

Abstract
Verification of candidate biomarker proteins in blood is typically done using multiple reaction monitoring (MRM) of peptides by LC-MS/MS on triple quadrupole MS systems. MRM assay development for each protein requires significant time and cost, much of which is likely to be of little value if the candidate biomarker is below the detection limit in blood or a false positive in the original discovery data. Here we present a new technology, accurate inclusion mass screening (AIMS), designed to provide a bridge from unbiased discovery to MS-based targeted assay development. Masses on the software inclusion list are monitored in each scan on the Orbitrap MS system, and MS/MS spectra for sequence confirmation are acquired only when a peptide from the list is detected with both the correct accurate mass and charge state. The AIMS experiment confirms that a given peptide (and thus the protein from which it is derived) is present in the plasma. Throughput of the method is sufficient to qualify up to a hundred proteins/week. The sensitivity of AIMS is similar to MRM on a triple quadrupole MS system using optimized sample preparation methods (low tens of ng/ml in plasma), and MS/MS data from the AIMS experiments on the Orbitrap can be directly used to configure MRM assays. The method was shown to be at least 4-fold more efficient at detecting peptides of interest than undirected LC-MS/MS experiments using the same instrumentation, and relative quantitation information can be obtained by AIMS in case versus control experiments. Detection by AIMS ensures that a quantitative MRM-based assay can be configured for that protein. The method has the potential to qualify large number of biomarker candidates based on their detection in plasma prior to committing to the time- and resource-intensive steps of establishing a quantitative assay.
The clinical importance of biomarkers has been well established (1). They can be used to screen healthy individuals to predict predisposition to disease or to detect the presence of asymptomatic disease (e.g. prostate-specific antigen for prostate cancer). Biomarkers can also be used to monitor the stage or severity of diseases, guide molecularly targeted therapy (e.g. Her2Neu status for breast cancer therapy), and assess response to treatment. In the biopharmaceutical industry biomarkers are used to stratify patients for initial assessment of new drug therapies and as surrogate end points in early phase drug trials. However, despite intensified interest and investment by both industry and academia, the rate of introduction of new protein biomarkers of disease has fallen dramatically to fewer than one/year since 1998 (2). The reasons for this serious discrepancy have been explored in several recent studies (3, 4) and reflect the long and difficult path that a biomarker must take from initial candidate “discovery” to clinical use (5).
In an attempt to increase the likelihood that MS-discovered biomarker candidates will advance into clinical validation, we have presented a notional pipeline for biomarker discovery that emphasizes the need to “verify” candidate markers coming from discovery efforts (5). A verification phase is essential for a number of reasons, all relating to the uncertainty of whether detected differences in candidate abundance are real and disease-specific, and whether the protein can be robustly quantified in blood. Abundant protein depletion combined with multidimensional fractionation at the peptide level prior to analysis of tissue, proximal fluid, or other biofluid samples by LC-MS/MS now routinely provides confident identification of thousands of proteins, hundreds of which appear to vary significantly between case and control samples. However, because these LC-MS/MS experiments are almost always underpowered with respect to the numbers of samples analyzed relative to the very high dimensionality of the data, many of the discoveries are likely to be false positives. In addition, many of these studies are carried out in tissues or proximal fluids (e.g. ovarian cyst fluid or nipple aspirate fluid). The presence of each candidate biomarker protein at detectable levels in blood (or other readily accessible biofluids such as urine) is thus not guaranteed and must be experimentally determined. Finally, proteomics data are not the only source of potential candidate biomarkers. The open literature and publicly available genomics data sets are among the other viable sources for candidates worthy of additional scrutiny (6, 7). Clearly it is essential to have a robust method to help prioritize these lengthy candidate lists before investing the significant resources required for quantitative assay development.
The goal of verification is to quantify protein biomarker candidates in a sufficient number of samples to determine which are best able, singly or in small combinations, to discriminate the presence or absence of disease. Measurements must have sufficient assay sensitivity, specificity, and precision to achieve this task. The core methodology for these measurements is stable isotope dilution-multiple reaction monitoring (MRM)1-mass spectrometry (8, 9). Stable isotope dilution-MRM-MS is predicated on measurement of “signature peptides” that uniquely represent the protein candidates of interest. In general, MRM-based assay development starts with three to five synthetic peptides per protein and ends with one to two configured assays for any given protein. The cost of configuring such an MRM-based assay for a protein is on the order of a few thousand dollars for reagents, instrument, and personnel costs. We estimate that the rate at which these assays can be developed is on the order of 100/year for an expert laboratory. Thus cost and capacity are significant impediments to using the MRM approach as a screen to determine which of the candidate proteins merit study in larger numbers of patient samples. In principal antibody-based measurements could be used. However, the required ELISA-grade antibodies with sufficient sensitivity and specificity exist for only a small number of the potential candidate biomarker proteins, and the cost of developing such an assay for a poorly credentialed candidate is prohibitive. An intermediate step between discovery and verification is clearly needed.
Here we present a method that uses targeted high mass accuracy MS to detect sequence-verified peptides from candidate protein biomarkers in plasma and translate them to MRM assays for quantitative verification of the candidate biomarkers. The technology, that we refer to as accurate inclusion mass screening or AIMS, is derived from earlier studies in our laboratory on the use of targeted MS methods to search for computationally predicted proteins in cell lysates (10) and to identify proteins whose levels were observed to change using pattern-based discovery methods (11). We describe the AIMS technology and evaluate its suitability for targeted detection of proteins in blood. AIMS can rapidly triage lengthy lists of candidate biomarker proteins, focusing effort on that subset detectable in blood at the low ng/ml range or above. Peptide precursors and fragment ions generated by this technique are subsequently used to facilitate development of sensitive and quantitative MRM assays on low resolution triple quadrupole MS systems. The potential of AIMS to serve as a bridge between unbiased discovery and quantitative assay development for verification studies is demonstrated.
EXPERIMENTAL PROCEDURES
Uniformly 15N-Labeled Proteins—
Uniformly 15N-labeled proteins were the kind gift of Dr. Lee Makowski of Argonne National Laboratory. Details concerning their cloning, expression, and purification are available upon request. 41 proteins (Supplement 1 includes the identities, accessions, and sequences) were used in this study. Analysis of these proteins by mass spectrometry demonstrated that >99.5 atom % of nitrogens were 15N.
