

Abstract
Glycinamide ribonucleotide transformylase (GAR Tfase) catalyzes the first of two formyl transfer steps in the de novo purine biosynthetic pathway that require folate cofactors and has emerged as a productive target for antineoplastic therapeutic intervention. The asymmetric synthesis and evaluation of the two diastereomers of 10-methylthio-DDACTHF (10R-3 and 10S-3) and related analogues as potential inhibitors of GAR Tfase are reported. This work, which defines the importance of the C10 stereochemistry for this class of inhibitors of GAR Tfase, revealed that both diastereomers are potent inhibitors of rhGAR Tfase (10R-3 Ki = 210 nM, 10S-3 Ki = 180 nM) that exhibit effective cell growth inhibition (CCRF-CEM IC50 = 80 and 50 nM, respectively) which is dependent on intracellular polyglutamation by folylpolyglutamate synthetase (FPGS), but not intracellular transport by the reduced folate carrier.
Introduction
Glycinamide ribonucleotide transformylase (GAR Tfase) is a folate-dependent enzyme central to the de novo purine biosynthetic pathway.1,2 GAR Tfase utilizes the cofactor (6R)-N10-formyltetrahydrofolic acid (10-formyl-THF) to transfer a formyl group to the primary amine of its substrate, β-glycinamide ribonucleotide (β-GAR) (Figure 1). This one carbon transfer provides the C-8 carbon of the purines and is the first of two formyl transfer reactions enlisted in the biosynthesis of purines. Inhibitors of folate dependent enzymes including GAR Tfase have provided important compounds for cancer chemotherapy as a result of their inhibition of the biosynthesis of nucleic acid precursors.3,4 Validation of GAR Tfase as a useful anticancer target emerged with the discovery of the first potent and selective inhibitor, 5,10-dideaza-5,6,7,8-tetrahydrofolic acid (DDATHF).5 Its selective toxicity has been attributed in part to the reliance of sensitive tumor cells on de novo purine synthesis, whereas the salvage pathway is an available and primary source of purines in normal untransformed cells.6,7
Figure 1.
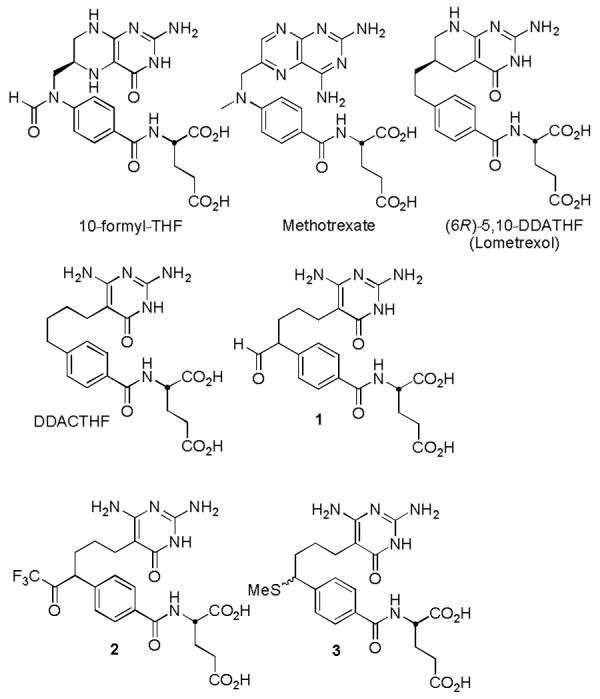
Cofactor and representative inhibitors of folate-dependent enzymes.
In previous studies, we reported the synthesis and biological evaluation of two unique folate-based inhibitors of GAR Tfase. 10-Formyl-DDACTHF8 (1) and 10-CF3CO-DDACTHF9 (2), bearing a nontransferable formyl or trifluoroacetyl group, proved to be potent inhibitors of GAR Tfase (1, Ki = 0.014 μM against rhGAR Tfase; 2, Ki = 0.015 μM against rhGAR Tfase). Both inhibitors have been shown through X-ray and NMR studies to bind GAR Tfase as the gem-diols.9,10 The formation of the gem-diol mimics the formyl transfer reaction tetrahedral intermediate and provides strong stabilizing interactions between the inhibitor and the active site catalytic residues of the protein. In both instances, the inhibitors C10 diastereomers rapidly interconvert preventing independent assessment of the individual diastereomers.
In a more recent study designed to explore the ability of 10-methanesulfonyl-DDACTHF to also mimic such active site binding interactions, we reported the synthesis and biological activity of its precursor 3, 10-methylthio-DDACTHF, which proved to be a surprisingly potent inhibitor of GAR Tfase (IC50 = 100 nM, CCRF-CEM; Ki = 250 nM, rhGAR Tfase).11 Although the activity of 3 was unexpected since it does not contain a C10 tetrahedral intermediate mimic, the thiomethyl moiety does incorporate a potential hydrogen bond acceptor and presents a soft hydrophobic substituent potentially stabilizing active site binding. The introduction of sulfur within inhibitors of GAR Tfase is well established. The known inhibitors LY309887 (4), AG2034 (5), and AG2037 (6) all incorporate sulfur into their structures although none do so in a manner analogous to 3 (Figure 2),12-14 and it has been reported that analysis of the GAR Tfase active site using the program GRID suggested that sulfur atoms should have particular affinity for two regions of the folate cofactor binding site.13
Figure 2.
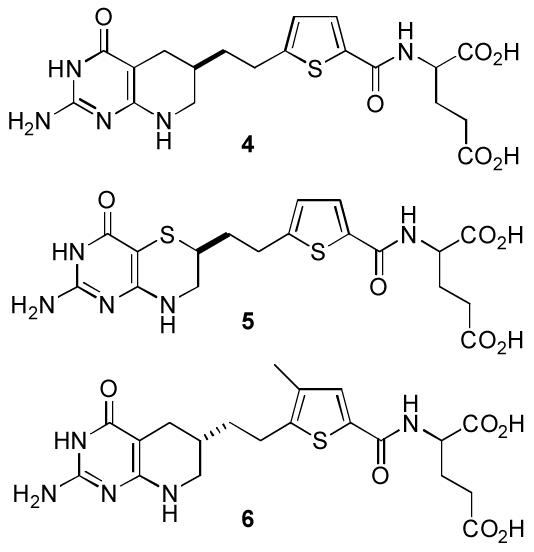
Inhibitors of GAR Tfase that contain sulfur atoms.
The original synthesis of 3 was racemic and the derivative was evaluated as an equi-mixture of C10-diastereomers. However, and unlike 1 and 2, the individual C10-diastereomers of 3 would not be expected to easily interconvert and could be anticipated to be individually evaluated revealing the importance of the C10 stereochemistry to the properties of the candidate inhibitors. Herein, we report an asymmetric synthesis of 10-methylthio-DDACTHF (3) permitting this investigation into the importance of the C10 stereochemistry and report the biological properties of each C10-diastereomer of 10-thiomethyl-DDACTHF (3) along with that of a series of key analogues that further define the importance of the C10 substituent itself: 10-hydroxy-DDACTHF (10R-7 and 10S-7) and 10-methoxy-DDACTHF (10R-8 and 10S-8) and C10 substituted dithiane 9.
Inhibitor Synthesis
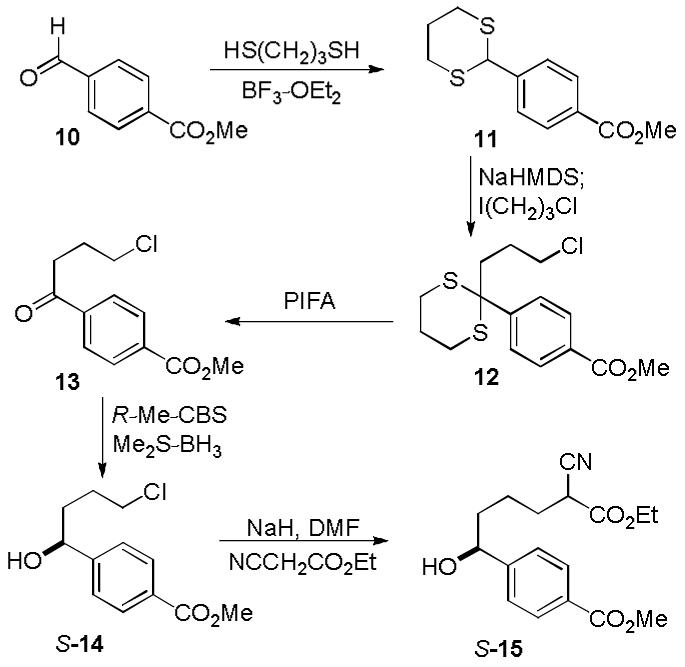
Starting from methyl 4-formylbenzoate, the aldehyde was converted to dithiane 11 upon reaction with 1,3-propanedithiol (Scheme 1). The resulting dithiane was alkylated with 1-chloro-3-iodopropane to give 12, and subsequent removal of the dithiane using bis(trifluoro)acetoxyiodobenzene (PIFA) provided 13. Ketone 13 was reduced using the R-Me-CBS15 catalyst in high yield (97%) and high enantiomeric excess (99% ee) to yield alcohol S-14. The absolute stereochemistry of S-14 and its subsequent derivatives were assigned based on the well-established stereochemical preferences of the reagent,16 and confirmed in X-ray structures of both 10R-3 and 10S-3 bound to recombinant human GAR Tfase. The details of these latter studies will be disclosed elsewhere. Similarly, the S-Me-CBS catalyst was utilized to provide the enantiomer of 14 (not shown) and characterization data for this enantiomeric series is provided in the Supporting Information.
