

Abstract
In response to DNA damage, the ATM protein kinase activates signal transduction pathways essential for coordinating cell cycle progression with DNA repair. In the human disease ataxia-telangiectasia, mutation of the ATM gene results in multiple cellular defects, including enhanced sensitivity to ionizing radiation. This phenotype highlights ATM as a potential target for novel inhibitors that could be used to enhance tumor cell sensitivity to radiotherapy. A targeted compound library was screened for potential inhibitors of the ATM kinase and CP466722 was identified. The compound is non-toxic and does not inhibit PI3K or PI3K-like protein kinase family members in cells. CP466722 inhibited cellular ATM-dependent phosphorylation events and disruption of ATM function resulted in characteristic cell cycle checkpoint defects. Inhibition of cellular ATM kinase activity was rapidly and completely reversed by removing CP466722. Interestingly, clonogenic survival assays demonstrated that transient inhibition of ATM is sufficient to sensitize cells to ionizing radiation and suggests that therapeutic radiosensitization may only require ATM inhibition for short periods of time. The ability of CP466722 to rapidly and reversibly regulate ATM activity provides a new tool to ask questions about ATM function that could not easily be addressed using genetic models or RNA interference technologies.
Keywords: ATM, ATR, Chk1, Chk2, DNA damage, IR-sensitivity, G2-arrest, Checkpoints
Introduction
Cells respond to DNA damage by activating a complex network of signal transduction pathways. These DNA damage response (DDR) pathways include sensors responsible for recognizing the genotoxic insult, transducers responsible for relaying/amplifying the signal, and effectors that induce the appropriate cellular response. Together these signaling cascades are responsible for coordinating cell cycle progression with DNA repair to facilitate maintenance of genomic stability (1).
The human autosomal recessive disease ataxia-telangiectasia (A-T) has a complex clinical phenotype including progressive cerebellar ataxia, oculocutaneous telangiectasias, immune deficiency, hypogonadism, growth retardation, premature aging, radiosensitivity and cancer predisposition (2). Cells obtained from A-T patients display DNA damage checkpoint defects in G1, S and G2-phases of the cell cycle, increased chromosomal instability, and radiosensitivity (3). The defective gene in A-T was identified as ATM (ataxia-telangiectasia, mutated) and encodes a 350kDa protein that belongs to the phosphatidylinositol 3-kinase family of proteins (4). Based on the phenotype displayed by A-T cells, it is not surprising that the ATM protein kinase has been characterized as a major regulator of the DDR pathways, along with the closely related family members ATR (Ataxia-Telangiectasia and Rad3-related kinase) and DNA-PK (DNA-dependent protein kinase) (5). In an unperturbed cell, ATM exists as an inactive dimer (or higher-order oligomer), but the introduction of DNA double strand breaks (DSBs) by ionizing radiation (IR) or other insults activates the ATM kinase by intermolecular autophosphorylation and dimer dissociation (6, 7). Once activated, ATM phosphorylates several downstream substrates that contribute to the proper regulation of IR-induced arrests in G1-phase (e.g. p53, Mdm2 and Chk2, refs: (8–12)), S-phase (e.g. Nbs1, Smc1, Brca1 and FancD2, refs: (13–16)), and G2-phase (e.g. Brca1 and Rad17, refs: (16, 17)) of the cell cycle.
Studies of cells that are functionally defective in different components of the DDR pathways (e.g. BRCA1/2, NBS1, ATM, Mre11, Fanconi Anemia proteins etc.) demonstrate cell cycle checkpoint defects, decreased ability to repair damaged DNA and an increased sensitivity to IR and other DNA damaging agents (2, 15, 18, 19). This latter observation highlights components of these DDR pathways as potential therapeutic targets for the development of small molecule inhibitors that could enhance the sensitivity of tumor cells to the cytotoxic effects of radio-/chemo-therapeutic agents (20–22).
The idea of using small molecule inhibitors to disrupt ATM function and sensitize tumor cells to radio-/chemo-therapeutic agents is not a novel concept (20–23). However, the most commonly used ATM inhibitors (caffeine and wortmannin) are neither specific nor useful in vivo, which has fueled an interest in identifying more specific and potent inhibitors and resulted in the recent identification of KU55933 (24). Using an in vitro kinase assay, we screened a targeted library of approximately 1500 small molecule compounds for potential ATM inhibitors and identified CP466722. This compound inhibited ATM kinase activity in vitro, but did not inhibit phosphatidylinositol-3-kinase (PI3K) or closely related PI3K-like protein kinase (PIKK) family members. The compound also inhibited the ATM signal transduction pathway in cells, disrupted cell cycle checkpoint function and sensitized tumor cells to IR. CP466722 is a rapidly reversible inhibitor of ATM function and transient exposure used in clonogenic survival assays suggests that short-term inhibition of ATM function is sufficient to sensitize cells to IR. This observation has potential implications for sensitization of tumor cells in vivo, where drug pharmacokinetics becomes an important consideration. Identification of CP466722 provides a novel chemical structure that inhibits ATM function in cells and can now be modified to generate more potent and specific agents that could be effective at enhancing tumor cell killing in vivo. In addition, the fact that ATM function can be rapidly turned-off and -on provides new opportunities for studying the ATM pathway.
Materials and Methods
Chemicals
Pfizer (Cambridge, MA) identified: 2-(6,7-dimethoxyquinazolin-4-yl)-5-(pyridin-2-yl)-2H-1,2,4-triazol-3-amine (CP466722). CP466722 (3mM), KU55933 & Wortmannin (10mM, Sigma, St Louis, MO) and Imatinib (10mM, TRC, On. Canada) were resuspended in DMSO. Caffeine (100mM, Sigma) was resuspended in dH2O. Aphidicolin (5mM, Sigma) was resuspended in methanol. Recombinant Human Insulin Growth Factor I (IGF-I, 0.1mg/ml, Invitrogen, Carlsbad, CA) was diluted in dH2O (0.1%w/v-BSA). Cells were routinely pretreated with: DMSO(vehicle control), CP466722 or Wortmannin(30min) and Caffeine or KU55933(1h).
Cell culture
Cells were plated (0.5 × 106cells/plate) 24h prior to treatment and maintained at 37°C in a humidified atmosphere (5%CO2). HeLa, normal diploid HFF, Mcf7 (ATCC, Manassas, VA), HFF(expressing hTERT, (25)) and A-T(GM02052 expressing hTERT, (26)) cells were cultured in DMEM (10%v/v-FBS, 10mM HEPES, penicillin 100units/ml and streptomycin 100ug/ml). Atm wild-type and deficient MEFs (Arf-deficient background) (27) were cultured in DMEM (10%v/v-FBS, 2mM Glutamine, 0.1mM NEAA, 55μM β-mercaptoethanol, penicillin 100units/ml and streptomycin 100ug/ml). Arf-deficient mouse pre-B cells expressing the human p185-BCR-ABL isoform (p185 +/Arf−/−, gift Dr R.Williams, St. Jude Children’s Research Hospital) were plated (1 × 106cells/ml) 24h prior to treatment and cultured as previously described (28). For radiation studies, IR from a 137Cs-source was delivered at a rate of 120cGy min−1 (Gammacell40-exactor, MDS Norton).