Construction of Test Mixtures—
Human female plasma was depleted of abundant proteins using an IgY-12 high capacity LC10 column (12.7 × 79 mm; GenWay Biotech, Inc., San Diego, CA) according to the manufacturer's directions. Two sets of mixtures were constructed by addition of 15N-labeled proteins into the depleted plasma.
The first set of mixtures was for a small scale pilot study (“Small Mixes”). The compositions of these mixtures are shown in Table I under “Results.” The four proteins used were betaine-homocysteine methyltransferase (BHMT; NCBI accession number gi|4502407), protein phosphatase 1G (PP1G; NCBI accession number gi|4505999), aldo-keto reductase family 1, member C1 (AKR1C1; NCBI accession number gi|5453543), and calreticulin (CALR; NCBI accession number gi|4757900).
Table I.
AIMS detection of targeted 15N-labeled proteins
Concentration values are the concentrations of the 15N-labeled proteins shown spiked into human female plasma. See “Footnote 1” for abbreviation translations.
| Protein | Small Mix 1 concentration | Peptides found Mix 1 | Small Mix 2 concentration | Peptides found Mix 2 | Total unique peptides found |
|---|---|---|---|---|---|
| ng/ml | ng/ml | ||||
| BHMT | 10 | 1 | 100 | 6 | 6 |
| PP1G | 33 | 1 | 66 | 6 | 6 |
| AKR1C1 | 66 | 2 | 33 | 1 | 3 |
| CALR | 100 | 3 | 10 | 0 | 3 |
The second set of mixtures (“Big Mixes”) utilized 40 of the 41 proteins shown in Supplement 1 (protein phosphatase 1G was omitted). Four Big Mixes were constructed, each with all 40 proteins at the following levels: 10, 30, 60, and 100 ng/ml. These were the final concentrations in the background of depleted human female plasma that had been concentrated using Vivaspin 15R concentrators (5000-dalton molecular mass cutoff; Vivascience, Hannover, Germany). The overall protein concentration after depletion and concentration was 3.3 mg/ml.
100 μl of each mixture was denatured with 6 m urea, reduced with 20 mm DTT for 30 min at 37 °C, and then alkylated with 50 mm iodoacetamide for 30 min at room temperature in the dark. The urea concentration was diluted 10-fold with water prior to the digestion by trypsin (sequencing grade modified; Promega, Madison, WI) overnight at 37 °C with gentle shaking at a protein-to-trypsin ratio of 50:1 (w/w). Digests were desalted using Oasis HLB 1-cc (30 mg) reversed phase cartridges (Waters, Milford, MA) and vacuum-concentrated to dryness.
SCX Fractionation of Mixtures—
Each lyophilized digest was resuspended in 25% acetonitrile, 0.05% formic acid (pH 3.0). The entire digest was subjected to strong cation exchange chromatography under the following conditions: ThermoFisher BioBasic SCX 1 × 250-mm column; buffer A, 25% acetonitrile, 0.05% formic acid (pH 3.0); buffer B, 25% acetonitrile, 250 mm ammonium formate, 4% formic acid (pH 3.0); flow rate, 50 μl/min; gradient program, hold 0% B 0–15 min, 0–20% B from 15 to 35 min, 20–40% B from 35 to 45 min, 40–100% B from 45 to 55 min, hold 100% B from 55 to 65 min, 100–0% B from 65 to 67 min, and hold 0% B from 67 to 85 min. Fractions were collected for 5-min intervals. Fractions were vacuum-concentrated to dryness. The 10 fractions collected during the 15–65-min window were further analyzed. Mixtures were processed sequentially starting with the lowest spike-in level and proceeding to the highest.
AIMS for Target Proteins and Peptides—
Directed LC-MS AIMS experiments were performed to detect peptides derived from the 15N-labeled proteins spiked into plasma. These experiments were accomplished by means of an “inclusion list” in the mass spectrometry instrument method. The inclusion list on the Orbitrap (software release LTQ Orbitrap 2.4, July 19, 2007) can have at least 2000 entries. The inclusion list consists of m/z, z, and retention time values for tryptic peptides predicted from the target proteins (retention time is optional and was not used in our experiments). The values placed on the list were governed by a few simple rules. 1) The peptide is fully tryptic and contains no missed cleavages. 2) The peptide can assume charges from z = 2 up to z = 4 given sufficient basic residues to accommodate such a charge. 3) The m/z of the peptide at a given charge is between 300 and 1500. 4) No modifications are considered save for full 15N metabolic labeling and carbamidomethylation of cysteines, which was an intentional modification performed in our laboratory. 5) No retention time prediction is associated with a given peptide; it may be observed at any point during the LC-MS experiment. When a mass spectral peak is detected by the instrument during acquisition that satisfies the criteria of any entry on the inclusion list, an MS/MS spectrum is automatically obtained for the associated precursor ion. The targets and values of our inclusion lists are given in Supplement 3 (all values were derived from the MSDigest functionality of SpectrumMill Proteomics Workbench (Agilent)).
These experiments were performed on a ThermoFisher Scientific Orbitrap mass spectrometer coupled to an Agilent 1100 nano-LC pump fitted with a 13.5-cm × 75-μm column pulled and packed in house with Reprosil C18-AQ 3-μm packing material. The following conditions were used for chromatography: Buffer A, 0.1% formic acid; Buffer B, 0.1% formic acid in 90% acetonitrile; gradient and flow program, 3% B from 0 to 20 min at 0.8 μl/min, 3–7.5% B from 20 to 22 min at 0.2 μl/min, 7.5–50% B from 22 to 60 min at 0.2 μl/min, 50–90% B from 60 to 65 min at 0.2 μl/min, 90% B from 65 to 75 min at 0.8 μl/min, and 3% B from 75 to 90 min at 0.8 μl/min. Amounts equivalent to 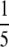 and
and 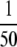 of an SCX fraction were injected onto the column in a volume of 2 μl.
of an SCX fraction were injected onto the column in a volume of 2 μl.