Scheme 1.
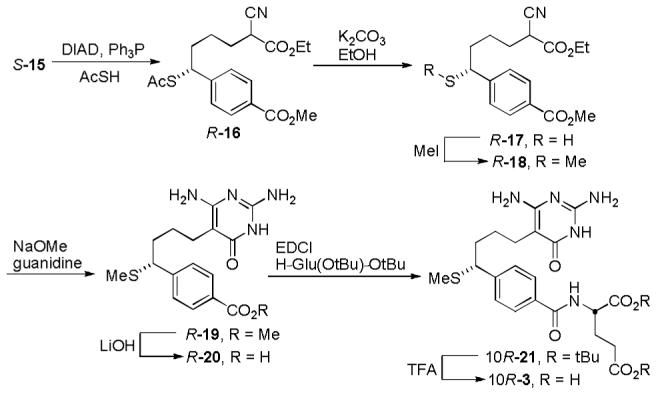
The preformed sodium salt of ethyl cyanoacetate was alkylated with S-14 to give the common intermediate S-15 for all three optically active inhibitors. Treatment of S-15 with thiolacetic acid under modified Mitsunobu conditions provided R-16 with inversion of C10 stereochemistry (Scheme 2). Compound R-16 was subsequently deprotected and the free thiol was methylated to yield R-18. Intermediate R-18 was treated with the free base of guanidine under basic conditions to form the pyrimidine R-19 and this intermediate was subsequently saponified to give R-20. Compound R-20 was coupled with di-tert-butyl l-glutamate and subsequently deprotected using trifluoroacetic acid to provide final inhibitor 10R-3. The C10 diastereomer 10S-3 was prepared using an analogous route employing the enantiomeric R-Me-CBS catalyst to provide R-14 (See Supporting Information).
Scheme 2.
The inhibitor 10S-7 bearing a C10-hydroxyl substituent, was prepared from the common intermediate S-15 (Scheme 3). Compound S-15 was treated with guanidine hydrochloride to give the corresponding pyrimidone S-22 which subsequently was saponified using lithium hydroxide to afford S-23. Intermediate S-23 was then coupled with dimethyl l-glutamate and deprotected under basic conditions to provide the final inhibitor 10S-7.
Scheme 3.
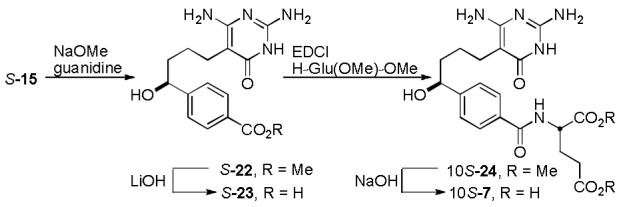
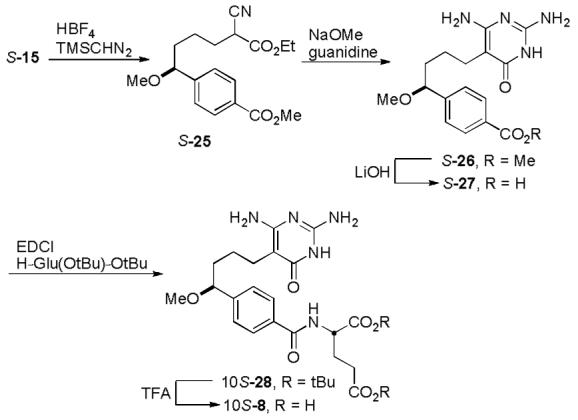
A key compound for comparison is the C10-methoxy derivative 8 where an oxygen replaces the sulfur of 3 as a probe for its unique importance. The synthesis of inhibitor 10S-8, bearing the C10-methoxy substituent, was accomplished by methylation of the common intermediate S-15 using TMSCHN2 under acidic conditions17 to give S-25 (Scheme 4). Intermediate S-25 was carried forward employing an analogous route, starting with formation of pyrimidone S-26 and saponification to give S-27. Compound S-27 was coupled with di-tert-butyl l-glutamate hydrochloride and deprotected using trifluoroacetic acid to provide 10S-8.
Scheme 4.
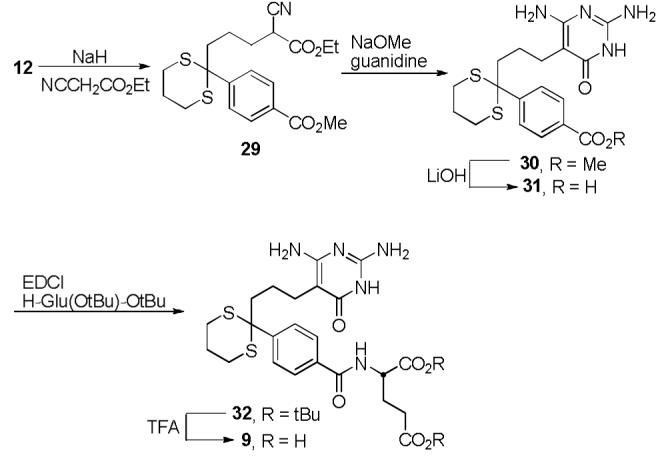
Finally, 9, which incorporates two C10 sulfur atoms and, in a sense, simultaneously incorporates both C10-diastereomers of 3, was judged as an attractive additional derivative to examine that avoids the presence of two C10-diastereomers. Synthesis of inhibitor 9 started with alkylation of 12 using ethyl cyanoacetate to give 29 (Scheme 5). Intermediate 29 was converted to 9 using the same route detailed for the previous inhibitors, starting with formation of pyrimidine 30 and saponification to give 31. Compound 31 was coupled with di-tert-butyl l-glutamate and deprotected using trifluoroacetic acid to provide 9.
Scheme 5.
Results and Discussion
Each diastereomer of 3, 7, 8, and 9 was examined for inhibition of rhGAR Tfase and aminoimidazole carboxamide ribonucleotide transformylase (rhAICAR Tfase), and the results are summarized in Table 1. Remarkably, both diastereomers of the thiomethyl derivative 3 exhibited potent activity against rhGAR Tfase with 10S-3 being slightly more potent than 10R-3 (Ki = 180 nM vs Ki = 210 nM, respectively). This nearly indistinguishable activity of the two C10-diastereomers of 3 is only consistent with both possessing near equivalent capabilities for binding at the GAR Tfase active site and suggests that the flexible linker region joining the pyrimidine and phenyl ring conformationally adjusts to allow a comparable positioning of the thiomethyl substituent in the formyl transfer pocket of the GAR Tfase active site. Inhibitors 10R-7 and 10S-7 displayed very modest activity (Ki = 1060 nM and 1210 nM, respectively) against rhGAR Tfase. This represents a 6-fold reduction in activity relative to 3 and again both diastereomers proved active, although it was the C10-R diastereomer that was slightly more potent than the corresponding C10-S diastereomer. Even more interestingly, methoxy derivatives 10R-8 and 10S-8 were found to inhibit rhGAR Tfase with potencies nearly equivalent to the corresponding alcohols (Ki = 850 nM and 1240 nM, respectively). Again, while both diastereomers exhibited near equivalent potencies, the C10-R diastereomer was slightly more effective, and the 4-6 fold distinction relative to 3 highlights the special nature of the thiomethyl versus methoxy interaction at the enzyme active site. Inhibitor 9 also exhibited a reduced binding to rhGAR Tfase (Ki = 1060 nM), illustrating that optimally one, but not two, thioalkyl substituents may enhance binding at the enzyme active site. These results define a clear role and remarkable stereochemical accommodation of the C10-substituents on DDACTHF with a unique stabilization attributable to the C10-thiomethyl group.
Table 1.
GAR and AICAR Tfase Inhibition (Ki, μM)
| Compound | rhGAR Tfase | rhAICAR Tfase |
|---|---|---|
| 10R-3 | 0.21 | > 100 |
| 10S-3 | 0.18 | > 100 |
| 10R-7 | 1.1 | > 100 |
| 10S-7 | 1.4 | > 100 |
| 10R-8 | 0.85 | > 100 |
| 10S-8 9 DDACTHF 2 |
1.2 1.1 1.7 0.03 |
> 100 20 20 > 100 |
| Lometrexol | 0.06a | > 100 |
Ref 18
All inhibitors were also examined for activity against rhAICAR Tfase. The only compound in the series to show any activity against the enzyme was dithiane 9 (Ki = 20 μM). Each diastereomer of the thiomethyl, hydroxyl, and methoxy inhibitors was inactive against rhAICAR Tfase (Ki >100 μM) illustrating that they disrupt de novo purine biosynthesis by selectively targeting GAR Tfase.