Cell viability
Cells were plated in triplicate (40,000cells/plate), incubated as required before culture media and trypsinsed cells were combined and viability determined: Vi-CELL™ XR cell viability analyzer (Beckman Coulter, Fullerton, CA).
Serum starvation and IGF-I stimulation
Cells were plated as normal, incubated for 24h before being removed from culture media, washed with and then cultured for 24h in normal or low serum DMEM (0.1%v/v-FBS, 10mM HEPES, penicillin 100units/ml and streptomycin 100ug/ml). Cells were stimulated by addition of IGF-I (10ng/ml) for 20min at 37°C prior to harvesting.
In vitro kinase assays
To screen for small molecule inhibitors of ATM kinase activity, an in vitro kinase assay was adapted (10, 29), and an ELISA assay developed which measured the phosphorylation status of the ATM downstream target p53. Recombinant GST-p53(1-101) and full-length Flag-tagged ATM & ATR were purified for use in the ELISA and in vitro kinase assays. Briefly, Nunc 96 well Maxisorp plates were coated overnight (4°C) with 2μg of purified, recombinant GST-p53(1-101) in PBS. All subsequent incubations were performed at room temperature. The plates were washed (0.05%v/v-Tween/PBS) before addition of purified recombinant full-length ATM kinase (30–60ng) in a final volume of 80μl of reaction buffer (20mM HEPES, 50mM NaCl2, 10mM MgCl2, 10mM MnCl2, 1mM DTT and 1μM ATP) in the presence or absence of compound. Compounds (10μM) were added to plates in duplicate and the kinase assay was incubated (90min). Plates were washed (0.05%v/v-Tween/PBS), blocked (1h, 1%w/v-BSA/PBS) and rinsed before anti-Phospho(Ser15)-p53 antibody (1:1000/PBS) was added to the plates and incubated (1h). To reduce non-specific binding plates were washed (0.05%v/v-Tween/PBS) prior to incubation (1h) with HRP-conjugated goat anti-rabbit IgG secondary antibody (1:5000/PBS, Promega, Madison, WI). Secondary antibody that was linked to the phosphorylated GST-p53(1–101) protein was detected with TMB substrate reagent (Pierce, Rockford, IL). Plates were developed (15–30min) and the reaction was stopped (1M H2SO4 final concentration) before absorbance was determined (λ450nm, AnalystAD plate-reader, LJL Systems). Compounds that inhibited ATM kinase activity in ELISA assays, were characterized with respect to inhibition of ATM/ATR kinases using in vitro kinase assays. Western blotting using the anti-Phospho(Ser15)-p53 antibody was used as a readout of ATM/ATR inhibition. Extended analysis of CP466722 (10μM) against a commercially available panel of kinases was performed by Upstate (Lake Placid, NY).
Western blotting
Cells were harvested, lysed (TGN buffer), quantitated and prepared for western blotting analysis as previously described (7). Antibodies were diluted 1:1000(5%w/v-BSA/TBST or 5%w/v-milk/TBST). Sigma (St Louis, MO): anti-β-actin. Santa-Cruz (Santa-Cruz, CA): anti-p53; anti-Chk1-G4. Cell Signaling Technology (Danvers, MA): PathScan-Bcr/Abl activity assay; anti-cAbl; anti-CrkL; anti-Phospho-(Ser15)-p53; anti-Phospho-(Thr68)-Chk2; anti-Phospho-(Thr387)-Chk2; anti-Phospho-(Ser345)-Chk1; anti-Phospho-(Ser308)-Akt; anti-Phospho-(Thr473)-Akt; anti-Akt. Millipore (Temecula, CA): anti-Histone H2A(acidic patch); anti-Phospho-(Ser139) H2AX. Bethyl Labs (Montgomery, TX): anti-SMC1. Miscellaneous: anti-Phospho-(Ser957)-SMC1 (30); anti-ATM (MAT3; gift Y.Shiloh, Tel-Aviv, Israel) and anti-Phospho-(Ser1981)-ATM (6). ImageJ(http://rsb.info.nih.gov/ij/) was used to quantitate band density on autoradiograms from western blotting and relative inhibition was calculated as percentage of control.
Flow cytometric analysis
Cell cycle analysis
Cells were harvested and fixed (4°C) with 70%v/v-Ethanol-PBS. Cells(1 × 106) were washed (PBS) and incubated (30min/dark) at room temperature in PBS(10μg/ml PI (Sigma), 250μg/ml RNaseA (Qiagen, Valencia, CA). DNA content was determined using a FACSCalibur (Becton Dickinson, San Jose, CA) and data analyzed (CellQuest software).
Immunofluorescent detection of phosphorylated-Histone H3
Cells were harvested 1h following IR and fixed (−20°C) with 70%v/v-Ethanol-PBS. Cells were stained and analyzed as previously described (31).
Clonogenic survival assay
HeLa or A-T (GM02052 expressing hTERT) cells were plated in triplicate (0.5 × 106cells/plate) and incubated for 24h. Cells were pre-treated: DMSO, CP466722 or KU55933 prior to IR (0-10Gy). Cells were incubated for 4h following IR before media was removed, cells washed (PBS), trypsinsed, counted and re-plated (2000cells/plate, 10cm plates) in the absence of drug and incubated for 10 days. Prior to colony counting, cells were washed (PBS), stained (PBS, 0.0037%v/v-formaldehyde, 0.1%w/v-crystal violet), rinsed (dH2O) and dried. Defined populations (>50cells) were counted as one surviving colony, data were calculated as percentage surviving colonies relative to control plates +/− SE.
Results
Identification of an in vitro inhibitor of the ATM kinase
Large amounts of purified protein would be required to run High Throughput Screens to identify small molecule inhibitors of ATM. Therefore, a directed screen based approach was adopted where a library of 1500 compounds was selected based on known kinase inhibitor templates and calculated kinase pharmacophores from the Pfizer proprietary chemical file. These compounds were screened using an in vitro ELISA assay, with potential inhibitors being identified by a decreased ability of purified ATM kinase to phosphorylate GST-p53(1–101) substrate (data not shown). Compounds identified by this assay were subjected to an in vitro kinase assay to screen out false positives (data not shown). This screening approach identified the compound CP466722 (Figure 1) as a candidate for characterization as an ATM inhibitor in tissue culture models. Though the ATM-related kinase, ATR, was not inhibited by CP466722 in vitro, inhibitory activities against abl and src kinases were noted in this in vitro screen.