The following parameters were used for inclusion list-dependent acquisition on the Orbitrap mass spectrometer. A single Orbitrap MS scan from m/z 300 to 1500 at resolution 60,000 was followed by up to three ion trap MS/MS scans at the normal scan rate. The top three most abundant precursors from the inclusion list (if present) were targeted for MS/MS spectrum acquisition over the course of a 90-min experiment. Preview mode and charge state screening were enabled for selection of precursors. The m/z tolerance around targeted precursors was ±7.5 ppm. Dynamic exclusion was also enabled with a repeat count of 2 with a repeat duration of 10 s and an exclusion duration of 20 s. Again the m/z tolerance for dynamic exclusion was ±7.5 ppm. The intensity threshold for triggering of a detected peak was set to 100, and collision energy was specified at 28% for all list members.
All raw data were analyzed using SpectrumMill Proteomics Workbench (Agilent and in-house). MS/MS peak lists were extracted from raw data files using default Orbitrap parameters and a 45-s scan merge tolerance. Extracted spectra were searched against the human RefSeq database (downloaded June 2007) that was verified to contain each of the 41 potentially targeted proteins. Search parameters were as follows: all masses considered monoisotopic; parent mass tolerance, ±0.035 Da; fragment mass tolerance, ±0.7 Da; no missed cleavages; and fixed modifications: carbamidomethylation, 15N/14N mixture. Autovalidation was performed as follows: protein grouping mode, aggregate score of 7.8 and all files grouped together; peptide rules: z = 2 score >5, SPI > 50%, ΔRank1–2 > 2; z = 3 score >6, SPI > 50%, ΔRank1–2 > 2; and z = 4 score >7, SPI > 55%, ΔRank1–2 > 2.
MRM-based Follow-up of AIMS Experiments—
MRM assays were configured for all peptides detected by the AIMS experiments. MS/MS spectra of each peptide were obtained during AIMS using the linear ion trap front end of the Orbitrap. The fragments observed in the trap were used to define the transitions to monitor on the triple quadrupole instrument. For peptides detected at lower charge state (typically 2 or 3) on Orbitrap but containing extra basic amino acids, the MRM assay was configured for both charge states (3 or 4). If Orbitrap MS/MS spectra were not observed for a particular charge state, the transitions were selected taking into account existing MS/MS for another charge state and also applying common rules for peptide fragmentation. To maximize specificity, five to seven transitions were selected and monitored for each peptide. MRM transition lists specific to each SCX fraction were constructed based on the Orbitrap results.
MRM experiments were performed on a 4000 Q Trap Hybrid triple quadrupole/linear ion trap mass spectrometer coupled to a Tempo LC system (Applied Biosystems, Framingham, MA). SCX fractions were diluted 1:3 and 1:10 prior to MRM analysis, and full loop injection of 1 μl of each dilution was performed on PicoFrit columns (75-μm inner diameter, 10-μm tip opening; New Objective, Woburn, MA) packed in house with 12–13 cm of ReproSil-Pur C18-AQ 3-μm reversed phase resin (Dr. Maisch, GmbH). Sample was eluted at 300 nl/min with a gradient of 3–20% solvent B for 3 min, 20–55% solvent B for 35 min, and 55–80% solvent B for 3 min. Data acquisition was performed with an ion spray voltage of 2200 V, curtain gas of 20 p.s.i., nebulizer gas of 3 p.s.i., and an interface heating temperature of 150 °C. MRM parameters were defined as follows. 1) Declustering potential was 50 for the precursors < m/z = 400, 70 for 400 < m/z < 800, and 100 for m/z > 800. 2) Collision energy was calculated for each precursor using equations in the MRM builder of Analyst software. 3) Collision cell exit potential was set to 10 for all of the transitions. 4) A dwell time of 10–12 ms was used to maximize number of transitions per MRM method. To avoid exceeding the cycle time of 1 s, most of the SCX fractions were analyzed using two or more MRM methods.
Data analysis was done manually. Extracted ion chromatograms (XICs) for all transitions of a given peptide were plotted for the 100 ng/ml spiked sample. Confirmation of the presence of a peptide was based upon the co-elution of all transitions and by the relative ratio of transitions for those peptides with MS/MS spectra obtained on the Orbitrap. XICs for all transitions were compared across mixtures to confirm the presence of a peptide at a lower concentration of spiked protein. Relative retention time information from the AIMS experiments combined with the differences in intensity level of the XICs in multiple mixtures helped to identify peptides despite matrix interferences observed even with five to seven transitions monitored.
RESULTS
The practical goals of the AIMS experiment in the context of biomarker discovery and verification are to 1) triage long lists of biomarker candidates obtained from proteomics discovery experiments or integrative genomics approaches down to those that are readily detectable in clinically relevant samples and 2) provide information of value for developing quantitative assays for the detected candidate biomarker proteins using MRM on triple quadrupole instrumentation. To accomplish these goals, an inclusion list is populated with the accurate masses of signature peptides derived from the high priority candidate proteins (Fig. 1). Masses on the inclusion list are monitored in each scan on the Orbitrap MS system, and MS/MS spectra are acquired only when a peptide from the list is detected with both the correct accurate mass and charge state. Peptide MS/MS data are automatically interpreted using standard software. The MS/MS fragmentation observed for signature peptides by AIMS is subsequently used to populate the parent-fragment transition list used to configure sensitive MRM-based quantification assays.
Fig. 1.

Schematic of AIMS-to-MRM experiment. In this approach, serum or plasma is depleted of abundant proteins and minimally processed with optional standard addition of labeled proteins. In parallel, a panel of candidate proteins for screening is analyzed to generate a list of target (m/z, z) pairs for AIMS. Note that some peptides may be represented by more than one target on the list if multiple charge states are expected. The boxed portion of the schematic illustrates the selectivity power of accurate mass screening and how the MS/MS spectra are integrated into MRM assay development. The shaded pink area indicates the ±7.5-ppm window used in the study.
Small Scale Pilot Study—
To initially test the utility of our methodology we constructed simple mixtures of four proteins and spiked them into human female plasma that had been depleted of abundant proteins at concentrations typically associated with candidate biomarkers (see Table I for concentrations). To aid in our evaluation and avoid possible confusion with endogenous levels of these proteins, we used recombinant proteins that were uniformly 15N-labeled (see “Experimental Procedures”). The mixtures were digested, coarsely fractionated by strong cation exchange, and then subjected to LC-MS/MS analysis using AIMS (Fig. 1).