The compounds were also examined for cytotoxic activity (growth inhibition) both in the presence (+) and absence (-) of added hypoxanthine (purine) or thymidine (pyrimidine) against the CCRF-CEM cell line (Table 2). All the inhibitors exhibited activity that paralleled their potency against rhGAR Tfase. Of all the inhibitors, only 10R-3 and 10S-3 exhibited potent activity in this cell-based assay (IC50 = 80 and 50 nM, respectively), being only 3-5 times less potent than 2, the most potent inhibitor of GAR Tfase disclosed to date. While both C10-diastereomers displayed this potent cytotoxic activity, the 10S-diastereomer proved to be slightly more active than the 10R-diastereomer, and both display a potency that slightly exceeds that observed on the isolated enzyme. Compounds 10R-7, 10S-7, 10R-8, and 10S-8 were all approximately ten times less potent than the thiomethyl inhibitors. All still showed modest activity and there was little distinction between the individual C10 diastereomers. Inhibitor 9, on the other hand, showed no activity in this cell-based assay. An especially interesting set of comparisons are the activities of the two diastereomers of 3 and the related inhibitors with DDACTHF lacking a C10 substituent. These comparisons indicate that each of the substituents actually enhance, or at least maintain, the GAR Tfase inhibition of DDACTHF and enhance and maintain its corresponding cell growth inhibitory activity. Thus, the differences observed among one another and relative to DDACTHF may be attributed to relative binding enhancements provided by the key C10 substituents and not to destabilizing interactions they introduce at the enzyme active site. Finally, in the presence of thymidine, a pyrimidine, all inhibitors retained their activity, whereas the inhibitors were inactive in the presence of added hypoxanthine. This indicates that these compounds, like 2, inhibit cell growth by selectively inhibiting an enzyme within the purine biosynthetic (not pyrimidine) pathway consistent with their potent inhibition of GAR Tfase.
Table 2.
In vitro Cytotoxic Activity
| Compound | CCRF-CEM (IC50, μM) | ||
|---|---|---|---|
| (-) T, (-) H | (+) T, (-) H | (-) T, (+) H | |
| 10R-3 | 0.08 | 0.08 | >10 |
| 10S-3 | 0.05 | 0.07 | >10 |
| 10R-7 | 2.4 | 5.7 | >10 |
| 10S-7 | 0.5 | 0.6 | >10 |
| 10R-8 | 0.5 | 0.7 | >10 |
| 10S-8 | 0.7 | 0.9 | >10 |
| 9 | >100 | >100 | >10 |
| DDACTHF 2 |
2.7 0.016 |
3.6 0.017 |
>10 >10 |
| Lometrexol | 0.2 | 0.2 | >10 |
All seven inhibitors were subsequently examined in mutant CCRF-CEM cell lines that lack folypolyglutamate synthetase (CCRF-CEM/FPGS-) or the reduced folate carrier (CCRF-CEM/MTX) (Table 3). Like the past inhibitors of GAR Tfase that we have examined, all inhibitors including 3 lost activity against the cell line that lacks FPGS (ca. 100-fold for 3), indicating that they benefit from intracellular polyglutamation. This observation suggests that the potent activity of both diastereomers of 3 in the cell growth assays, which exceed their inhibition of GAR Tfase roughly 3-fold, may be attributed to their FPGS polyglutamation and the resulting intracellular accumulation or enhanced target affinity. In contrast, none of the inhibitors disclosed herein exhibited altered activity against the cell line lacking the reduce folate carrier indicating its transport of the inhibitors is not essential to their activity. Thus, cell lines and tumors that derive their resistance to antifolates through down regulation of the reduced folate carrier would be expected to remain sensitive to the two diastereomers of 3.
Table 3.
In vitro Cytotoxic Activity in Mutant Cell Lines
| Compound | IC50, μM [(-)T, (-)H] | ||
|---|---|---|---|
| CCRF-CEM | CCRF-CEM/FPGS- | CCRF-CEM/MTX | |
| 10R-3 | 0.09 | 5.5 | 0.07 |
| 10S-3 | 0.06 | 5.0 | 0.06 |
| 10R-7 | 5.50 | >10 | 4.4 |
| 10S-7 | 0.60 | >10 | 0.6 |
| 10R-8 | 0.60 | 6.0 | 0.6 |
| 10S-8 | 0.80 | >10 | 0.9 |
| 9 | >100 | nd | nd |
| DDACTHF 2 |
2.7 0.06 |
>10 >10 |
>100 nd |
| Lometrexol | 0.20 | >10 | nd (>100)a |
Ref 18
Conclusions
Examination of the importance of the C10 stereochemistry of DDACTHF-based inhibitors was addressed with the asymmetric synthesis of the two diastereomers of 10-methylthio-DDACTHF (3) and a series of key analogues incapable of in situ racemization of the C10 center. Remarkably, the activities of the C10-substituted DDACTHF derivatives proved to be essentially independent of the C10 stereochemistry and those bearing a C10-thiomethyl substituent were surprisingly potent and efficacious. Presumably this reflects the conformational flexibility of the linking region between the pyrimidine and phenyl group which contains the C10-substituent that allows a comparable positioning of the thiomethyl group in the formyl transfer pocket of the GAR Tfase active site. In addition to being substantially more potent than the corresponding C10-methoxy derivatives highlighting the unique properties embodied in 3, the two diastereomers of 3 proved especially efficacious in inhibiting cell growth (CCRF-CEM IC50 = 80 and 50 nM for 10R-3 and 10S-3, respectively) that could be rescued by added purine (but not pyrimidine) and that relies on intracellular FPGS polyglutamation, but not intracellular transport by the reduced folate carrier. Similarly, both diastereomers of 3 were found to be potent and essentially indistinguishable inhibitors of rhGAR Tfase (Ki = 210 and 180 nM for 10R-3 and 10S-3, respectively), albeit with potencies that are slightly lower than their actual cell growth inhibitory activity. This latter observation suggests that the required intracellular polyglutamation observed with 3 functionally enhances its potency in the cell-based assays.
Experimental Section
Characterization of the compounds with the alternative C10-stereochemistry is provided in the Supporting Information. Our use of “concentrated” in the following experimental refers to evaporation to dryness on a rotary evaporator.
Methyl 4-(1,3-Dithian-2-yl)benzoate (11)
A solution of methyl 4-formylbenzoate (14.10 g, 85.9 mmol) in anhydrous CH2Cl2 (282 mL) was treated with 1,3-propanedithiol (9.48 mL, 94.5 mmol, 1.1 equiv) and the mixture was stirred for 1 h at room temperature. The solution was cooled to 0 °C and boron trifluoride diethyl etherate was added (11.9 mL, 94.5 mmol, 1.1 equiv). The reaction mixture was stirred and allowed to warm to room temperature overnight. The mixture was diluted with CH2Cl2 and quenched with the addition of saturated aqueous NaHCO3. The organic layer was washed with H2O and saturated aqueous NaCl, dried over Na2SO4, and concentrated (evaporated to dryness on a rotary evaporator). Column chromatography (SiO2, 25-50% EtOAc/hexanes gradient) yielded 11 (21.0 g, 96%) as a white solid: mp 132-134°C; 1H NMR (500 MHz, CDCl3) δ 8.02 (d, 2H, J = 8.3 Hz), 7.55 (d, 2H, J = 8.3 Hz), 5.22 (s, 1H), 3.92 (s, 3H), 3.10-3.04 (m, 2H), 2.96-2.92 (m, 2H), 2.22-2.19 (m, 1H), 1.99-1.95 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 166.8, 144.2, 130.3 (2C), 130.2, 128.1 (2C), 52.4, 51.3, 32.1 (2C), 25.2; IR (film) νmax 2903, 1707, 1272, 1108 cm-1 ; ESI-TOF m/z 255.0507 (M + H+ , C12H14O2S2 requires 255.0508).
Methyl 4-(2-(3-Chloropropyl)-1,3-dithian-2-yl)benzoate (12)
A solution of 11 (20.96 g, 82.4 mmol) in THF (629 mL) cooled to -78 °C was treated with NaHMDS (2 M in THF, 51.5 mL, 103 mmol, 1.25 equiv) dropwise. The solution was stirred for 30 min followed by the addition of 1-chloro-3-iodopropane (44.3 mL, 412 mmol, 5.0 equiv). The reaction mixture was allowed to warm to room temperature overnight and was quenched with the addition of saturated aqueous NH4Cl. The mixture was diluted with EtOAc and subsequently washed with saturated aqueous NaCl, dried over Na2SO4, and concentrated. Column chromatography (SiO2, 0-10% EtOAc/hexanes gradient) yielded 12 (21.73 g, 80%) as a white solid: mp 75-77 °C; 1H NMR (500 MHz, CDCl3) δ 8.05 (d, 2H, J = 8.7 Hz), 8.00 (d, 2H, J = 8.7 Hz), 3.92 (s, 3H), 3.40 (t, 2H, J = 6.4 Hz), 2.75-2.60 (m, 4H), 2.17-2.14 (m, 2H), 1.96-1.93 (m, 2H), 1.75-1.70 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 166.8, 147.2, 130.0 (2C), 129.2, 129.0 (2C), 58.1, 52.3, 44.7, 42.3, 27.8 (2C), 27.3, 25.0; IR (film) νmax 2944, 1723, 1277 cm-1 ; ESI-TOF m/z 331.0587 (M + H+ , C15H19ClO2S2 requires 331.0588).