Figure 1.
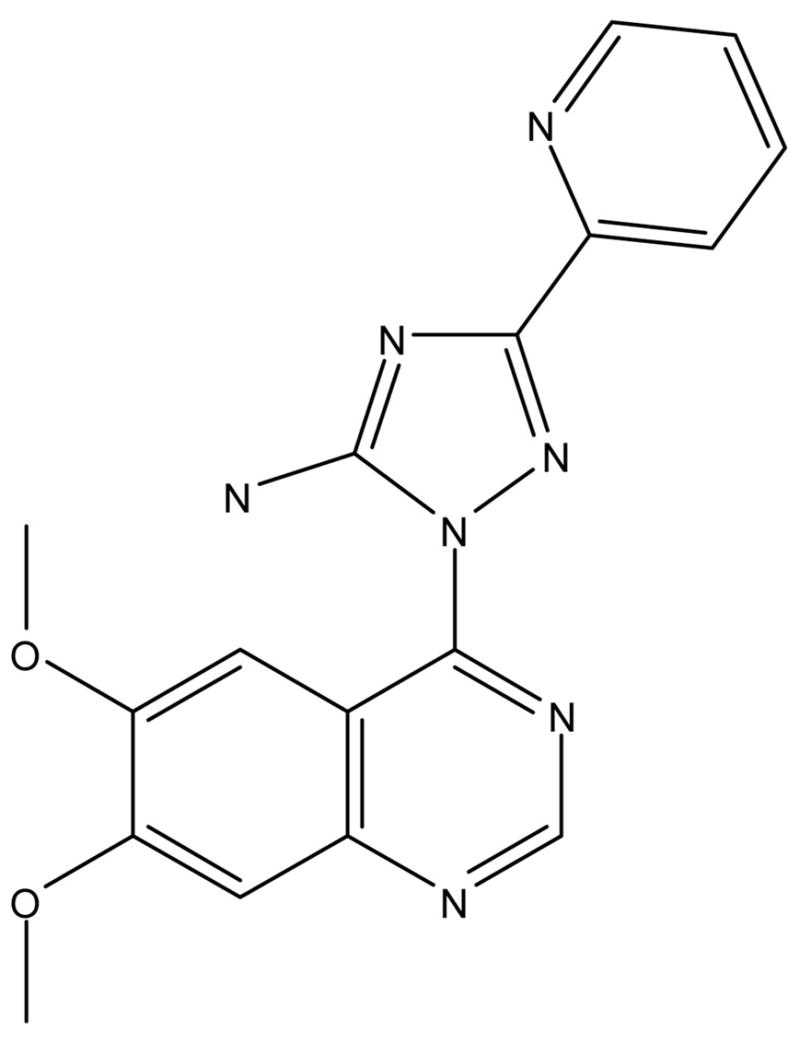
Chemical structure of 2-(6,7-dimethoxyquinazolin-4-yl)-5-(pyridin-2-yl)-2H-1,2,4-triazol-3-amine (CP466722).
Lack of toxicity and inhibition of ATM kinase activity in human and mouse cells
As an initial assessment of cellular effects of exposure to CP466722, no adverse effects on cell viability were observed in primary and hTERT-immortalized human diploid fibroblasts (Supplemental Figure 1) or in a variety of human tumor cell lines (data not shown), even after continuous exposure for 72 hours. To establish whether CP466722 could inhibit ATM kinase activity in cells and to determine an effective concentration for inhibition, HeLa cells were exposed to IR in the presence of varying concentrations of the inhibitor and phosphorylation of ATM targets was assessed. The established ATM inhibitor KU55933 was used as a positive control for ATM inhibition (24). IR-induced ATM kinase activity resulted in the expected increases in ATM-dependent phosphorylation events and CP466722 treatment inhibited all of these events. Virtually complete disruption of ATM cellular activity was noted at doses of 6μM and above (Figure 2). Disruption of ATM-dependent phosphorylation events as well as inhibition of ATM-dependent p53 induction were also observed in MCF-7 human breast cancer cells and primary and immortalized diploid human fibroblasts (Supplementary Figure 2a and 2b). Overall, the response to IR in cells treated with CP466722 was similar to that seen in cells lacking ATM (Supplementary Figure 2a).
Figure 2. CP466722 inhibits ATM kinase activity in cells in response to IR-induced DNA damage.
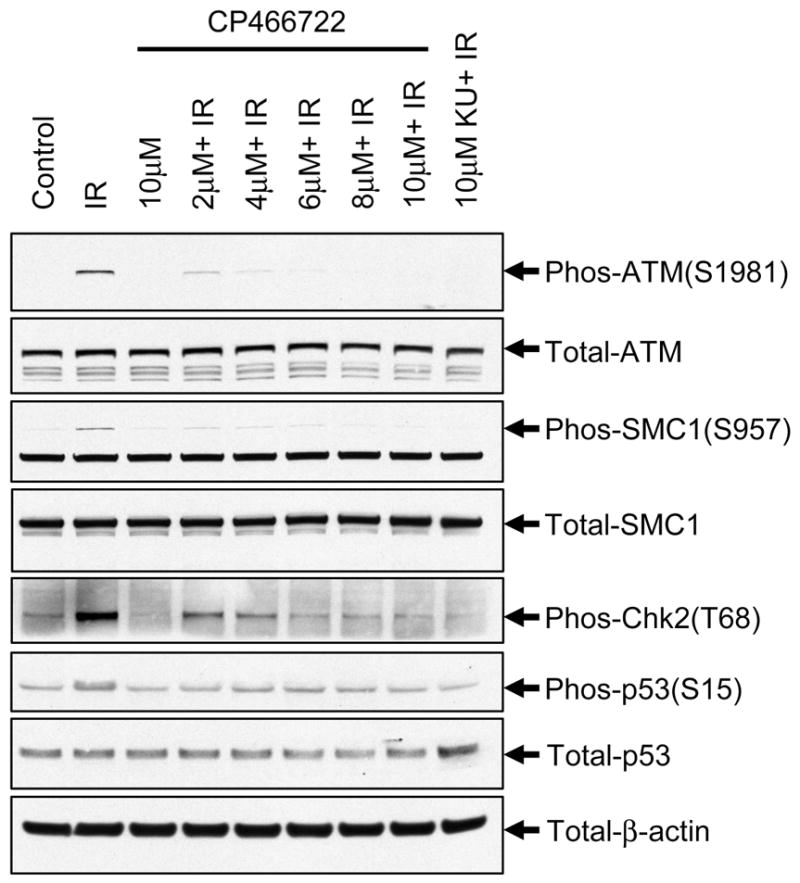
HeLa cells were preincubated with varying concentrations of CP466722, DMSO or 10μM KU55933 (KU) prior to mock-IR (control) or IR (2Gy) followed by incubation at 37°C for 30min before being harvested. To determine the effect of CP466722 on ATM kinase activity, ATM-intermolecular autophosphorylation at Serine 1981 and phosphorylation of downstream ATM-targets were monitored by western blotting analysis (representative of several repeat experiments).