We were able to detect multiple peptides from all four of the targeted proteins (Table I) on the Orbitrap via AIMS. All validated peptide spectra for these proteins were interpreted as being fully 15N-labeled, and no peptides were detected from these proteins without 15N labels. Labeling provided additional confidence in the interpretation of the data for these proof-of-principle experiments but is not necessary for routine application of AIMS. There were 155 unique (m/z, z) pairs (representing 102 unique peptide sequences) targeted in this study (note that z = 4 peptides were not targeted in the initial study). Every (m/z, z) pair was triggered at least once during the analysis of the fractions. This was not unexpected given the background of other human proteins present in the sample that could give rise to peptides satisfying the requirements for triggering within the tolerances specified (see “Discussion” for more details on this). In fact, peptides derived from proteins other than those targeted were also detected (including peptides from other common serum proteins like complements, transferrin, and apolipoproteins). However, the four targeted proteins had the top four aggregate protein scores (sum of all peptide scores) in our analysis by SpectrumMill, and no other validated peptides were interpreted as bearing the 15N label.
One may also note that the number of peptides detected per protein seems to scale with protein abundance. This has long been recognized in proteomics experiments and might be used as the basis for crude relative quantification. An even better approach is to leverage the high resolution MS spectra obtained on the Orbitrap to generate XICs for each peptide detected. These chromatograms serve as a more truly quantitative measure of peptide abundance and could be used to approximate the difference in candidate biomarker levels between case and control samples in verification experiments. For instance, in our small scale study, there were two peptides that were identified at multiple spike levels. In both of these cases, XIC-based quantification yielded an estimate of the relative difference between the two levels that was within 2-fold of the actual difference (data not shown).
Large Scale Targeting of Protein Targets for Biomarker Verification—
We sought to extend this method to triage much larger numbers of proteins expected to be submitted as candidate biomarkers from discovery studies. To test this, we made mixtures of 40 15N-labeled proteins and used the exact same methodology described above. Summaries of the small and large scale experiments are given in Table II and Fig. 2. We were able to detect peptides for 35 of the 40 proteins on our target list by AIMS. Again we could be quite confident in our identifications because of the incorporation of 15N labels and subsequent interpretation of peptide spectra as bearing this label. Given the expanded target list, we expected (and found) many additional proteins besides the ones targeted. In fact, 551 of the 645 peptides that we targeted had potentially conflicting peptides at the same (m/z, z) values given the tolerances we selected (over 13,500 in silico digested peptides in the human RefSeq database matched to one or more (m/z, z) pairs). Nevertheless the proteins that we targeted clustered near the top of the list of all proteins when rank-ordered by aggregate protein score (Fig. 3) with 32 of the 35 proteins detected in the top half of the ranked list.
Table II.
Summary of experimental results from small and large scale AIMS scouting experiments
Results were aggregated from both the 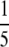 and
and 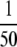 SCX fraction amount-injected levels. n/c, not calculated. See Fig. 2 for an illustration of the meaning of “triggered” and “untriggered” targets and peptides.
SCX fraction amount-injected levels. n/c, not calculated. See Fig. 2 for an illustration of the meaning of “triggered” and “untriggered” targets and peptides.
| Small | Large | |
|---|---|---|
| Experimental setup | ||
| Number of proteins targeted | 4 | 40 |
| Number of (m/z, z) pairs on list | 155 | 1,161 |
| Number of unique peptides | 102 | 645 |
| Peptides with potential interference | n/c | 551 |
| Triggering data | ||
| Number of triggers | ∼19,000 | ∼86,000 |
| Targets triggered | 155 | 1,047 |
| Peptides triggered | 102 | 636 |
| Targets untriggered | 0 | 114 |
| Peptides untriggered | 0 | 9 |
| Success rates | ||
| Total target proteins detected/attempted (cumulative) | 4/4 | 35/40 |
| At 10 ng/ml | n/c | 3 |
| At 30 ng/ml | n/c | 18 |
| At 60 ng/ml | n/c | 25 |
| At 100 ng/ml | n/c | 35 |
| Other proteins detected | 20 | 340 |
Fig. 2.

Illustration of the meaning of triggered and untriggered targets and peptides in Table II. Boxed entries are triggered. Xed boxes are untriggered.
Fig. 3.

Target proteins rank near the top of all identified proteins. Tick marks show the rank list position of targeted proteins.
Given the specificity afforded by the 15N labeling, we could explore the space of peptide and protein interpretation scores in SpectrumMill that would generate valid protein identifications. We were surprised that scores lower than those accepted in routine practice could generate perfectly reasonable peptide and protein interpretation. Of course, lowering score thresholds also introduces increased numbers of false positive identifications. This is evidenced by the observation that peptides belonging to proteins other than those targeted in the experiment were being interpreted as bearing the 15N label. In total, 306 spectra were interpreted as 145 unique 15N-labeled peptides at the score thresholds that we chose. 196 of the 306 spectra (64%) represented 87 unique peptides and were unique to proteins we had targeted in the experiment. The average SpectrumMill interpretation score was 11.8 for these spectra where the maximum obtainable score is 25, and a score of 13 is considered unambiguous for single peptide identifications. The root mean square (RMS) mass error for the peptides was 5.2 ppm. The remaining 110 spectra represented 58 unique peptides but had an average SpectrumMill interpretation score of only 7.4 and a root mean square mass error of 16.5 ppm. Clearly we could utilize tighter mass tolerances during both acquisition and interpretation to reduce the number of false positives (see “Discussion”). However, the central message is that even low interpretation scores for peptides targeted in an AIMS experiment are likely to be correct. Any number of factors could also help to explain the spurious 15N-labeled interpretations, such as impure proteins spiked into the experiment that carry some other 15N-labeled proteins from the expression system. Indeed a significant number of these spectra scored higher against peptides from proteins that might be present in the bacterial expression system when re-searched against a database containing bacterial proteins even while retaining the 15N label in their interpretation. Another significant fraction of these “spurious” 15N-labeled spectra scored higher against other peptide sequences not bearing the 15N label when certain tolerances (such as number of missed cleavages allowed and database size) were relaxed.