Methyl 4-(4-Chlorobutanoyl)benzoate (13)
A solution of 12 (10.79 g, 32.6 mmol) in 9:1 MeCN/H2O (163 mL) was treated with bis(trifluoro)acetoxyiodobenzene (21.0 g, 48.9 mmol, 1.5 equiv) and allowed to stir for 30 min. The reaction mixture was quenched with the addition of saturated aqueous NaHCO3 and diluted with EtOAc. The mixture was extracted with H2O (2×) and saturated aqueous NaCl, dried over Na2SO4, and concentrated. Column chromatography (SiO2, 20% EtOAc/hexanes) provided 13 (6.37 g, 81%) as a white solid: mp 45-46 °C; 1H NMR (500 MHz, CDCl3) δ 8.15 (d, 2H, J = 8.3Hz), 8.04 (d, 2H, J = 8.2 Hz), 3.96 (s, 3H), 3.70 (t, 2 H, J = 6.3 Hz), 3.22 (t, 2H, J = 6.9 Hz), 2.28-2.22 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 198.6, 166.3, 140.0, 134.1, 130.0 (2C), 128.0 (2C), 52.6, 44.6, 35.8, 26.7; IR (film) νmax 2944, 1723, 1682, 1272, 1108 cm-1 ; ESI-TOF m/z 241.0624 (M + H+ , C12H13ClO3 requires 241.0626).
Methyl (S)-4-(4-Chloro-1-hydroxybutyl)benzoate (S-14)
A solution of R-Me-CBS (1 M in toluene, 1.09 mL, 1.09 mmol, 0.1 equiv) in anhydrous THF (10.1 mL) cooled to 0 °C was treated with borane dimethysulfide complex (2 M in THF, 6.80 mL, 13.6 mmol, 1.25 equiv) and stirred for 15 min at 0 °C. A solution of 13 (2.62 g, 10.9 mmol) in THF (54.4 mL) was added dropwise to the reaction mixture and stirred for 1 h, allowing the reaction mixture to warm to room temperature. The reaction was quenched with the addition of MeOH and allowed to stir for 30 min at room temperature. The mixture was diluted with EtOAc and washed with 1 N HCl (3×), H2O (2×), dried over Na2SO4, and concentrated. Column chromatography (SiO2, 40% EtOAc/hexanes) afforded S-14 (2.55 g, 97%) as a clear oil: 1H NMR (500 MHz, CDCl3) δ 8.04 (d, 2H, J = 8.2 Hz), 7.44 (d, 2H, J = 8.4 Hz), 4.81 (t, 1H, J = 5.9 Hz), 3.92 (s, 3H), 3.60-3.53 (m, 2H), 1.97-1.82 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 167.0, 149.6, 130.0 (2C), 129.5, 125.8 (2C), 73.4, 52.2, 45.0, 36.3, 28.8; IR (film) νmax 2923, 1748, 1277 cm-1 ; ESI-TOF m/z 243.0781 (M + H+ , C12H15ClO3 requires 243.0782); [α]D -30 (c 1.0, CHCl3). Enantiomeric excess was determined (99% ee) using a Chiralcel AD-H analytical column (250 mm × 4.6 mm, 3% i-PrOH/hexane, 1 mL/min, α = 1.07).
Methyl 4-((1S)-5-Cyano-6-ethoxy-1-hydroxy-6-oxohexyl)benzoate (S-15)
A suspension of NaH (60% dispersion, 1.11 g, 27.7 mmol, 3.0 equiv) in anhydrous DMF (27.7 mL) was treated with ethyl cyanoacetate (2.96 mL, 27.7 mmol, 3.0 equiv) at 0 °C. The reaction mixture was stirred for 30 min while allowing the mixture to warm to room temperature, forming the sodium salt as a clear solution. A solution of S-14 (2.24 g, 9.24 mmol) in DMF (18.5 mL) was added at room temperature and the reaction mixture was stirred at 60 °C for 12 h. The reaction mixture was cooled to room temperature and quenched with the addition of saturated aqueous NH4Cl. The mixture was concentrated and then diluted with EtOAc. The organic layer was washed with H2O and saturated aqueous NaCl, dried over Na2SO4, and concentrated. Column chromatography (SiO2, 50% EtOAc/hexanes) provided S-15 (1.76 g, 60%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.04 (d, 2H, J = 8.2 Hz), 7.43 (d, 2H, J = 8.2 Hz), 4.78 (t, 1H, J = 6.0 Hz), 4.26/4.25 (two q, 2H, J = 7.1 Hz), 3.92 (s, 3H), 3.49/3.48 (two t, 1H, J = 6.9 Hz), 2.00-1.97 (m, 2H), 1.85-1.65 (m, 4H), 1.31/1.30 (two t, 3H, J = 7.1 Hz); 13C NMR (125 MHz, CDCl3) δ 167.0, 166.1, 149.6, 130.0 (2C), 129.6, 125.8 (2C), 116.5, 73.6, 63.0, 52.2, 38.1, 37.6, 29.8, 23.2, 14.1; IR (film) νmax 3467, 2923, 1744, 1713, 1282, 1195, 1113 cm-1; ESI-TOF m/z 342.1309 (M + Na+, C17H21NO5 requires 342.1312).
Methyl 4-((1R)-1-(Acetylthio)-5-cyano-6-ethoxy-6-oxohexyl)benzoate (R-16)
A cooled (0 °C) solution of triphenylphosphine (358 mg, 1.36 mmol, 2.0 equiv) in THF (3.4 mL) was treated with DIAD (270 μL, 1.36 mmol, 2.0 equiv). The reaction mixture was stirred at 0 °C for 30 min before a solution of S-15 (218 mg, 0.68 mmol) and thiolacetic acid (121 uL, 1.36 mmol, 2.0 equiv) in THF (1.7 mL) was added dropwise. The mixture was stirred for 1 h, while allowing it to warm to room temperature. The reaction mixture was diluted with EtOAc, washed with saturated aqueous NaHCO3 (3×), dried over Na2SO4, and concentrated. Column chromatography (SiO2, 30% EtOAc/hexanes) yielded R-16 (246 mg, 95%) as a yellow oil: 1H NMR (500 MHz, CDCl3) δ 8.00 (d, 2H, J = 8.3 Hz), 7.36 (d, 2H, J = 8.2 Hz), 4.60 (t, 1H, J = 7.7 Hz), 4.25/4.24 (two q, 2H, J = 7.1 Hz), 3.91 (s, 3H), 3.47-3.43 (m, 1H), 2.31 (s, 3H), 2.02-1.94 (m, 4H), 1.54-1.43 (m, 2H), 1.29/1.28 (two t, 3H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3) δ 194.4, 166.8, 165.9, 146.6, 130.2 (2C), 129.5, 127.7 (2C), 116.4, 63.0, 52.3, 47.2, 37.4, 35.1, 30.6, 29.4, 24.7, 14.1; IR (film) νmax 3733, 2974, 1733, 1641, 1256, 1108 cm-1 ; ESI-TOF m/z 400.1191 (M + Na+ , C19H23NO5S requires 400.1189).
Methyl 4-((1R)-5-Cyano-6-ethoxy-1-mercapto-6-oxohexyl)benzoate (R-17)
A solution of R-16 (2.01 g, 5.3 mmol) in EtOH (26.6 mL) was treated with K2CO3 (3.68 g, 26.6 mmol, 5.0 equiv) and stirred for 12 h. The reaction mixture was diluted with EtOAc and washed with H2O and saturated aqueous NaCl, dried over Na2SO4, and concentrated. Column chromatography (SiO2, 30% EtOAc/hexanes) yielded the unstable free thiol R-17 (291 mg, 16%) as a colorless oil: 1H NMR (600 MHz, CDCl3) δ 8.00 (d, 2H, J = 7.8 Hz), 7.38 (d, 2H, J = 8.2 Hz), 4.26-4.21 (m, 2H), 4.01-3.98 (m, 1H), 3.91 (s, 3H), 3.47-3.43 (m, 1H), 1.99-1.92 (m, 4H), 1.96 (d, 1H, J = 5.6 Hz), 1.66-1.58 (m, 1H), 1.49-1.40 (m, 1H), 1.30-1.26 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 166.7, 165.9, 149.4, 130.3 (2C), 129.4, 126.9 (2C), 116.4, 63.0, 52.2, 43.4, 38.6, 37.4, 29.4, 25.2, 14.1; IR (film) νmax 2933, 1718, 1436, 1271, 1184, 1102 cm-1 ; ESI-TOF m/z 336.1267 (M + H+ , C17H21NO4S requires 336.1264).
Methyl 4-((1R)-5-cyano-6-ethoxy-1-(methylthio)-6-oxohexyl)benzoate (R-18)
Free thiol R-17 (291 mg, 0.87 mmol) dissolved in MeOH (8.68 mL) was treated with MeI (542 μL, 8.68 mmol, 10 equiv) and NaHCO3 (80 mg, 0.97 mmol, 1.1 equiv) and the mixture was stirred for 3 h at room temperature. The reaction mixture was diluted with EtOAc and washed with H2O and saturated aqueous NaCl, dried over Na2SO4, and concentrated. Column chromatography (SiO2, 30% EtOAc/hexanes) provided R-18 (238 mg, 79%) as a colorless oil: 1H NMR (600 MHz, CDCl3) δ 8.01 (d, 2H, J = 7.3 Hz), 7.37 (d, 2H, J = 7.8 Hz), 4.26-4.20 (m, 2H), 3.91 (s, 3H), 3.69 (t, 1H, J = 7.4 Hz), 3.46-3.41 (m, 1H), 1.99-1.87 (m, 3H), 1.85 (s, 3H), 1.64-1.56 (m, 1H), 1.50-1.45 (m, 1H), 1.31-1.26 (m, 3H); 13C NMR (150 MHz, CDCl3) δ 166.9, 166.0, 147.4, 130.1 (2C), 129.3, 127.9 (2C), 116.4, 63.0, 52.2, 50.9, 37.5, 35.1, 29.6, 25.1, 14.4, 14.1; IR (film) νmax 3358, 2923, 1718, 1431, 1267, 1097, 1010 cm-1 ; ESI-TOF m/z 372.1252 (M + Na+ , C18H23NO4S requires 372.1240).