Since one future goal is to characterize the ability of CP466722 to sensitize tumors to radiation or chemotherapeutic agents in murine models in vivo, it was important to know if CP466722 was effective at inhibiting Atm kinase in mouse cells. The ATM signaling pathway is conserved from human to mouse and ATM kinase activity can be monitored by analyzing similar downstream events (30). An exception is phosphorylation of Chk2 on threonine 68 which is difficult to detect in mouse cells. Therefore, we examined phosphorylation of the conserved residue threonine 387 of Chk2, which is an ATM-dependent event in human cells (32, 33). Atm wild-type and deficient MEFs were exposed to IR in the presence or absence of CP466722 or KU55933. In Atm wild-type MEFs, ATM kinase activity was induced by IR and there were strong increases in phosphorylation of SMC1 (Ser957), Chk2 (Thr387) and p53 (Ser15) relative to control (Supplementary Figure 3). These phosphorylation events were ATM-dependent as no IR-induced increases in phosphorylation were detected in Atm-deficient MEFs. As with human cells, both CP466722 and KU55933 inhibited p53 induction and all of these ATM-dependent phosphorylation events in mouse cells (Supplementary Figure 3).
CP466722 does not inhibit PI3K, PI3K-like protein kinases or Abl kinase in cells
The ATR kinase is also activated by DNA damage and other cellular stresses (e.g. UV and aphidicolin) and phosphorylates many of the same substrates as ATM (1). While ATM is preferentially activated by DSBs and phosphorylates Chk2 on threonine 68, ATR is preferentially activated by stalled replication forks and phosphorylates serine 345 of Chk1 (34, 35). Though CP466722 did not affect ATR kinase activity in vitro, we examined the ability of the compound to affect ATR kinase activity in cells. hTERT-immortalized human fibroblasts were treated for 1h with the replication inhibitor aphidicolin in the presence or absence of CP466722 (36). ATR-dependent phosphorylation of Chk1 (Ser345) was not inhibited by CP466722 (Figure 3a), even though ATM-dependent phosphorylation of Chk2 (Thr68) was blocked in these cells. Failure to inhibit aphidicolin-induced Chk1 (Ser345) phosphorylation in cells lacking ATM provided even more definitive evidence that CP466722 does not inhibit ATR kinase in cells (Figure 3a).
Figure 3. PI3K and PIKK family members are not inhibited by CP466722 in cells.
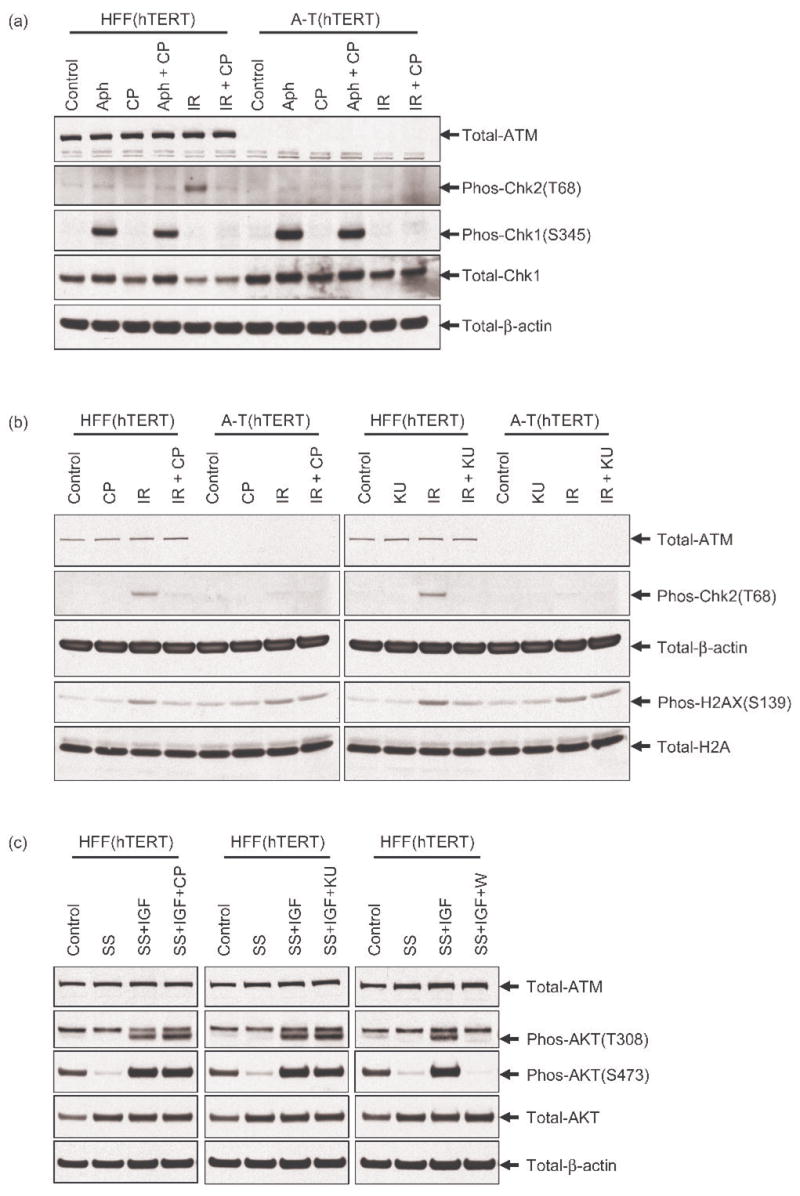
HFF(hTERT) and A-T(hTERT) fibroblasts were used to establish the effects of CP466722 on PI3K and PIKK family members. (a) Cells were preincubated with DMSO or 6μM of CP466722 (CP) before being mock-treated or treated with 20μM aphidicolin (Aph) for a 1h period at 37°C prior to harvesting. As a control, cells were preincubated with DMSO or 6μM of CP466722 before being exposed to IR (2Gy) followed by incubation at 37°C for 30min and harvesting. To determine the effect of CP466722 on the ATM- and ATR- checkpoint pathways in response to aphidicolin and IR, Phospho(Ser345)-Chk1 and Phospho(Thr68)-Chk2 were monitored by western blotting analysis. (b) Cells were preincubated with DMSO, 6μM CP466722 (CP) or 10μM KU55933 (KU) prior to mock-IR (control) or IR (2Gy) followed by incubation at 37°C for 30min and harvesting. As a measure of ATM/DNA-PK kinase activity Phospho(Ser139)-H2AX was monitored by western blotting analysis. (c) Cells were cultured under normal (Control) or serum starved (SS) conditions for a 24h period before being preincubated with DMSO, 6μM CP466722 (CP), 10μM KU55933 (KU) or 200nM Wortmannin (W). Cells were then subject to growth factor stimulation with IGF-I (10ng/ml) for a 20min period at 37°C before being harvested. The activity of PI3K and PIKK family members were assessed by monitoring Phospho(Thr308)-Akt and Phospho(Ser473)-Akt by western blotting analysis (representative of several repeat experiments).