We also evaluated the usefulness of the method as compared with undirected data-dependent sampling. We chose to resample the set of fractions with 100 ng/ml spikes (at the 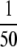 amount-injected level; see “Experimental Procedures”) using an equivalent Top 3 methodology but without use of an inclusion list. This condition was probably the most favorable for regular data-dependent sampling in that the spikes were at their highest level, but the column was not overloaded with peptides as it was at the
amount-injected level; see “Experimental Procedures”) using an equivalent Top 3 methodology but without use of an inclusion list. This condition was probably the most favorable for regular data-dependent sampling in that the spikes were at their highest level, but the column was not overloaded with peptides as it was at the 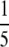 amount-injected level (data not shown). In the inclusion list experiment, 67 spectra corresponding to 28 of the targeted proteins were detected, whereas only 15 spectra corresponding to 11 of the targeted peptides were found in the undirected data-dependent experiment. The >400% increase in the number of spectra detected is especially important considering the goal of downstream MRM assay configuration. Obtaining more spectra for the relevant peptides offers more empirical information about the fragments one could expect to use for transitions. As we demonstrate below, the fragments observed in the screening experiments are highly concordant with those observable in the MRM experiments.
amount-injected level (data not shown). In the inclusion list experiment, 67 spectra corresponding to 28 of the targeted proteins were detected, whereas only 15 spectra corresponding to 11 of the targeted peptides were found in the undirected data-dependent experiment. The >400% increase in the number of spectra detected is especially important considering the goal of downstream MRM assay configuration. Obtaining more spectra for the relevant peptides offers more empirical information about the fragments one could expect to use for transitions. As we demonstrate below, the fragments observed in the screening experiments are highly concordant with those observable in the MRM experiments.
Rapid Configuration and Evaluation of MRM Assays—
Successful MRM assay configuration is highly dependent upon signature peptides derived from target proteins by enzymatic digestion. Selection of signature peptides takes into account their observed and/or predicted LC retention, sequence homology (i.e. uniqueness), molecular weight, and predicted or observed charge state with preference for moderately hydrophobic peptides likely to produce doubly or triply charged ions in the detectable mass range of the mass spectrometer. Although priority is generally given to peptides detected in unbiased discovery experiments, peptides can also be selected from in silico analysis of the protein sequence, a step that is necessary if the candidate protein was obtained from sources other than proteomics experiments. Our usual goal in peptide selection is to synthesize three to five peptides per target protein both because different peptides from the same protein can vary widely in their MS response and recovery from sample processing, and because of the high potential for interference in plasma. The necessity of multiple signature peptides per target protein increases (but does not guarantee) the likelihood that a specific and sensitive MRM assay will be developed. However, despite the careful and thoughtful selection process, extensive optimization of MRM parameters and evaluation of LC retention, chromatographic peak shape, and interferences from the plasma matrix are required for each signature peptide before the final MRM assay is constructed. Depending upon the number of MRM assays being configured for a given verification study, this process could range from days to several weeks.
For MRM assay configuration here, we relied exclusively on experimental data collected in the AIMS experiments. From our small scale pilot study, we were able to immediately configure MRM assays on an Applied Biosystems 4000 Q Trap triple quadrupole mass spectrometer for the 18 peptides observed on the Orbitrap (Table I). This is a significant reduction in complexity (102 → 18 targets) for MRM assay formulation. We selected the most intense fragment ions from the ion trap MS/MS spectra as the basis for transitions on the 4000 Q Trap, choosing up to seven/peptide where possible (Table III and supplemental data). Aliquots of the same sample fractions used for screening on the Orbitrap were injected on the 4000 Q Trap without further sample preparation (although dilutions of concentrated fractions were made in some cases). We only needed to analyze fractions where the peptides had already been detected on the Orbitrap, reducing the total instrument time. We also tailored MRM methods to each fraction, allowing more transitions for fewer peptides that were known to be present in the fraction, again based on Orbitrap data. It should be noted that several of the peptides observed might not be considered “canonical” MRM assay peptides (such as those containing cysteine or methionine, or very long peptides), but we view this as an advantage of letting the empirical data guide the experimental process.
Table III.
Peptides, observed transitions, and average limits of detection for peptides detected via AIMS by MRM-based methods
“Attempted” transitions were derived from the Orbitrap linear ion trap MS/MS spectra, and “observed” transitions were the number actually seen on the 4000 Q Trap. The average (Avg) peptide limit of detection (LOD) was computed by adding the individual peptide limits of detection for each protein and dividing by the total number of peptides observed for that protein.
| Small scale experiment
| ||||
|---|---|---|---|---|
| Protein name | Number of peptides detected | Total transitions observed vs. attempted at 100a or 66b ng/ml | Total transitions observed vs. attempted at 10a or 33b ng/ml | Avg peptide LOD |
| ng/ml | ||||
| AKRF1C1a | 2 | 8/11 | 8/11 | 33 |
| BHMTb | 5 | 24/29 | 24/29 | 10 |
| Calreticulin precursora | 2 | 10/13 | 10/13 | 10 |
| PP1Gb | 6 | 29/36 | 25/36 | 33 |
| Large scale experiment
| ||||
|---|---|---|---|---|
| Protein name | Number of peptides detected | Total transitions observed vs. attempted at 100 ng/ml | Total transitions observed vs. attempted at 10 ng/ml | Avg peptide LOD |
| ng/ml | ||||
| ADP-ribosylation factor 6 | 1 | 6/6 | 6/6 | 10 |
| AKRF1C1 | 4 | 19/24 | 11/24 | 32.5 |
| AKRF1C2 | 2 | 8/12 | 2/12 | 55 |
| PDZ and LIM domain 1 (elfin) | 3 | 11/15 | 4/15 | 40 |
| S100 calcium-binding protein A1 | 2 | 9/11 | 6/11 | 10 |
| S100 calcium-binding protein, β | 3 | 15/15 | 14/15 | 10 |
| Annexin I | 5 | 25/30 | 14/30 | 46 |
| Annexin IV | 2 | 9/12 | 9/12 | 10 |
| Brain creatine kinase | 3 | 12/15 | 8/15 | 40 |