Methyl (R)-4-(4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-(methylthio)butyl)benzoate (R-19)
A solution of NaOMe (60 mg, 1.12 mmol, 3.0 equiv) in anhydrous MeOH (3 mL) was treated with guanidine hydrochloride (107 mg, 1.12 mmol, 3.0 equiv) at room temperature. After the reaction mixture was stirred for 30 min, a solution of R-18 (130 mg, 0.372 mmol) in MeOH (2.10 mL) was added. The reaction mixture was warmed at reflux for 16 h before being quenched with acetic acid after cooling to room temperature. After concentration, column chromatography (SiO2, 10% MeOH/CH2Cl2) gave R-19 (39 mg, 29%) as a white solid: mp 180-185 °C; 1H NMR (500 MHz, CD3OD) δ 7.96 (d, 2H, J = 8.4 Hz), 7.43 (d, 2H, J = 8.4 Hz), 3.89 (s, 3H), 3.81 (t, 1H, J = 6.7 Hz), 2.29 (t, 2H, J = 7.2 Hz), 1.97-1.86 (m, 2H), 1.84 (s, 3H), 1.53-1.47 (m, 1H), 1.42-1.34 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 168.4, 165.2, 164.1, 154.8, 150.2, 130.6 (2C), 129.8, 129.2 (2C), 89.8, 52.6, 52.2, 36.3, 27.2, 23.1, 14.2; IR (film) νmax 3353, 2933, 1713, 1610, 1431, 1282, 1108 cm-1 ; ESI-TOF m/z 363.1489 (M + H+ , C17H22N4O3S requires 363.1485); [α]D +70 (c 0.1, MeOH).
(R)-4-(4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-(methylthio)butyl) benzoic Acid (R-20)
A solution of R-19 (39 mg, 0.11 mmol) in MeOH (1.5 mL) was treated with LiOH monohydrate (23 mg, 0.54 mmol, 5 equiv) in water (840 μL), and the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was diluted with H2O, washed with EtOAc, acidified to pH 4 by the addition of aqueous 1 N HCl, and concentrated. The resulting white solid, R-20 (38 mg, 100%), was used directly in the next step: mp 210 °C (dec); 1H NMR (500 MHz, CD3OD) δ 7.97 (d, 2H, J = 8.3 Hz), 7.43 (d, 2H, J = 8.4 Hz), 3.81 (t, 1H, J = 6.8 Hz), 2.35-2.31 (m, 2H), 1.97-1.87 (m, 2H), 1.85 (s, 3H), 1.55-1.48 (m, 1H), 1.43-1.36 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 169.6, 164.3, 153.4, 152.6, 149.8, 130.8 (2C), 130.5, 129.1 (2C), 89.6, 52.0, 36.2, 26.9, 22.5, 14.3; IR (film) νmax3356, 1569, 1405, 1241 cm-1; ESI-TOF m/z 349.1336 (M + H+ , C16H20N4O3S requires 349.1329); [α]D +30 (c 0.1, MeOH).
Di-tert-butyl (S)-2-(4-((R)-4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-(methylthio)butyl)benzamido)pentanedioate (10R-21)
A solution of R-20 (38 mg, 0.11 mmol), di-tert-butyl l-glutamate hydrochloride (48 mg, 0.16 mmol, 1.5 equiv) and NaHCO3 (27 mg, 0.32 mmol, 3.0 equiv) in DMF (1.08 mL) was treated with EDCI (62 mg, 0.32 mmol, 3.0 equiv). The reaction mixture was stirred for 24 h at room temperature before the addition of CHCl3. The resulting solution was washed with saturated aqueous NaHCO3 (2×), dried over Na2SO4, and concentrated. Column chromatography (SiO2, 10% MeOH/CH2Cl2) provided the protected glutamate intermediate 10R-21 (27 mg, 43%) as a white solid: mp 108-112 °C; 1H NMR (500 MHz, CD3OD) δ 7.80 (d, 2H, J = 8.3 Hz), 7.43 (d, 2H, J = 8.3 Hz), 4.52-4.49 (m, 1H), 3.81 (t, 1H, J = 6.8 Hz), 2.40 (t, 2H, J = 7.4 Hz), 2.30 (t, 2H, J = 7.6 Hz), 2.24-2.17 (m, 1H), 2.06-1.99 (m, 1H), 1.97-1.86 (m, 2H), 1.84 (s, 3H), 1.53-1.50 (m, 1H), 1.49 (s, 9H), 1.44 (s, 9H), 1.41-1.36 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 173.9, 172.7, 170.2, 165.1, 164.2, 154.8, 148.6, 133.7, 129.2 (2C), 128.6 (2C), 89.8, 83.0, 81.9, 54.5, 52.1, 36.4, 32.9, 28.3 (6C), 27.6, 27.3, 23.1, 14.2; IR (film) νmax 2913, 1733, 1631, 1149 cm-1 ; ESI-TOF m/z 590.2999 (M + H+ , C29H43N5O6S requires 590.3007); [α]D +36 (c 0.1, MeOH).
(S)-2-(4-((R)-4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-(methylthio)butyl)benzamido)pentanedioic Acid (10R-3)
A solution of 10R-21 (21 mg, 0.035 mmol) in CHCl3 (1.26 mL) was treated with trifluoroacetic acid (1 mL) at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight. The solution was concentrated and triturated with Et2O to give 10R-3 (20 mg, 100%) as a white solid: mp 190 °C (dec); 1H NMR (500 MHz, CD3OD) δ 7.82 (d, 2H, J = 8.4 Hz), 7.42 (d, 2H, J = 8.3 Hz), 4.66-4.63 (m, 1H), 3.81 (t, 1H, J = 6.6 Hz), 2.49 (t, 2H, J = 7.5 Hz), 2.35-2.28 (m, 3H), 2.15-2.07 (m, 1H), 1.98-1.91 (m, 1H), 1.88-1.82 (m, 1H), 1.84 (s, 3H), 1.54-1.47 (m, 1H), 1.42-1.33 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 176.6, 175.1, 170.2, 164.6, 154.8, 153.0, 148.4, 133.8, 129.2, 128.6, 89.6, 53.8, 52.0, 36.3, 31.5, 27.6, 26.9, 22.6, 14.3; IR (film) νmax 2964, 1653, 1544, 1210 cm-1 ; ESI-TOF m/z 478.1752 (M + H+ , C21H27N5O6S requires 478.1755); [α]D +32 (c 0.1, MeOH). Agilent 1100 LC/MS: reverse phase (2-40% acetonitrile/water/0.1% formic acid, flow rate 0.75 mL/min); tR, 12.0 min; purity, 99.2%.
Methyl (S)-4-(4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-hydroxybutyl)benzoate (S-22)
A solution of NaOMe (270 mg, 5.01 mmol, 3.0 equiv) in anhydrous MeOH (13.8 mL) was treated with guanidine hydrochloride (478 mg, 5.01 mmol, 3.0 equiv) at room temperature. After the reaction mixture was stirred for 30 min, a solution of S-15 (533 mg, 1.67 mmol) in MeOH (9.4 mL) was added. The reaction mixture was warmed at reflux for 16 h and quenched with acetic acid after cooling to room temperature. After concentration, column chromatography (SiO2, 10% MeOH/CH2Cl2) gave S-22 (336 mg, 61%) as a white solid: mp 120 °C (dec); 1H NMR (500 MHz, CD3OD) δ 7.97 (d, 2H, J = 8.4 Hz), 7.46 (d, 2H, J = 8.2 Hz), 4.73 (t, 1H, J = 7.4 Hz), 3.89 (s, 3H), 2.35-2.32 (m, 2H), 1.77-1.74 (m, 2H), 1.61-1.52 (m, 1H), 1.50-1.42 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 168.0, 164.7, 163.7, 152.1, 152.0, 130.0 (2C), 129.9, 126.6 (2C), 89.5, 74.1, 52.0, 39.1, 24.9, 22.7; IR (film) νmax 3333, 2912, 1718, 1615, 1436, 1287, 1102 cm-1 ; ESI-TOF m/z 333.1562 (M + H+ , C16H20N4O4 requires 333.1557); [α]D -8 (c 0.1, MeOH).
(S)-4-(4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-hydroxybutyl)benzoic Acid (S-23)
A solution of S-22 (36 mg, 0.11 mmol) in MeOH (1.5 mL) was treated with LiOH monohydrate (23 mg, 0.54 mmol, 5 equiv) in water (840 μL), and the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was diluted with H2O, washed with EtOAc, acidified to pH 4 by the addition of aqueous 1 N HCl, and concentrated. The resulting white solid, S-23 (35 mg, 100%), was used directly in the next step: mp 220 °C (dec); 1H NMR (500 MHz, CD3OD) δ 7.98 (d, 2H, J = 8.3 Hz), 7.45 (d, 2H, J = 8.4 Hz), 4.73 (t, 1H, J = 7.3 Hz), 2.36 (t, 2H, J = 7.4 Hz), 1.78-1.73 (m, 2H), 1.61-1.55 (m, 1H), 1.50-1.44 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 169.8, 165.2, 163.5, 158.4, 152.2, 130.8 (2C), 130.5, 127.0 (2C), 89.9, 74.6, 39.4, 25.2, 22.8; IR (film) νmax 3374, 2923, 1636, 1390 cm-1; ESI-TOF m/z 319.1405 (M + H+ , C15H18N4O4 requires 319.1401); [α]D -12 (c 0.1, MeOH).