DNA-PK is another PIKK family member that contributes to damage-induced signaling and both ATM and DNA-PK can phosphorylate histone H2AX on Serine139 following IR (37). To investigate potential effects of CP466722 on DNA-PK, phosphorylation of histone H2AX (Ser139) was assessed in wild-type and A-T cells since DNA-PK phosphorylates this site in the absence of ATM kinase activity (24, 37). While H2AX (Ser139) phosphorylation following IR was inhibited by CP466722 or KU55933 in wild-type cells, these ATM inhibitors failed to inhibit IR-induced H2AX phosphorylation in A-T cells (Figure 3b), demonstrating a lack of detectable effects on DNA-PK.
In response to growth factor stimulation, AKT is activated by phosphorylation of threonine 308 by the PI3K-pathway and serine 473 by other PIKK family members (e.g. mTOR, DNA-PK and ATM) (38). To demonstrate that CP466722 was not inhibiting PI3K or PIKK family members, human fibroblasts were serum starved for 24h before being stimulated with IGF-I either in the presence or absence of CP466722, KU55933 or Wortmannin (Figure 3c). Serum starvation resulted in an almost complete loss of AKT(Thr308/Ser473) phosphorylation. These phosphorylation events were strongly induced upon addition of IGF-I to serum starved cells and, as expected, were strongly inhibited by the known PI3K inhibitor wortmannin. No inhibition was noted with CP466722 or KU55933 treatment. Taken together, these results indicate that CP466722 inhibits ATM kinase, but does not affect the cellular activity of PI3K or PIKK family members.
Abl and Src kinases were identified in the initial in vitro screens as potential targets of CP466722. To address whether CP466722 inhibits cellular Abl and Src kinases, we utilized a mouse pre-B cell (p185 +/Arf−/−) model (28). In this system, the BCR-Abl (p185) fusion protein is constitutively active, driving autophosphorylation of residue tyrosine 245 (BCR-Abl & endogenous Abl) and phosphorylation of a downstream target CrkL on tyrosine 207 (Supplementary Figure 4a) (39–41). Src kinase undergoes intermolecular autophosphorylation of residue tyrosine 416 on its activation loop to become fully activated (42, 43). In cells expressing BCR-Abl, SRC kinases are activated and increased levels of Src (Tyr416) phosphorylation have been reported suggesting that Src is active and undergoing autophosphorylation (Supplementary Figure 4a) (41, 44).
As a control, CP466722 and KU55933 were shown to inhibit ATM kinase activity in the mouse pre-B cells as demonstrated by disruption of p53 (Ser15) phosphorylation and p53 stabilization in response to IR (Supplementary Figure 4b). To establish whether the inhibitors affected Abl and Src kinase activity, the mouse pre-B cells were treated with CP466722, KU55933 or Imatinib (Abl kinase inhibitor) as a positive control (45). As expected, autophosphorylation of BCR-Abl (Tyr245), endogenous Abl (Tyr245), and Abl-dependent phosphorylation of CrkL (Tyr207) were all detected in control mouse pre-B cells (Supplementary Figure 4a). Imatinib inhibited all these phosphorylation events, while, CP466722 or KU55933 failed to inhibit BCR-Abl kinase activity or phosphorylation of downstream targets. Although imatinib is not reported to directly inhibit Src kinase activity, cellular Src (Tyr416) autophosphorylation was prevented by imatinib under these experimental conditions (41, 45, 46). Treatment with both CP466722 and KU55933 resulted in decreased Src (Tyr416) autophosphorylation relative to the control cells (Supplementary Figure 4a). This data indicates that at doses capable of inhibiting ATM, CP466722 and KU55933 do not inhibit Abl kinase activity in cells, however, both compounds have inhibitory effects on Src kinase activity in this system (84% and 73% inhibition at 10uM respectively).
CP466722 disrupts ATM-dependent cell cycle checkpoints in cells
Small molecule disruption of the ATM signal transduction pathway should recapitulate the A-T cellular phenotypes, including characteristic cell cycle checkpoint defects. Cells lacking ATM exhibit pronounced G2-accumulation over time following IR due to a failure to arrest in S-phase (31). In response to IR, HeLa cells treated with either KU55933 or CP466722 resulted in an enhanced proportion of cells with G2/M DNA content and a decreased proportion of cells with G1-phase DNA content relative to DMSO treated cells (Figure 4a). In the absence of IR-induced DNA damage, these doses of CP466722 and KU55933 had no effect on cell cycle distribution during this time frame (Figure 4ai and aii).
Figure 4. CP466722 inhibits ATM function in response to IR-induced DNA damage.

(a) An asynchronous population of HeLa cells were preincubated with DMSO, 6μM CP466722 or 10μM KU55933 prior to mock-IR or IR (5Gy). Following irradiation cells were incubated at 37°C for 16h before being harvested, fixed and stained with propidium iodide for cell cycle analysis by flow cytometry. DNA content profiles are representative of several repeat experiments (i). The data displayed by the DNA content profiles were analyzed and the cell cycle phase information is represented graphically (ii). (b) An asynchronous population of HeLa cells were preincubated with DMSO, 6μM CP466722, 2mM caffeine or 10μM KU55933 prior to mock-IR or IR (2Gy). Following irradiation cells were incubated at 37°C for 1h before being harvested, fixed and stained for Phospho(Ser10)-Histone H3 and propidium iodide. DNA content and Phospho(Ser10)-Histone H3 positivity were determined by flow cytometry and the data displayed are representative of several repeat experiments.
To establish whether CP466722 and KU55933 treatment disrupted the ATM-dependent G2/M checkpoint (31), asynchronous populations of HeLa cells were pretreated with either DMSO, caffeine, CP466722, or KU55933 before being exposed to mock-IR or IR. A decrease in the percentage of mitotic cells following IR in the presence of DMSO indicated an IR-induced G2 arrest, while both KU55933 and CP466722 prevented this IR-induced decrease (Figure 4b). In contrast to the effects seen with the less specific ATM/ATR inhibitor, caffeine, neither compound affected G2/M progression in the absence of DNA damage (Figure 4b). Taken together the results demonstrate that CP466722 is capable of disrupting ATM function and recapitulates checkpoint defects reported for A-T cells.