| Calreticulin precursor | 3 | 13/20 | 2/20 | 40 |
| Chromogranin A precursor | 5 | 23/30 | 16/30 | 28 |
| Cystatin B | 1 | 6/6 | 5/6 | 10 |
| Enolase 2 | 3 | 10/17 | 2/17 | 70 |
| Fibroblast growth factor 1 (acidic) isoform 1 precursor | 1 | 5/5 | 3/5 | 10 |
| Fructose-bisphosphate aldolase C | 2 | 9/12 | 1/12 | 55 |
| Glucose-phosphate isomerase | 6 | 28/36 | 27/36 | 10 |
| Glutathione S-transferase M1 isoform 1 | 1 | 5/5 | 1/5 | 10 |
| Glutathione S-transferase M2 | 1 | 5/6 | 5/6 | 10 |
| Glutathione S-transferase M3 | 3 | 13/16 | 10/16 | 40 |
| Growth arrest and DNA damage-inducible, γ | 2 | 11/11 | 11/11 | 10 |
| Heat shock 27-kDa protein 1 | 4 | 19/23 | 8/23 | 55 |
| Inhibitor of DNA binding 2 | 1 | 5/5 | 4/5 | 10 |
| Interleukin 18 proprotein | 1 | 4/5 | 4/5 | 10 |
| Non-metastatic cells 1, protein (NM23A) isoform b | 3 | 12/16 | 12/16 | 10 |
| Peroxiredoxin 2 isoform a | 2 | 10/11 | 7/11 | 10 |
| Phosphoserine aminotransferase isoform 1 | 3 | 15/16 | 13/16 | 10 |
| Ser (or Cys) proteinase inhibitor, clade B, member 3 | 1 | 4/5 | 0/5 | 100 |
| Spermidine/spermine N1-acetyltransferase | 2 | 8/11 | 5/11 | 55 |
| Superoxide dismutase 1, soluble | 2 | 10/10 | 6/10 | 10 |
| Transgelin | 1 | 3/5 | 2/5 | 10 |
| Tyr 3-/Trp 5-monooxygenase activation protein, ε | 4 | 21/21 | 16/21 | 10 |
| Ubiquitin-conjugating enzyme E2C isoform 2 | 2 | 10/10 | 10/10 | 10 |
| Ubiquitin-conjugating enzyme E2I | 4 | 23/24 | 20/24 | 10 |
| Ubiquitin-conjugating enzyme E2N | 6 | 28/31 | 27/31 | 10 |
15 of the 18 peptides detected in the small scale pilot experiment on the Orbitrap generated suitable MRM assay performance on the 4000 Q Trap. “Suitable assay performance” is defined by the ability to observe multiple transitions from a peptide that unambiguously identify the target with a signal-to-noise ratio of 3 or more. In the absence of a stable isotopically labeled analog of each peptide, true quantitative assays could not be configured. We were able to achieve an increase in sensitivity in many cases by porting the assay to the MRM platform as shown in Table III (and supplemental data). Strikingly there was a high degree of concordance between the MS/MS fragmentation spectra obtained in the linear ion trap of the Orbitrap and the transitions we were able to observe in the 4000 Q Trap. For the 15 peptides that we were able to observe on the 4000 Q Trap, the majority of the product ions observed as fragments on the Orbitrap were also observed as transitions on the 4000 Q Trap (average of 80%/protein; median of 100%/protein). The CID spectra of only one peptide from the triple quadrupole MS system exhibited fewer than three of the seven product ions observed in the ion trap data.
We subsequently applied this methodology to the larger scale AIMS study. Using the MS/MS spectra acquired on the Orbitrap mass spectrometer, we selected five to seven transitions for 86 of the 87 peptides observed by AIMS (one was omitted by mistake; 645 peptides were initially targeted; Table II and Fig. 2). These peptides represent 34 of the 40 proteins spiked into plasma. Of 16 SCX fractions collected we analyzed only eight fractions by MRM representing the fractions where those peptides were detected by the AIMS approach on the Orbitrap mass spectrometer. We were able to detect all 86 peptides from 34 proteins spiked into plasma at 100 ng/ml and 70 peptides at 10 ng/ml by the MRM assay. 12 of the 16 peptides not detected at 10 ng/ml were detected at 30 ng/ml. The results again showed a high degree of correlation between the two different instrument platforms suggesting that we can accelerate MRM assay design by performing AIMS on an Orbitrap mass spectrometer and leverage the observed peptides and their fragmentation behavior to configure MRM assays on triple quadrupole MS systems.
Although not important for successful use of the AIMS technology, the use of uniformly 15N-labeled proteins enabled us to determine that the natural concentration of calreticulin in plasma is ∼100 ng/ml. We created XICs for the assumed naturally 14N-labeled version of the peptides FYALSASFEPFSNK, GTWIHPEIDNPEYSPDPSIYAYDNFGVLGLDLWQVK, and IDDPTDSKPEDWDKPEHIPDPDAK. The concentration was estimated based on comparison of the 14N XIC peak area with the 15N XIC peak area from the peptides derived from the protein spiked at 100 ng/ml. We initially made 15N and 14N XICs of all peptides targeted in the small scale study to see whether there were any detectable endogenous levels of the proteins. The calreticulin result was also borne out by MRM assays constructed for the “light” versions of these peptides in parallel with the 15N-labeled version.
DISCUSSION
The explosion of proteomics biomarker discovery studies and technologies has yielded lengthy lists of candidate markers in a wide variety of diseases. However, many if not all of these candidates remain just that, candidates, because of the daunting task of following up results in a systematic and efficient manner. The significant gap between unbiased discovery methods for candidate biomarker discovery and validation, recently characterized as a “tar pit” (7), has been described as the chief obstacle to effective proteomics biomarker development (5). In principle, antibody reagents, especially ELISA grade antibodies, could be used to bridge this gap, but highly specific and sensitive antibodies have yet to be generated for the vast majority of proteins and their modifications of interest. Clearly alternatives to immunodetection that have faster assay development time, higher capacity, and lower cost are needed.
The AIMS technology we developed is specifically designed to help bridge the gap between unbiased discovery experiments or integrative genomics approaches to candidate biomarker list development and verification of those candidates in blood or other complex biofluids (Fig. 4). Of course the AIMS methodology can also be used to confirm detection or do targeted discovery in any sample source (e.g. cells, proximal fluids, or tissue). Successful detection of peptides associated with candidate biomarkers in a sample matrix (like plasma) that might not be the same as that which was used for the discovery of the candidate (like primary tissue) is clearly a powerful winnowing agent. Thus we propose that those proteins that are readily observable in clinically accessible samples should be prioritized as biomarker candidates for further study. It is important to note that lack of observation of a candidate protein by AIMS does not mean that the candidate protein is not present in blood and potentially detectable by other means. Such candidates, particularly if additionally credentialed as potential biomarkers of that disease by literature precedent, observation in orthogonal data sets (e.g. microarray), etc., would be deprioritized for MRM assay development but might be prioritized higher for moving directly to protein or peptide immunoaffinity enrichment methods (7, 12–14).