Dimethyl (S)-2-(4-((S)-4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-hydroxybutyl)benzamido)pentanedioate (10S-24)
A solution of S-23 (29 mg, 0.090 mmol), dimethyl l-glutamate hydrochloride (29 mg, 0.14 mmol, 1.5 equiv) and NaHCO3 (23 mg, 0.27 mmol, 3.0 equiv) in DMF (0.90 mL) was treated with EDCI (52 mg, 0.27 mmol, 3.0 equiv). The reaction mixture was stirred for 24 h at room temperature before being concentrated. Column chromatography (SiO2, 20% MeOH/CH2Cl2) provided 10S-24 (31 mg, 72%) as a white solid: mp 105-110 °C; 1H NMR (600 MHz, CD3OD) δ 7.82 (d, 2H, J = 8.4 Hz), 7.46 (d, 2H, J = 7.8 Hz), 4.73 (t, 1H, J = 6.0 Hz), 4.65-4.63 (m, 1H), 3.75 (s, 3H), 3.65 (s, 3H), 2.50 (t, 2H, J = 7.2 Hz), 2.35-2.27 (m, 3H), 2.14-2.09 (m, 1H), 1.79-1.74 (m, 2H), 1.57-1.54 (m, 1H), 1.46-1.43 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 174.9, 173.7, 170.3, 165.2, 164.3, 154.8, 151.2, 133.5, 128.5 (2C), 127.1 (2C), 90.0, 74.7, 60.4, 53.7, 52.8, 52.2, 39.7, 31.3, 27.4, 25.5, 23.3; IR (film) νmax 3364, 1723, 1631, 1441 cm-1 ; ESI-TOF m/z 476.2142 (M + H+ , C22H29N5O7 requires 476.2140); [α]D -18 (c 0.1, MeOH).
(S)-2-(4-((S)-4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-hydroxybutyl)benzamido)pentanedioic Acid (10S-7)
A solution of 10S-24 (26 mg, 0.055 mmol) in MeOH (0.55 mL) was treated with aqueous 1 N NaOH (0.22 mL, 4 equiv). The reaction mixture was stirred for 18 h and then acidified with the addition of Dowex 50X8-200. The solution was filtered and concentrated to give 10S-7 (21 mg, 87%) as a white solid: mp 200 °C (dec); 1H NMR (500 MHz, CD3OD) δ 7.82 (d, 2H, J = 8.3 Hz), 7.45 (d, 2H, J = 8.3 Hz), 4.72 (t, 1H, J = 6.1 Hz), 4.64-4.61 (m, 1H), 2.40-2.24 (m, 5H), 2.17-2.08 (m, 1H), 1.79-1.74 (m, 2H), 1.59-1.54 (m, 1H), 1.49-1.42 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 176.7, 175.1, 174.3, 170.3, 163.7, 163.6, 151.0, 133.8, 128.5 (2C), 127.1 (2C), 89.9, 74.6, 53.8, 39.4, 35.4, 31.0, 25.2, 23.0; IR (film) νmax 3333, 1600, 1451, 1405, 1318, 1108 cm-1 ; ESI-TOF m/z 448.1836 (M + H+ , C20H25N5O7 requires 448.1827); [α]D +4 (c 0.1, MeOH). Agilent 1100 LC/MS: reverse phase (2-40% acetonitrile/water/0.1% formic acid, flow rate 0.75 mL/min); tR, 9.9 min; purity, 99.5%.
Methyl 4-((1S)-5-Cyano-6-ethoxy-1-methoxy-6-oxohexyl)benzoate (S-25)
A solution of S-15 (246 mg, 0.77 mmol), 49% aqueous fluoroboric acid (97 μL, 1.54 mmol, 2 equiv) in CH2Cl2 (3.08 mL) was cooled to 0 °C. TMSCHN2 (2 M in hexanes, 0.385 mL, 0.77 mmol, 1 equiv) was added to the mixture and stirring was continued at 0 °C. Three further portions of TMSCHN2 (0.193 mL (0.5 equiv), 0.097 mL (0.25 equiv), and 0.097 mL (0.25 equiv)) were added dropwise at intervals of 20 min. The reaction mixture was stirred a further 30 min, poured into H2O and extracted with EtOAc. The organic layer was washed with H2O, dried over Na2SO4, filtered, and concentrated. Column chromatography (SiO2, 30% EtOAc/hexanes) gave S-25 (110 mg, 43%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 8.04 (d, 2H, J = 8.2 Hz), 7.36 (d, 2H, J = 8.1 Hz), 4.28-4.22 (m, 2H), 4.18-4.15 (m, 1H), 3.92 (s, 3H), 3.49-3.45 (m, 1H), 3.22 (s, 1.5H), 3.21 (s, 1.5H), 1.98-1.93 (m, 2H), 1.82-1.78 (m, 1H), 1.71-1.63 (m, 2H), 1.54-1.48 (m, 1H), 1.32-1.28 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 166.9, 166.0, 147.3, 129.9 (2C), 129.7, 126.5 (2C), 116.5, 83.1, 62.8, 57.0, 52.1, 37.6, 37.2, 29.7, 23.2, 14.0; IR (film) νmax 3405, 1708, 1272, 1103 cm-1 ; ESI-TOF m/z 356.1465 (M + Na+ , C18H23NO5 requires 356.1468).
Methyl (S)-4-(4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-methoxybutyl)benzoate (S-26)
A solution of NaOMe (44 mg, 0.81 mmol, 3.0 equiv) in anhydrous MeOH (2.2 mL) was treated with guanidine hydrochloride (77 mg, 0.81 mmol, 3.0 equiv) at room temperature. After the reaction mixture was stirred for 30 min, a solution of S-25 (90 mg, 0.27 mmol) in MeOH (1.53 mL) was added. The reaction mixture was warmed at reflux for 16 h and quenched with acetic acid after cooling to room temperature. After concentration, column chromatography (SiO2, 10% MeOH/CH2Cl2) gave S-26 (27 mg, 29%) as a white solid: mp 160-164 °C ; 1H NMR (600 MHz, CD3OD) δ 8.00 (d, 2H, J = 8.4 Hz), 7.41 (d, 2H, J = 7.8 Hz), 4.28-4.25 (m, 1H), 3.90 (s, 3H), 3.20 (s, 3H), 2.30 (t, 2H, J = 7.5 Hz), 1.84-1.78 (m, 1H), 1.68-1.62 (m, 1H), 1.56-1.52 (m, 1H), 1.43-1.36 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 168.4, 165.1, 164.0, 154.8, 149.3, 130.7 (2C), 130.4, 127.9 (2C), 89.8, 84.4, 57.1, 52.6, 38.5, 25.4, 23.2; IR (film) νmax 3354, 2933, 1610, 1431, 1282 cm-1 ; ESI-TOF m/z 347.1729 (M + H+ , C17H22N4O4 requires 347.1714); [α]D -30 (c 0.1, MeOH).
(S)-4-(4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-methoxybutyl)benzoic Acid (S-27)
The intermediate pyrimidine S-26 (30 mg, 0.086 mmol) in MeOH (1.2 mL) was treated with LiOH monohydrate (18 mg, 0.43 mmol, 5 equiv) in water (675 μL), and the reaction mixture was stirred at room temperature for 24 h. The reaction mixture was diluted with H2O, washed with EtOAc, acidified to pH 4 by the addition of 1 N aqueous HCl, and concentrated. The resulting white solid, S-27 (29 mg, 100%), was used directly in the next step: mp 175 °C (dec); 1H NMR (600 MHz, CD3OD) δ 7.95 (d, 2H, J = 8.2 Hz), 7.32 (d, 2H, J = 8.2 Hz), 4.24 (t, 1H, J = 5.8 Hz) 3.19 (s, 3H), 2.30 (t, 2H, J = 7.5 Hz), 1.84-1.78 (m, 1H), 1.70-1.63 (m, 1H), 1.56-1.52 (m, 1H), 1.42-1.36 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 173.2, 165.1, 164.0, 154.9, 147.3, 135.4, 130.6 (2C), 127.4 (2C), 90.0, 85.2, 57.0, 38.5, 25.5, 23.3; IR (film) νmax 3385, 2923, 1626, 1395, 1246, 1087 cm-1 ; ESI-TOF m/z 333.1563 (M + H+ , C16H20N4O4 requires 333.1557); [α]D -20 (c 0.1, MeOH).