Chemical inhibition of ATM can be rapidly and completely reversed
KU55933 displays strong inhibition of ATM for at least 4h in tissue culture (24). To determine whether CP466722 could inhibit ATM for prolonged periods of time in tissue culture, HeLa cells were incubated with either DMSO, KU55933 or CP466722 for various times and then exposed to IR and harvested after a 30min recovery period. Relative to control cells, the results demonstrate that ATM was activated by IR to the same degree in the presence of DMSO at all time points tested (Figure 5a left panel). Similar to KU55933, IR fails to induce ATM activation and downstream signaling in the presence of CP466722 and inhibition of the ATM-dependent phosphorylation events are maintained over the 8h time course of the experiment (Figure 5a center and right panel). These results demonstrate that CP466722 strongly inhibits ATM kinase pactivity for at least an 8h period in tissue culture.
Figure 5. CP466722 can be used as a molecular switch to regulate cellular ATM kinase activity.

(a) CP466722 potently inhibits ATM for at least 8h in culture. HeLa cells were preincubated with DMSO, 6μM CP466722 or 10μM KU55933 to reach the experimental start point (i.e. 0h). After preincubation, cells were exposed to mock-IR (control) or IR (2Gy) at the indicated time points (0–8h). Following irradiation cells were incubated at 37°C for 30min before being harvested (the times displayed in the figure represent the time of IR-exposure and do not include the 30min recovery time). To determine whether these compounds displayed a limited half-life with respect to inhibition of ATM kinase activity, ATM-intermolecular autophosphorylation at Serine 1981 and phosphorylation of Chk2 (Thr68) were determined by western blotting analysis (representative of several repeat experiments). (b) CP466722 and KU55933 display rapid and complete reversibility of ATM kinase inhibition in culture. HeLa cells were preincubated with DMSO, 6μM CP466722 or 10μM KU55933 to reach the experimental start point (i.e. 0h). After preincubation, the compounds were either left on the cells (+DMSO, +CP466722 & + KU55933) or removed (−DMSO, −CP466722 & − KU55933) and fresh media added. Following wash off, the cells were exposed to mock-IR (control) or IR (2Gy) at the indicated times (0–4h). Irradiated cells were incubated at 37°C for 30min before being harvested (the times displayed in the figure represent the time of IR-exposure and do not include the 30min recovery time). To determine whether the inhibition of ATM kinase activity had been reversed by removal of the compounds, ATM-intermolecular autophosphorylation at Serine1981 and phosphorylation of downstream ATM-targets were determined by western blotting analysis (representative of several repeat experiments).
As part of the characterization of CP466722 we were interested in the reversibility of the ATM inhibition. To address this question, HeLa cells were pretreated with either DMSO, CP466722 or KU55933 and then washed with addition of fresh culture media in the absence of any compounds. Cells were subsequently exposed to IR at various times (Figure 5b). In the presence of DMSO, the IR-induced ATM-dependent phosphorylation events were easily detected both before and after wash-off. In contrast, the presence of CP466722 or KU55933 strongly inhibited these ATM-dependent phosphorylation events in response to IR (Figure 5b). However, all ATM-dependent phosphorylation events were detected within the first 30 minutes following removal of the inhibitors (Figure 5b: 0h –CP466722 or –KU55933) and inhibition was reversed completely within 1 hour after wash-off (Figure 5b: 30min –CP466722 or –KU-5933). Taken together these results demonstrate that the ATM pathway can be rapidly inhibited; however, following removal of these compounds, the inhibition can be rapidly and completely reversed.
Transient inhibition of ATM sensitizes cells to IR induced DNA damage
One characteristic feature of cells deficient in functional ATM is their increased sensitivity to IR-induced DNA damage. This has been demonstrated genetically using A-T cells, which have permanently disrupted ATM function or by chemical inhibition (KU55933), where ATM function has been disrupted for prolonged periods of time in cells (24). Based on the results indicating that inhibition of ATM kinase activity by these compounds was rapidly reversible, we were interested in whether transient inhibition of ATM could sensitize cells to IR. Following pretreatment of HeLa cells with either DMSO, CP466722 or KU55933 the cells were exposed to the indicated doses of IR and allowed to recover for a period of 4h in the presence of DMSO or the inhibitors. The cells were then replated and incubated for a period of 10 days to allow for colony formation in the absence of inhibitors. Similar plating efficiencies were achieved in the presence (41% +/− 3.7 and 39% +/− 1.7) or absence (42% +/−4.2 and 40% +/−2.0) of CP466722 and KU55933 respectively, suggesting that neither compound affected cell plating nor cell viability. Transient exposure to either CP466722 or KU55933 sensitized cells to IR (Figure 6). Since the compounds were only present for a 4h period and since the ATM pathway is reactivated rapidly upon removal of these compounds (Figure 5b), it appears that a transient (4 hour or less) inhibition of ATM is sufficient to enhance the sensitivity of HeLa cells to IR. Importantly, no differences in clonogenic survival of cells from A-T patients were noted in the presence or absence of CP466722 (Supplementary Figure 5), demonstrating that the radiosensitization caused by this compound was in fact due to ATM inhibition and not any off-target effects.
Figure 6. Transient inhibition of ATM kinase activity is able to sensitize cells to IR-induced DNA damage.

HeLa cells were plated in triplicate and incubated for 24h. Cells were preincubated with DMSO, 6μM CP466722 or 10μM KU55933 before being exposed to a range of doses of IR (0-10Gy). Cells were incubated for 4h following irradiation before being re-plated in fresh media in the absence of drug and incubated for a period of 10 days to allow for colony formation. To determine the effect of transient ATM inhibition by CP466722 and KU55933 in response to IR the surviving colonies were counted and the data presented on a log axis (represents the mean of three independent experiments +/−SE).
Discussion
Mammalian cells are constantly at risk from potentially lethal or mutagenic genomic lesions from both endogenous (e.g. free oxygen radicals) and exogenous (e.g. UV, IR) sources. As a result eukaryotic cells have developed an intricate network of signal transduction pathways (DDR pathways) that allow them to sense and repair damaged DNA (1). Loss of function of critical proteins from these pathways can leave cells with enhanced sensitivity to DNA damaging agents (47). The ATM kinase is an important component of these DDR pathways and cells deficient for ATM (A-T) display hypersensitivity to certain DNA damaging agents (3). Based on these observations it has been proposed that specific inhibition of ATM function in combination with current radio-/chemo-therapeutic treatments may result in enhanced cancer cell killing (20, 22–24). This principal has been demonstrated by the ability of specific antisense/siRNA to attenuate ATM function and sensitize certain cancer cell lines to IR (48–50). Furthermore, the recent identification and characterization of the ATM inhibitor KU55933 has strengthened this hypothesis and demonstrated that specific small molecule inhibition of ATM in vitro is capable of sensitizing human cancer cell lines to IR and topoisomerase poisons (24). Our aim in this study was to identify and characterize a novel inhibitor of the ATM protein kinase with a future goal of modifying this small molecule for characterization and use with in vivo models. In this paper we identified the non-toxic compound CP466722 as an inhibitor of ATM and offer a comparison to the established ATM inhibitor KU55933 (24).