Fig. 4.

Proposed biomarker pipeline and the location of AIMS. ID, identification; TP, throughput; Rel., relative.
We are beginning to use the AIMS approach to triage large number of biomarker candidates in ongoing biomarker studies of cancer and cardiovascular disease. We expect the optimized AIMS approach to evaluate hundreds of biomarker candidates per month. This throughput is compatible with the scale of lists typically generated in discovery experiments. Although the addition of AIMS to the pipeline initially incurs costs in time and resources, the overall efficiency gained by focusing efforts on detectable candidate biomarkers is likely to be substantial. The AIMS methodology can also be used to confirm detection in the original discovery samples (e.g. proximal fluids or tissue).
The AIMS approach is also suitable for any number of applications where coverage of a specific set of protein or peptide targets is desired. One could imagine using AIMS experiments for stable isotope labeling by amino acids in cell culture (SILAC)- or isobaric tags for relative and absolute quantitation (iTRAQ)-based quantitative methods, phosphopeptide mapping (or other post-translational modification mapping), or cross-link detection. We have previously shown its utility in verification of predicted mitochondrial proteins (10) in cell lysates, and Picotti et al. (15) have used a variation of the approach to identify minor proteolytic fragments in digests of simple protein mixtures.
The Orbitrap, with its high mass accuracy and high resolution, provides a sensitive and highly specific way to interrogate a sample for targeted analytes. To try to quantify what we mean by high specificity, in the present study we programmed the instrument to search for 1161 m/z targets (with specific z states) within a tolerance of 7.5 ppm. There are over 216,000 7.5-ppm “channels” in the m/z range 300–1500 that can be interrogated by AIMS, which means that we were interrogating only 0.5% of the m/z space. Coupled with associated charge state information for each potential precursor, this represents a tremendous amount of selectivity during data acquisition. Although in principle it is possible to implement an inclusion list-based approach on nearly all MS/MS-capable MS instrument types (a similar approach termed MIDAS (multiple reaction monitoring-initiated detection and sequencing) has been described for use on triple quadrupole MS systems (16)), the key to obtaining such high selectivity is the ability to trigger MS/MS in an accurate mass-dependent manner.
Selectivity of the method can be further increased by tightening the m/z tolerance required to trigger an MS/MS spectrum. Part of our motivation in using a relatively wide 7.5-ppm tolerance was the fact that we operated the instrument by using the “preview scan,” or first FT transient, of the Orbitrap to guide AIMS triggering. The preview scan may not be as accurate as the final scan (averaged accumulated transients), and thus we provided some latitude in triggering. We could disable preview scan-based triggering at the expense of overall duty cycle. As an illustration of the power of mass selectivity, there were over 13,500 potentially conflicting peptides in the human RefSeq database with the set of 1161 (m/z, z) pairs that we specified at 7.5 ppm. Fig. 5 shows how this value decreases with more stringent m/z tolerance. Further improvements in instrument mass accuracy and stability may likely allow for further tightening of mass tolerances without loss of information.
Fig. 5.

Number of interfering peptides in the human RefSeq database by ppm tolerance. This graph shows the number of peptides present in the human RefSeq database (as a function of ppm tolerance) that have (m/z, z) parameters that could cause a trigger event based on our list of 1161 specifically targeted (m/z, z) pairs. The ratio above each point shows the average number of untargeted peptides that could trigger an MS/MS scan to a specific target for each tolerance level.
Another factor governing the utility of this method is sample processing and throughput. Here we minimally fractionated digested plasma samples for the purpose of screening for spiked protein standards, simulating a biomarker discovery experiment and permitting limits of detection to be unambiguously determined. We were able to screen for 40 proteins at four different spike-in levels in duplicate (at 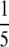 and
and 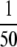 the amount of each fraction) in less than 1 week using the AIMS methods on the Orbitrap. As a result of this screening, MRM analysis could be applied selectively to fractions of interest resulting in the design and completion of MRM experiments in about another week. If desired, deeper coverage might be obtained by increasing the granularity of fractionation and/or tightening of m/z-based tolerances for data-dependent selection.
the amount of each fraction) in less than 1 week using the AIMS methods on the Orbitrap. As a result of this screening, MRM analysis could be applied selectively to fractions of interest resulting in the design and completion of MRM experiments in about another week. If desired, deeper coverage might be obtained by increasing the granularity of fractionation and/or tightening of m/z-based tolerances for data-dependent selection.
Observation in the high performance experiments generates information in the form of observed MS/MS fragments that is of considerable value for further follow-up via MRM assay. We and others have already demonstrated the sensitive and specific nature of MRM-based quantification in clinical sample settings. Its ability to multiplex quantification to tens or hundreds of analytes in a single experiment fulfills a requirement for rapid biomarker candidate verification. Until now, in the absence of experimental data, it has been common practice to select MRM assay peptide targets, both precursors and transitions, by inspection of the primary protein sequence plus educated guessing and use of computer prediction algorithms. For quantitative assay development, both heavy and light versions of the peptides are required to establish calibration curves (8). However, in cases where MRM is being used primarily to interrogate samples for the presence/absence of specific peptides (and modified versions thereof) rather than to derive quantitative information, synthetic peptides are not necessarily required if correct information regarding which fragments to evaluate by MRM is available. We have shown that the AIMS Orbitrap experiments can provide that information. The evidence gained by AIMS may also obviate the need to synthesize multiple peptides to configure a single successful MRM assay.
Given that the AIMS portion of the experiment is carried out on high resolution/high mass accuracy instrumentation, highly specific abundance information (in the form of XIC peak areas) can also be derived from full-scan MS spectra. This information might be used as the basis for label-free relative quantitation (11) that has come into wide practice with high performance mass spectrometers. An additional filter could be placed in the biomarker qualification pipeline in addition to mere detection in plasma: the requirement of a differential between individual or pooled case and control samples.
