive in their avascular environment. Our data suggest that this defect in autophagy is caused, in part, by the engulfment of lysosomes by undigested GAGs that leads to an impairment of the autophagosome–lysosome fusion (Settembre et al. 2008).
Growth plate cartilage is a hypoxic structure in which the transcription factor Hif1α is required for chondrocytes survival. The fact that hypoxia stimulate autophagy (Mizushima 2007) suggests that autophagy could be one pathway through which Hif1α allows chondrocytes to survive. Moreover, chondrocytes starvation does not increase the autophagosome number as commonly observed in other cellular lines, suggesting that in this cell type autophagy is a constitutive rather than an adaptive pathway.
Our study also shows that proteoglycan desulfation is a negative regulator of Fgf18 signaling during endochondral ossification. This may be a direct consequence of the level of desulfation of certain proteoglycans such as heparan sulphate proteoglycan (HSPG) rather than a secondary effect of the engulfment of lysosomes with GAGs. Our results are in agreement with the notion that, in cell culture, ECM proteoglycans, and particularly HSPG bind members of FGF family (Bishop et al. 2007). It is likely that the two main sulfatases involved in this regulatory pathways are Sulf1 and Sulf2, which are strongly expressed in chondrocytes and whose substrate is HSPG (Lum et al. 2007). Together, our data show that proteoglycan desulfation eventually affects several aspects of chondrocyte biology during skeletal development not only by determining lysosomal function but also by modulating growth factor signaling.
Materials and methods
Animals
Sumf1−/−, Fgf18−/−, and GFP-LC3 transgenic mice were described previously (Liu et al. 2002; Mizushima et al. 2004; Settembre et al. 2007). Genotyping was performed by genomic PCR.
Skeletal preparation
Skeletons were fixed in 100% ethanol overnight and stained with alcian blue and alizarin red according to standard protocols. At least three mice of each genotype were analyzed per stage.
LacZ staining and BrdU labeling
Embryos were fixed in 1% paraformaldehyde, 0.2% glutalaldehyde, and stained overnight with X-Gal (Roche). Specimens were embedded in paraffin, sectioned at 6 μm, and counterstained with eosin (Sigma). Embryos were labeled with BrdU by injecting pregnant females with 500 μL of 10 mM BrdU 1 h before harvest. BrdU incorporation was detected using a Zymed BrdU staining kit (Invitrogen). Three to five embryos/mice were analyzed for each genotype and age group. Statistical significance was assessed by the Student’s test. ([*] P < 0.05; [**] P < 0.01).
Light and electron microscopy
Tibia were fixed in a 4% paraformaldehyde/1% glutaraldehyde fixative solution (pH 7.4), in 0.1 M sodium cacodylate buffer, dehydrated in ethanol, and embedded in plastic resin. Ultrathin sections (80 nm) were examined with a transmission electron microscope operated at 80 kV.
Cell cultures, Western blot analyses, and in situ hybridization
See the Supplemental Material.
Isolation of radiolabeled GAGs
Primary chondrocytes were cultured in presence of 10 μCi/mL Na35SO4 (25–40 Ci/mg) and 20 μCi/mL D-[6-3H]glucosamine (40 Ci/mmol) for 3 d in sulfate and glucose-free medium. GAG extraction, purification, and analysis were performed as described previously (Bame and Esko 1989).
Acknowledgments
We are indebted to Drs. D.M. Ornitz and N. Mizushima for providing Fgf18-deficient mice and GFP-LC3 transgenic mice. C.S thanks M.P. Cosma, P. Ducy, A. Fraldi, and G. Parenti for their critical reading of the manuscript. C.S is the recipient of a predoctoral fellowship of the European School of Molecular Medicine (SEMM). This work was supported by grants from the NIH to G.K.
Footnotes
Supplemental material is available at http://www.genesdev.org.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.1711308.