In response to IR, ATM initiates a signaling cascade and phosphorylates downstream targets (e.g. p53; Chk2 and SMC1) on characteristics sites which can be used as a measure of cellular ATM kinase activity (8, 10, 11, 13). CP466722 disrupts these cellular phosphorylation events in a dose-dependent manner in several different cell types and recapitulates the signaling defects observed in A-T cells (3). Closely related kinases (DNA-PK and ATR) share some downstream targets with ATM and phosphorylate common sites on these substrates (29, 35, 37), however we found that CP466722 does not inhibit ATR kinase activity in vitro or the kinase activities of ATR or DNA-PK in cells. Furthermore, unlike the pan-PI3K inhibitor wortmannin, CP466722 does not inhibit PI3K activity in cells. Interestingly, phosphorylation of Akt at serine 473 is reported to be regulated by several PIKK family members including DNA-PK, ATM and mTOR (38). Although, Akt (Ser473) phosphorylation was inhibited by wortmannin, neither CP466722 nor KU55933 affected this modification. This implies that ATM is not required for this phosphorylation event under these experimental conditions and could indicate that these inhibitors do not affect additional PI3K-like protein kinases such as mTOR (38). Similar to KU55933, these results highlight CP466722 as a relatively specific inhibitor of ATM and a marked improvement on previous compounds used to inhibit ATM, such as wortmannin and caffeine.
Extended analysis of CP466722 indicated that Abl and Src kinase activity were inhibited in vitro. However, BCR-Abl kinase activity was not affected in cells treated with this compound at doses that inhibit ATM suggesting Abl is not a cellular target of CP466722. In contrast, autophosphorylation of Src (Tyr416) was reduced by both CP466722 and KU55933 although it is not clear whether these effects are direct or due to inhibition of signal transduction pathways that lead to Src kinase activation. This demonstrates that there is still a need to modify and improve the specificity of these ATM inhibitors and further characterization is required to identify and understand any potential off-target effects. It is noted that the lack of radiosensitization of A-T cells by CP466722 suggests that the inhibition of Src is not contributing to the radiosensitization induced by the drug.
Inhibition of ATM activity with CP466722 induced cellular effects indistinguishable from those seen in cells lacking ATM, including cell cycle checkpoint defects and radiosensitization (3). Similar to KU55933, CP466722 rapidly and potently inhibits ATM over a period of several hours demonstrating reasonable stability in tissue culture (24). However, upon removal of either CP466722 or KU55933 from tissue culture media, ATM kinase activity and the subsequent phosphorylation of downstream targets could be completely and rapidly restored. This ability to transiently inhibit ATM function followed by reactivation within such a short time frame is novel and opens new avenues for study of the ATM pathway. In effect, these inhibitors can be used as molecular switches to influence the immediate ATM-dependent DNA damage response and the subsequent repair process that contribute to cell survival.
Transient small molecule inhibition of ATM in vitro recapitulates the cellular A-T phenotype of increased sensitivity to IR, while causing no additional sensitivity in an A-T cell line. However, the sensitization induced by these short-term exposures do not completely reflect the characteristic low-dose hypersensitivity phenotype of A-T cells, which could highlight a difference between long- and short-term inhibition. In the study by Hickson et al (2004), long-term small molecule inhibition of ATM demonstrates enhanced sensitivity to IR at low doses (1-2Gy). Taken together, these results suggest that during and for a short period of time following IR, ATM plays an essential role in ensuring cellular survival that is not compensated for by other DDR pathways and can not be rescued by reactivation of ATM. This concept is consistent with the proposed critical role of ATM activation and activity in the earliest steps of DSB repair (7). Further characterization of this observation with these inhibitors is still required to understand the role of ATM at these early time points. It could be informative to investigate the effects of transient inhibition and reactivation of ATM in future studies and determine how this influences cellular responses to DNA breakage, including which damage response proteins are recruited to DSBs and the kinetics of repair (7).
Since CP466722 can inhibit the ATM-signal transduction pathway in murine cells, it may be possible to use mouse models to begin to explore the effects of this compound in vivo. The observation that transient inhibition of ATM in tissue culture causes measurable hypersensitivity to IR could imply that stable and prolonged inhibition of ATM may not be needed to provide a therapeutic window. This concept requires further investigation and will require careful studies on drug delivery, distribution, stability and activity in vivo.
In summary, we have identified and characterized a new inhibitor of ATM which can be utilized to further characterize the function of the ATM-signaling pathway and the immediate molecular response to IR. In addition, this compound provides us with a novel chemical structure that can be modified to enhance potency, specificity and ensure that second generation compounds can be taken forward into in vivo models. Further characterization of these inhibitors will help us to understand whether disruption of ATM function in vivo is a plausible approach for enhancing therapeutic potential.
Supplementary Matierals
Acknowledgments
This work was performed in collaboration with Pfizer (Cambridge MA) and supported by grants from the National Institutes of Health (CA71387, CA93632, CA21765) and by the American Lebanese Syrian Associated Charities (ALSAC) of St. Jude Children’s Research Hospital. We thank Dr Richard Williams for providing advice and cells for this study, Dr Christopher Bakkenist for his input with this research and all members of the Kastan laboratory for insightful suggestions throughout the course of this work. We would also like to acknowledge Mukta Bagul, Yan Zhang and Paul Bauer (Pfizer’s Research and Technology Center) for their contributions to this project.