Di-tert-butyl (S)-2-(4-((S)-4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-methoxybutyl)benzamido)pentanedioate (10S-28)
A solution of S-27 (29 mg, 0.086 mmol), di-tert-butyl l-glutamate hydrochloride (38 mg, 0.13 mmol, 1.5 equiv) and NaHCO3 (22 mg, 0.26 mmol, 3.0 equiv) in DMF (0.86 mL) was treated with EDCI (50 mg, 0.26 mmol, 3.0 equiv). The reaction mixture was stirred for 24 h at room temperature before the addition of CHCl3. The resulting solution was washed with saturated aqueous NaHCO3 (2×), dried over Na2SO4, and concentrated. Column chromatography (SiO2, 10% MeOH/CH2Cl2) provided 10S-28 (14 mg, 28%) as a white solid: mp 120-125 °C; 1H NMR (600 MHz, CD3OD) δ 7.83 (d, 2H, J = 7.8 Hz), 7.40 (d, 2H, J = 7.8 Hz), 4.51-4.49 (m, 1H), 4.27 (t, 1H, J = 7.4 Hz), 3.21 (s, 3H), 2.40 (t, 2H, J = 7.2 Hz), 2.30 (t, 2H, J = 7.2 Hz), 2.24-2.18 (m, 1H), 2.05-1.99 (m, 1H), 1.85-1.79 (m, 1H), 1.70-1.64 (m, 1H), 1.56-1.52 (m, 1H), 1.49 (s, 9H), 1.44 (s, 9H), 1.41-1.37 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 173.9, 172.6, 170.3, 165.2, 164.3, 154.8, 148.1, 134.4, 128.6 (2C), 127.9 (2C), 90.0, 85.0, 83.0, 81.9, 57.0, 54.5, 38.5, 32.9, 28.3 (6C), 27.5, 25.4, 23.3; IR (film) νmax 3344, 2923, 1723, 1626, 1446, 1369, 1149 cm-1 ; ESI-TOF m/z 574.3234 (M + H+ , C29H43N5O7 requires 574.3235); [α]D -48 (c 0.1, MeOH).
(S)-2-(4-((S)-4-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)-1-methoxybutyl)benzamido)pentanedioic Acid (10S-8)
The protected glutamate 10S-28 (8 mg, 0.015 mmol) in CHCl3 (0.52 mL) was treated with trifluoroacetic acid (0.5 mL) at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight. The solution was concentrated and triturated with Et2O to give 10S-8 (6 mg, 91%) as a white solid: mp 130 °C (dec); 1H NMR (600 MHz, CD3OD) δ 7.84 (d, 2H, J = 8.4 Hz), 7.40 (d, 2H, J = 8.4 Hz), 4.65-4.63 (m, 1H), 4.26 (t, 1H, J = 7.2 Hz), 3.20 (s, 3H), 2.50-2.47 (m, 2H), 2.33-2.30 (m, 3H), 2.14-2.08 (m, 1H), 1.85-1.79 (m, 1H), 1.70-1.64 (m, 1H), 1.58-1.51 (m, 1H), 1.44-1.37 (m, 1H); 13C NMR (150 MHz, CD3OD) δ 176.7, 175.1, 170.2, 165.0, 159.3, 154.0, 147.9, 134.4, 128.6 (2C), 127.9 (2C), 90.0, 85.0, 57.0, 53.8, 38.3, 31.6, 27.6, 25.1, 23.0; IR (film) νmax 3354, 2933, 1636, 1426, 1205 cm-1 ; ESI-TOF m/z 462.1997 (M + H+ , C21H27N5O7 requires 462.1983); [α]D -22 (c 0.1, MeOH). Agilent 1100 LC/MS: reverse phase (2-40% acetonitrile/water/0.1% formic acid, flow rate 0.75 mL/min); tR, 10.9 min; purity, 97.1%.
Methyl 4-(2-(4-Cyano-5-ethoxy-5-oxopentyl)-1,3-dithian-2-yl)benzoate (29)
A suspension of NaH (60% dispersion, 9.19 g, 230 mmol, 3.5 equiv) in anhydrous DMF (202 mL) was treated with ethyl cyanoacetate (24.5 mL, 230 mmol, 3.5 equiv) at 0 °C. The reaction mixture was stirred for 30 min while allowing the mixture to warm to room temperature, forming the sodium salt as a clear solution. A solution of 12 (21.7 g, 65.7 mmol) in DMF (122 mL) was added at room temperature and the reaction mixture was stirred at 60 °C for 12 h. The reaction mixture was cooled to room temperature and quenched with the addition of saturated aqueous NH4Cl. The mixture was concentrated and then diluted with EtOAc. The organic layer was washed with H2O and saturated aqueous NaCl, dried over Na2SO4, and concentrated. Column chromatography (SiO2, 15-50% EtOAc/hexanes gradient) followed by vacuum distillation provided 29 (14.06 g, 53%) as a white solid: mp 54-56 °C; 1H NMR (500 MHz, CDCl3) δ 8.06 (d, 2H, J = 8.6 Hz), 8.00 (d, 2H, J = 8.6 Hz), 4.21 (q, 2H, J = 7.1 Hz), 3.93 (s, 3H), 3.37 (t, 1H, J = 7.6 Hz), 2.70-2.63 (m, 4H), 2.03 (t, 2H, J = 8.3 Hz), 1.97-1.93 (m, 2H), 1.84-1.81 (m, 2H), 1.57-1.42 (m, 2H), 1.26 (t, 3H, J = 7.2 Hz); 13C NMR (125 MHz, CDCl3) δ 166.8, 165.9, 130.1 (2C), 129.2, 129.0 (2C), 116.3, 62.9, 58.4, 52.3, 44.2, 37.4, 29.8, 27.8 (2C), 25.0, 21.6, 14.1; IR (film) 2924, 2360, 1719, 1276, 1105, 1017 cm-1 ; ESI-FTMS m/z 408.1292 (M + H+ , C20H25NO4S2 requires 408.1298).
Methyl 4-(2-(3-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)propyl)-1,3-dithian-2-yl)benzoate (30)
A solution of NaOMe (3.26 g, 60.4 mmol, 3.5 equiv) in anhydrous MeOH (166 mL) was treated with guanidine hydrochloride (5.77 g, 60.4 mmol, 3.5 equiv) at room temperature. After the reaction mixture was stirred for 30 min, a solution of 29 (7.03 g, 17.25 mmol) in MeOH (97.5 mL) was added. The reaction mixture was warmed at reflux for 16 h and quenched with acetic acid after cooling to room temperature. After concentration, column chromatography (SiO2, 10% MeOH/CH2Cl2) gave 30 (6.04 g, 83%) as a white solid: mp dec 150 °C; 1H NMR (500 MHz, CD3OD) δ 8.00 (s, 4H), 3.91 (s, 3H), 2.74-2.59 (m, 4H), 2.19 (t, 2H, J = 7.2 Hz), 2.10-2.05 (m, 2H), 1.94-1.88 (m, 2H), 1.42-1.36 (m, 2H); 13C NMR (125 MHz, DMSO) δ 165.9, 162.3, 161.4, 152.9, 147.7, 129.3 (2C), 128.7 (2C), 128.1, 86.8, 58.1, 52.1, 43.2, 27.0 (2C), 24.6, 22.9, 21.9; IR (film) νmax 3361, 1711, 1646, 1435, 1282, 1111 cm-1; ESI-FTMS m/z 421.1364 (M + H+ , C19H24N4O3S2 requires 421.1363).
4-(2-(3-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)propyl)-1,3-dithian-2-yl)benzoic Acid (31)
A solution of 30 (2.01 g, 4.78 mmol) in MeOH (36 mL) was treated with LiOH monohydrate (1.00 g, 23.9 mmol, 5 equiv) in water (12 mL), and the reaction was stirred at room temperature for 24 h. The reaction mixture was diluted with H2O, washed with Et2O, acidified to pH 4 by the addition of aqueous 1 N HCl, and concentrated. The resulting white solid, 31 (1.48 g, 76%), was used directly in the next step: 1H NMR (500 MHz, CD3OD) δ 8.04 (d, 4H, J = 16.6 Hz), 2.72-2.62 (m, 4H), 2.22 (t, 2H, J = 7.3 Hz), 2.07-2.04 (m, 2H), 1.95-1.89 (m, 2H), 1.41-1.38 (m, 2H); 13C NMR (125 MHz, DMSO) δ 167.8, 167.1, 151.0, 151.4, 147.2, 129.7 (2C), 129.5, 128.6 (2C), 87.2, 58.0, 42.9, 27.2 (2C), 24.7, 22.6, 21.3.
Di-tert-butyl (S)-2-(4-(2-(3-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)propyl)-1,3-dithian-2-yl)benzamido)pentanedioate (32)
Compound 31 (1.48 g, 3.65 mmol), di-tert-butyl l-glutamate hydrochloride (1.62 g, 5.48 mmol, 1.5 equiv) and NaHCO3 (675 mg, 8.03 mmol, 2.2 equiv) in DMF (32 mL) were treated with EDCI (1.40 g, 7.30 mmol, 2.0 equiv). The reaction mixture was stirred for 24 h at room temperature before the addition of CHCl3. The resulting solution was washed with saturated aqueous NaHCO3 (2×), dried over Na2SO4, and concentrated. Column chromatography (SiO2, 10% MeOH/CH2Cl2) provided 32 (1.76 g, 75%) as a white solid: mp 120-124 °C; 1H NMR (500 MHz, CD3OD) δ 8.00 (d, 2H, J = 8.5 Hz), 7.84 (d, 2H, J = 8.5 Hz), 4.53-4.50 (m, 1H), 2.72-2.62 (m, 4H), 2.41 (t, 2H, J = 6.7 Hz), 2.37-2.30 (m, 2H), 2.19 (t, 2H, J = 7.5 Hz), 2.10-2.06 (m, 2H), 1.97-1.91 (m, 2H), 1.49 (s, 9H), 1.45 (s, 9H), 1.42-1.36 (m, 2H); 13C NMR (125 MHz, DMSO) δ 171.5, 171.0, 166.4, 161.4, 157.1, 153.0, 145.5, 132.5, 128.2 (2C), 127.6 (2C), 86.8, 80.6, 79.7, 58.2, 52.5, 43.3, 31.4, 30.9, 27.7 (3C), 27.6 (3C), 27.0, 25.9, 23.0, 22.0; IR (film) νmax 3349, 2961, 1728, 1617, 1440, 1364, 1183 cm-1 ; ESI-FTMS m/z 648.2890 (M + H+ , C31H45N5O6S2 requires 648.2884).