References
- Bame K.J., Esko J.D. Undersulfated heparan sulfate in a Chinese hamster ovary cell mutant defective in heparan sulfate N-sulfotransferase. J. Biol. Chem. 1989;264:8059–8065. [PubMed] [Google Scholar]
- Bishop J.R., Schuksz M., Esko J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Bulow H.E., Hobert O. The molecular diversity of glycosaminoglycans shapes animal development. Annu. Rev. Cell Dev. Biol. 2006;22:375–407. doi: 10.1146/annurev.cellbio.22.010605.093433. [DOI] [PubMed] [Google Scholar]
- Cosma M.P., Pepe S., Annunziata I., Newbold R.F., Grompe M., Parenti G., Ballabio A. The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell. 2003;113:445–456. doi: 10.1016/s0092-8674(03)00348-9. [DOI] [PubMed] [Google Scholar]
- Diez-Roux G., Ballabio A. Sulfatases and human disease. Annu. Rev. Genomics Hum. Genet. 2005;6:355–379. doi: 10.1146/annurev.genom.6.080604.162334. [DOI] [PubMed] [Google Scholar]
- Esko J.D., Selleck S.B. Order out of chaos: Assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziel L., Kunath M., Kelly O.G., Vortkamp A. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev. Cell. 2004;6:801–813. doi: 10.1016/j.devcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Kronenberg H.M. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Li Z., Yasuda Y., Li W., Bogyo M., Katz N., Gordon R.E., Fields G.B., Bromme D. Regulation of collagenase activities of human cathepsins by glycosaminoglycans. J. Biol. Chem. 2004;279:5470–5479. doi: 10.1074/jbc.M310349200. [DOI] [PubMed] [Google Scholar]
- Liu Z., Xu J., Colvin J.S., Ornitz D.M. Coordination of chondrogenesis and osteogenesis by fibroblast growth factor 18. Genes & Dev. 2002;16:859–869. doi: 10.1101/gad.965602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum J.J., Bauer D.E., Kong M., Harris M.H., Li C., Lindsten T., Thompson C.B. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Lum D.H., Tan J., Rosen S.D., Werb Z. Gene trap disruption of the mouse heparan sulfate 6-O-endosulfatase gene, Sulf2. Mol. Cell. Biol. 2007;27:678–688. doi: 10.1128/MCB.01279-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: Process and function. Genes & Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S., Balmes G., McKinney S., Zhang Z., Givol D., de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes & Dev. 2004;18:290–305. doi: 10.1101/gad.1179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld E., Muenzer J. The metabolic and molecular bases of inherited disease. McGraw-Hill; New York: 2001. [Google Scholar]
- Olsen B.J., Reginato A.M., Wang W. Bone development. Annu. Rev. Cell Dev. Biol. 2000;16:191–220. doi: 10.1146/annurev.cellbio.16.1.191. [DOI] [PubMed] [Google Scholar]
- Ornitz D.M., Marie P.J. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes & Dev. 2002;16:1446–1465. doi: 10.1101/gad.990702. [DOI] [PubMed] [Google Scholar]
- Perrimon N., Hacker U.2004Wingless, hedgehog and heparan sulfate proteoglycans Development 131 :2509–2511. [DOI] [PubMed] [Google Scholar]
- Schipani E., Ryan H.E., Didrickson S., Kobayashi T., Knight M., Johnson R.S. Hypoxia in cartilage: HIF-1α is essential for chondrocyte growth arrest and survival. Genes & Dev. 2001;15:2865–2876. doi: 10.1101/gad.934301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Annunziata I., Spampanato C., Zarcone D., Cobellis G., Nusco E., Zito E., Tacchetti C., Cosma M.P., Ballabio A. Systemic inflammation and neurodegeneration in a mouse model of multiple sulfatase deficiency. Proc. Natl. Acad. Sci. 2007;104:4506–4511. doi: 10.1073/pnas.0700382104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C., Fraldi A., Jahreiss L., Spampanato C., Venturi C., Medina D., de Pablo R., Tacchetti C., Rubinsztein D.C., Ballabio A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008;17:119–129. doi: 10.1093/hmg/ddm289. [DOI] [PubMed] [Google Scholar]
- Yamamoto A., Tagawa Y., Yoshimori T., Moriyama Y., Masaki R., Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]