References
- 1.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 2.Shiloh Y. Ataxia-telangiectasia and the Nijmegen breakage syndrome: related disorders but genes apart. Annu Rev Genet. 1997;31:635–62. doi: 10.1146/annurev.genet.31.1.635. [DOI] [PubMed] [Google Scholar]
- 3.Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–86. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 4.Savitsky K, Bar-Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 5.Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–68. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- 6.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 7.Berkovich E, Monnat RJ, Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–90. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 8.Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–7. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 9.Canman CE, Wolff AC, Chen CY, Fornace AJ, Jr, Kastan MB. The p53-dependent G1 cell cycle checkpoint pathway and ataxia-telangiectasia. Cancer Res. 1994;54:5054–8. [PubMed] [Google Scholar]
- 10.Canman CE, Lim DS, Cimprich KA, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–9. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 11.Matsuoka S, Rotman G, Ogawa A, et al. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A. 2000;97:10389–94. doi: 10.1073/pnas.190030497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maya R, Balass M, Kim ST, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–77. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–70. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim DS, Kim ST, Xu B, et al. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature. 2000;404:613–7. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 15.Taniguchi T, Garcia-Higuera I, Xu B, et al. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002;109:459–72. doi: 10.1016/s0092-8674(02)00747-x. [DOI] [PubMed] [Google Scholar]
- 16.Xu B, O’Donnell AH, Kim ST, Kastan MB. Phosphorylation of serine 1387 in Brca1 is specifically required for the Atm-mediated S-phase checkpoint after ionizing irradiation. Cancer Res. 2002;62:4588–91. [PubMed] [Google Scholar]
- 17.Bao S, Tibbetts RS, Brumbaugh KM, et al. ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature. 2001;411:969–74. doi: 10.1038/35082110. [DOI] [PubMed] [Google Scholar]
- 18.Petrini JH. The mammalian Mre11-Rad50-nbs1 protein complex: integration of functions in the cellular DNA-damage response. Am J Hum Genet. 1999;64:1264–9. doi: 10.1086/302391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature. 2000;408:429–32. doi: 10.1038/35044000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choudhury A, Cuddihy A, Bristow RG. Radiation and new molecular agents part I: targeting ATM-ATR checkpoints, DNA repair, and the proteasome. Semin Radiat Oncol. 2006;16:51–8. doi: 10.1016/j.semradonc.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 21.Eastman A. Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J Cell Biochem. 2004;91:223–31. doi: 10.1002/jcb.10699. [DOI] [PubMed] [Google Scholar]
- 22.Sarkaria JN, Eshleman JS. ATM as a target for novel radiosensitizers. Semin Radiat Oncol. 2001;11:316–27. doi: 10.1053/srao.2001.26030. [DOI] [PubMed] [Google Scholar]
- 23.Kastan M. Ataxia-telangiectasia--broad implications for a rare disorder. N Engl J Med. 1995;333:662–3. doi: 10.1056/NEJM199509073331014. [DOI] [PubMed] [Google Scholar]
- 24.Hickson I, Zhao Y, Richardson CJ, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 25.Bakkenist CJ, Drissi R, Wu J, Kastan MB, Dome JS. Disappearance of the telomere dysfunction-induced stress response in fully senescent cells. Cancer Res. 2004;64:3748–52. doi: 10.1158/0008-5472.CAN-04-0453. [DOI] [PubMed] [Google Scholar]
- 26.Wood LD, Halvorsen TL, Dhar S, et al. Characterization of ataxia telangiectasia fibroblasts with extended life-span through telomerase expression. Oncogene. 2001;20:278–88. doi: 10.1038/sj.onc.1204072. [DOI] [PubMed] [Google Scholar]
- 27.Kamijo T, van de Kamp E, Chong MJ, et al. Loss of the ARF tumor suppressor reverses premature replicative arrest but not radiation hypersensitivity arising from disabled atm function. Cancer Res. 1999;59:2464–9. [PubMed] [Google Scholar]
- 28.Williams RT, den Besten W, Sherr CJ. Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev. 2007;21:2283–7. doi: 10.1101/gad.1588607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538–43. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- 30.Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–38. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu B, Kim ST, Lim DS, Kastan MB. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol. 2002;22:1049–59. doi: 10.1128/MCB.22.4.1049-1059.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rainey MD, Black EJ, Zachos G, Gillespie DA. Chk2 is required for optimal mitotic delay in response to irradiation-induced DNA damage incurred in G2 phase. Oncogene. 2008;27:896–906. doi: 10.1038/sj.onc.1210702. [DOI] [PubMed] [Google Scholar]
- 33.Lee CH, Chung JH. The hCds1 (Chk2)-FHA domain is essential for a chain of phosphorylation events on hCds1 that is induced by ionizing radiation. J Biol Chem. 2001;276:30537–41. doi: 10.1074/jbc.M104414200. [DOI] [PubMed] [Google Scholar]
- 34.Bakkenist CJ, Kastan MB. Initiating cellular stress responses. Cell. 2004;118:9–17. doi: 10.1016/j.cell.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 35.Shiloh Y. The ATM-mediated DNA-damage response: taking shape. Trends Biochem Sci. 2006;31:402–10. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Hammond EM, Green SL, Giaccia AJ. Comparison of hypoxia-induced replication arrest with hydroxyurea and aphidicolin-induced arrest. Mutat Res. 2003;532:205–13. doi: 10.1016/j.mrfmmm.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 37.Stucki M, Jackson SP. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 2006;5:534–43. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 38.Lees-Miller SP. PIKK-ing a new partner: a new role for PKB in the DNA damage response. Cancer Cell. 2008;13:379–80. doi: 10.1016/j.ccr.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 39.Brasher BB, Van Etten RA. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J Biol Chem. 2000;275:35631–7. doi: 10.1074/jbc.M005401200. [DOI] [PubMed] [Google Scholar]
- 40.Jilani I, Kantarjian H, Gorre M, et al. Phosphorylation levels of BCR-ABL, CrkL, AKT and STAT5 in imatinib-resistant chronic myeloid leukemia cells implicate alternative pathway usage as a survival strategy. Leuk Res. 2007 doi: 10.1016/j.leukres.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 41.Hu Y, Swerdlow S, Duffy TM, et al. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A. 2006;103:16870–5. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470–80. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 43.Roskoski R., Jr Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005;331:1–14. doi: 10.1016/j.bbrc.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 44.Danhauser-Riedl S, Warmuth M, Druker BJ, Emmerich B, Hallek M. Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res. 1996;56:3589–96. [PubMed] [Google Scholar]
- 45.Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–53. doi: 10.1182/blood-2004-08-3097. [DOI] [PubMed] [Google Scholar]
- 46.Warmuth M, Simon N, Mitina O, et al. Dual-specific Src and Abl kinase inhibitors, PP1 and CGP76030, inhibit growth and survival of cells expressing imatinib mesylate-resistant Bcr-Abl kinases. Blood. 2003;101:664–72. doi: 10.1182/blood-2002-01-0288. [DOI] [PubMed] [Google Scholar]
- 47.Kennedy RD, D’Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol. 2006;24:3799–808. doi: 10.1200/JCO.2005.05.4171. [DOI] [PubMed] [Google Scholar]
- 48.Collis SJ, Swartz MJ, Nelson WG, DeWeese TL. Enhanced radiation and chemotherapy-mediated cell killing of human cancer cells by small inhibitory RNA silencing of DNA repair factors. Cancer Res. 2003;63:1550–4. [PubMed] [Google Scholar]
- 49.Fan Z, Chakravarty P, Alfieri A, et al. Adenovirus-mediated antisense ATM gene transfer sensitizes prostate cancer cells to radiation. Cancer Gene Ther. 2000;7:1307–14. doi: 10.1038/sj.cgt.7700242. [DOI] [PubMed] [Google Scholar]
- 50.Guha C, Guha U, Tribius S, et al. Antisense ATM gene therapy: a strategy to increase the radiosensitivity of human tumors. Gene Ther. 2000;7:852–8. doi: 10.1038/sj.gt.3301174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.