(S)-2-(4-(2-(3-(2,4-Diamino-6-oxo-1,6-dihydropyrimidin-5-yl)propyl)-1,3-dithian-2-yl)benzamido)pentanedioic Acid (9)
A solution of 32 (54 mg, 0.083 mmol) in CHCl3 (833 μL) was treated with trifluoroacetic acid (1 mL) at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight. The solution was concentrated and triturated with Et2O to give 9 (41 mg, 91%) as a white solid: mp dec 125-130 °C; 1H NMR (500 MHz, CD3OD) δ 7.98 (d, 2H, J = 8.6 Hz), 7.84 (d, 2H, J = 8.5 Hz), 4.68-4.65 (m, 1H), 2.72-2.62 (m, 4H), 2.51-2.48 (m, 2H), 2.43-2.39 (m, 2H), 2.19 (t, 2H, J = 7.1 Hz), 2.15-2.11 (m, 2H), 1.94-1.98 (m, 2H), 1.40-1.37 (m, 2H); 13C NMR (125 MHz, CD3OD) δ 176.9, 175.8, 170.0, 164.9, 154.0 (2C), 147.7, 133.7, 130.2 (2C), 128.5 (2C), 87.9, 59.5, 53.9, 44.8, 31.1, 28.6 (2C), 27.6, 26.9, 23.2, 22.7; IR (film) νmax 3380, 2961, 1718, 1634 cm-1 ; ESI-FTMS m/z 536.1625 (M + H+ , requires C23H29N5O6S2 536.1632). Agilent 1100 LC/MS: reverse phase (2-40% acetonitrile/water/0.1% formic acid, flow rate 0.75 mL/min); tR, 13.7 min; purity, 99.5%.
GAR and AICAR Tfase Assay
Enzyme assays for rhGAR and rhAICAR were performed as described previously.8,9 Kinetics of the enzyme reactions were monitored for 2 min after initiation of the reaction. Ki’s of the inhibitors were calculated using Dixon plots.
Cytotoxic Activity
The cytotoxic activity of the compounds was measured using the CCRF-CEM human leukemia cell lines as described previously.8,9,19
Supplementary Material
Acknowledgement
We gratefully acknowledge the financial support of the National Institutes of Health and the Skaggs Institute for Chemical Biology.
Abbreviations
- GAR Tfase
glycinamide ribonucleotide transformylase
- rhGAR Tfase
recombinant human GAR Tfase
- FPGS
folypolyglutamate synthetase
- 10-formyl-THF
10-formyltetrahydrofolic acid
- MTX
methotrexate
- AICAR
aminoimidazole carboxamide ribonucleotide transformylase
- rhAICAR Tfase
recombinant human AICAR Tfase
- DDACTHF
dideaza-acyclotetrahydrofolic acid
References
- (1).Warren L, Buchanan JM. 2-Amino-N-ribosylacetamide 5′-phosphate (glycinamide ribotide) transformylase. J. Biol. Chem. 1957;229:1979–1992. [PubMed] [Google Scholar]
- (2).Dev IK, Harvey RJ. N10-Formyltetrahydrofolate is the formyl donor for glycinamide ribonucleotide transformylase in Escherichia coli. J. Biol. Chem. 1978;253:4242–4244. [PubMed] [Google Scholar]
- (3).Newell DR. Clinical pharmokinetics of antitumor antifolates. Semin. Oncol. 1999;26:74–81. [PubMed] [Google Scholar]
- (4).Takimoto CH. Antifolates in clinical development. Semin. Oncol. 1997;24:A18-40–S18-51. [PubMed] [Google Scholar]
- (5).Taylor EC, Harrington PJ, Fletcher SR, Beardsley GP, Moran RG. Synthesis of the antileukemic agents 5,10-dideazaaminopterin and 5,10-dideaza-5,6,7,8-tetrahydroaminopterin. J. Med. Chem. 1985;28:914–921. doi: 10.1021/jm00145a012. [DOI] [PubMed] [Google Scholar]
- (6).Chen ZH, Olopade OI, Savarese TM. Expression of methylthioadenosine phosphorylase cDNA in p16-, MTAP- malignant cells: restoration of methylthioadenosine phosphorylase-dependent salvage pathways and alterations of sensitivity to inhibitors of purine de novo synthesis. Mol. Pharmacol. 1997;52:903–911. doi: 10.1124/mol.52.5.903. [DOI] [PubMed] [Google Scholar]
- (7).Jackson RC, Harkrader RJ. The contributions of de novo and salvage pathways of nucleotide biosynthesis in normal and malignant cells. In: Tattersall MHN, Fox RM, editors. Nucleosides and Cancer Treatment. Academic Press; Sydney: 1981. pp. 18–31. [Google Scholar]
- (8).Marsilje TH, Labroli MA, Hedrick MP, Jin Q, Desharnais J, Baker SJ, Gooljarsingh LT, Ramcharan J, Tavassoli A, Zheng Y, Wilson IA, Beardsley GP, Benkovic SJ, Boger DL. 10-Formyl-5,10-dideaza-acyclic-5,6,7,8-tetrahydrofolic acid (10-formyl-DDACTHF): A potent cytotoxic agent acting by selective inhibition of human GAR Tfase and the de novo purine biosynthetic pathway. Bioorg. Med. Chem. 2002;10:2739–2749. doi: 10.1016/s0968-0896(02)00102-5. [DOI] [PubMed] [Google Scholar]
- (9).Zhang Y, Desharnais J, Marsilje TH, Li C, Hedrick MP, Gooljarsingh LT, Tavassoli A, Benkovic SJ, Olson AJ, Boger DL, Wilson IA. Rational design, synthesis, evaluation, and crystal structure of a potent inhibitor of human GAR Tfase: 10-trifluoroacetyl-5,10-dideaza-acyclic-5,6,7,8-tetrahydrofolic acid. Biochemistry. 2003;42:6043–6056. doi: 10.1021/bi034219c. [DOI] [PubMed] [Google Scholar]
- (10).Greasley SE, Yamashita MM, Cai H, Benkovic SJ, Boger DL, Wilson IA. New insights into inhibitor design from the crystal structure and NMR studies of Escherichia coli GAR transformylase in complex with β-GAR and 10-formyl-5,8,10-trideazafolic acid. Biochemistry. 1999;38:16783–16793. doi: 10.1021/bi991888a. [DOI] [PubMed] [Google Scholar]
- (11).Cheng H, Chong Y, Hwang I, Tavassoli A, Zhang Y, Wilson IA, Benkovic SJ, Boger DL. Design, synthesis, and biological evaluation of 10-methanesulfonyl-DDACTHF, 10-methanesulfonyl-5-DACTHF, and 10-methylthio-DDACTHF as potent inhibitors of GAR Tfase and the de novo purine biosynthetic pathway. Bioorg. Med. Chem. 2005;13:3593–3599. doi: 10.1016/j.bmc.2004.12.004. [DOI] [PubMed] [Google Scholar]
- (12).Mendelsohn LG, Shih C, Schultz RM, Worzalla JF. Biochemistry and pharmacology of glycinamide ribonucleotide formyltransferase inhibitors: LY309887 and lometrexol. Invest. New Drugs. 1996;14:287–294. doi: 10.1007/BF00194532. [DOI] [PubMed] [Google Scholar]
- (13).Boritzki TJ, Bartlett CA, Zhang C, Howland EF, Margosiak SA, Palmer CL, Romines WH, Jackson RC. AG2034: a novel inhibitor of glycinamide ribonucleotide formyltransferase. Invest. New Drugs. 1996;14:295–303. doi: 10.1007/BF00194533. [DOI] [PubMed] [Google Scholar]
- (14).Kisliuk RL. Deaza analogs of folic acid as antitumor agents. Curr. Pharm. Design. 2003;9:2615–2625. doi: 10.2174/1381612033453695. [DOI] [PubMed] [Google Scholar]
- (15).Corey EJ, Bakshi RK, Shibata S, Chen C-P, Singh VK. A stable and easily prepared catalyst for the enantioselective reduction of ketones. Applications to multistep syntheses. J. Am. Chem. Soc. 1987;109:7925–7926. [Google Scholar]
- (16).Corey EJ, Helal CJ. Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: a new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew. Chem. Int. Ed. 1998;37:1986–2012. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- (17).Aoyama T, Shioiri T. Trimethylsilyldiazomethane: A convenient reagent for the O-methylation of alcohols. Tetrahedron Lett. 1990;31:5507–5508. [Google Scholar]
- (18).Habeck JL, Leitner TA, Shackelford KA, Gossett LS, Schultz RM, Andis SL, Shih C, Grindey GB, Mendelsohn LG. A novel class of monoglutamated antifolates exhibits tight-binding inhibition of human glycinamide ribonucleotide formyltransferase and potent activity against solid tumors. Cancer Res. 1994;54:1021–1026. [PubMed] [Google Scholar]
- (19).Bigham EC, Hodson SJ, Mallory WR, Wilson D, Duch DS, Smith GK, Ferone R. Synthesis and biological activity of open-chain analogues of 5,6,7,8-tetrahydrofolic acid-potential antitumor agents. J. Med. Chem. 1992;35:1399–1410. doi: 10.1021/jm00086a008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.