In conclusion, we demonstrated that AIMS is well suited to candidate biomarker triage as well as any other applications involving detection of predefined proteins or peptides in complex biological samples. The technology provides a rapid way to screen for the presence of large numbers of candidate proteins in complex samples and has sensitivity similar to MRM on triple quadrupole using optimized sample preparation methods (low tens of ng/ml in plasma). The throughput of the method is sufficient to qualify up to a hundred proteins/week. Most importantly, AIMS enables prioritization of large numbers of biomarker candidates based on their detection in plasma prior to committing to the time- and resource-intensive steps of establishing a quantitative assay. We believe the approach has significant potential to accelerate and increase the overall efficiency of biomarker discovery and verification studies. We hope that these methods will be an important link from basic science to clinical practice for the proteomics field as a whole.
Footnotes
Published, MCP Papers in Press, June 4, 2008, DOI 10.1074/mcp.M800218-MCP200
The abbreviations used are: MRM, multiple reaction monitoring; AIMS, accurate inclusion mass screening; BHMT, betaine-homocysteine methyltransferase; PP1G, protein phosphatase 1G; AKR1C1, aldo-keto reductase family 1, member C1; CALR, calreticulin; SCX, strong cation exchange; XIC, extracted ion chromatogram; SPI, scored peak intensity.
This work was supported, in whole or in part, by National Institutes of Health Grants 1U24 CA126476-02 from the NCI as part of the NCI's Clinical Proteomic Technologies Initiative and U01-HL081341 from the NHLBI (to S. A. C.). This work was also supported by the Entertainment Industry Foundation (to S. A. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.mcponline.org) contains supplemental material.
REFERENCES
- 1.Etzioni, R., Urban, N., Ramsey, S., McIntosh, M., Schwartz, S., Reid, B., Radich, J., Anderson, G., and Hartwell, L. ( 2003) The case for early detection. Nat. Rev. Cancer 3, 243–252 [DOI] [PubMed] [Google Scholar]
- 2.Anderson, N. L., and Anderson, N. G. ( 2002) The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics 1, 845–867 [DOI] [PubMed] [Google Scholar]
- 3.Anderson, N. L. ( 2005) The roles of multiple proteomic platforms in a pipeline for new diagnostics. Mol. Cell. Proteomics 4, 1441–1444 [DOI] [PubMed] [Google Scholar]
- 4.Gutman, S., and Kessler, L. G. ( 2006) The US Food and Drug Administration perspective on cancer biomarker development. Nat. Rev. Cancer 6, 565–571 [DOI] [PubMed] [Google Scholar]
- 5.Rifai, N., Gillette, M. A., and Carr, S. A. ( 2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 24, 971–983 [DOI] [PubMed] [Google Scholar]
- 6.Gortzak-Uzan, L., Ignatchenko, A., Evangelou, A. I., Agochiya, M., Brown, K. A., St Onge, P., Kireeva, I., Schmitt-Ulms, G., Brown, T. J., Murphy, J., Rosen, B., Shaw, P., Jurisica, I., and Kislinger, T. ( 2008) A proteome resource of ovarian cancer ascites: integrated proteomic and bioinformatic analyses to identify putative biomarkers. J. Proteome Res. 7, 339–351 [DOI] [PubMed] [Google Scholar]
- 7.Paulovich, A. G., Whiteaker, J. R., Hoofnagle, A. N., and Wang, P. ( 2008) The interface between biomarker discovery and clinical validation: the tar pit of the protein biomarker pipeline. Proteomics Clin. Appl., in press [DOI] [PMC free article] [PubMed]
- 8.Keshishian, H., Addona, T., Burgess, M., Kuhn, E., and Carr, S. A. ( 2007) Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 6, 2212–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whiteaker, J. R., Zhang, H., Zhao, L., Wang, P., Kelly-Spratt, K. S., Ivey, R. G., Piening, B. D., Feng, L. C., Kasarda, E., Gurley, K. E., Eng, J. K., Chodosh, L. A., Kemp, C. J., McIntosh, M. W., and Paulovich, A. G. ( 2007) Integrated pipeline for mass spectrometry-based discovery and confirmation of biomarkers demonstrated in a mouse model of breast cancer. J. Proteome Res. 6, 3962–3975 [DOI] [PubMed] [Google Scholar]
- 10.Calvo, S., Jain, M., Xie, X., Sheth, S. A., Chang, B., Goldberger, O. A., Spinazzola, A., Zeviani, M., Carr, S. A., and Mootha, V. K. ( 2006) Systematic identification of human mitochondrial disease genes through integrative genomics. Nat. Genet. 38, 576–582 [DOI] [PubMed] [Google Scholar]
- 11.Jaffe, J. D., Mani, D. R., Leptos, K. C., Church, G. M., Gillette, M. A., and Carr, S. A. ( 2006) PEPPeR, a platform for experimental proteomic pattern recognition. Mol. Cell. Proteomics 5, 1927–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson, N. L., Anderson, N. G., Haines, L. R., Hardie, D. B., Olafson, R. W., and Pearson, T. W. ( 2004) Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res. 3, 235–244 [DOI] [PubMed] [Google Scholar]
- 13.Berna, M., Ott, L., Engle, S., Watson, D., Solter, P., and Ackermann, B. ( 2008) Quantification of NTproBNP in rat serum using immunoprecipitation and LC/MS/MS: a biomarker of drug-induced cardiac hypertrophy. Anal. Chem. 80, 561–566 [DOI] [PubMed] [Google Scholar]
- 14.Whiteaker, J. R., Zhao, L., Zhang, H. Y., Feng, L. C., Piening, B. D., Anderson, L., and Paulovich, A. G. ( 2007) Antibody-based enrichment of peptides on magnetic beads for mass-spectrometry-based quantification of serum biomarkers. Anal. Biochem. 362, 44–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Picotti, P., Aebersold, R., and Domon, B. ( 2007) The implications of proteolytic background for shotgun proteomics. Mol. Cell. Proteomics 6, 1589–1598 [DOI] [PubMed] [Google Scholar]
- 16.Unwin, R. D., Griffiths, J. R., Leverentz, M. K., Grallert, A., Hagan, I. M., and Whetton, A. D. ( 2005) Multiple reaction monitoring to identify sites of protein phosphorylation with high sensitivity. Mol. Cell. Proteomics 4, 1134–1144 [DOI] [PubMed] [Google Scholar]