

Abstract
Oncolytic cancer therapy using herpes simplex viruses (HSV) that have direct tumoricidal effects and cancer immunotherapy using the cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF) have each been effective in preclinical testing. NV1034 is a multi-mutated oncolytic HSV carrying the gene for murine GM-CSF that attempts to combine these two anticancer strategies. The purpose of this study was to compare NV1034 to NV1023, the parent HSV mutants lacking GM-CSF, in order to determine if such combined oncolytic and immunotherapy using a single vector has advantages over oncolytic therapy alone. In vitro, expression GM-CSF did not alter the infectivity, in vitro cytotoxicity, or replication of NV1034 compared to the non-cytokine secreting control. Tumors infected with NV1034 produced GM-CSF in picogram quantities. In vivo efficacy of the viruses against murine colorectal carcinoma CT26 and murine hepatoma Hepa l–6 was then tested in subcutaneous tumors in syngeneic Balb/c and C57 L/J mice respectively. In these immune competent models, NV1034 or NV1023 each demonstrated potent antitumor activity. Treatment with NV1034 had significantly better antitumor effect compared to treatment with NV1023. Furthermore, in mice depleted of CD4+ and CD8+ T-lymphocytes, there was no difference in the antitumor efficacy of these viruses. Viral vectors combining oncolytic and immunotherapy are promising agents in treatment of colorectal carcinoma and hepatoma.
Keywords: Colorectal tumor, Hepatoma, HSV, Immunotherapy, Oncolytic therapy
INTRODUCTION
Replication-competent herpes simplex viruses (HSV) have been shown to have efficacy in models of neurologic(1;2) as well as non-neurologic malignancies including those of the breast, prostate, stomach, colon, lung, and head and neck(3–6). The herpes viruses being tested for oncolytic therapy have been engineered to attenuate their neurovirulence while maintaining their natural ability to infect and lyse tumor cells.
Another therapeutic strategy against tumors has been to enhance host antitumor immunity. In this regard, investigators have attempted to produce tumor vaccines by altering tumor cells ex-vivo to secrete immunostimulatory cytokines, and by reinfusing such cells to elicit a host anti-tumor immune response(7;8). The development of efficient viral vectors for gene transfer(9;10) has improved upon this strategy by allowing in vivo transduction of tumor cells with genes coding for immunostimulatory molecules. Intratumoral injection of vectors carrying the genes for various cytokines can result in efficient gene transfer and in sufficient cytokine production in proximity to tumor to result in antitumor efficacy(11;12).
Investigators have attempted to combine oncolytic and immunotherapy in order to boost the antitumor effect of oncolytic therapy. It has been demonstrated that the antitumor actions of intratumoral injection using a cytotoxic virus was greatly enhanced by the addition of a replication-defective HSV vector encoding interleukin-12, and the combination results in local and systemic antitumor immunity(13). Recently, replication-competent oncolytic HSV viruses carrying cytokine genes have demonstrated benefit when directly injected into experimental squamous cell cancers of the head and neck(14). The goal of the current study was to determine if a replication-competent HSV can act both as an oncolytic agent and as a vehicle for GM-CSF gene transfer to produce antitumor effects against colorectal and hepatocellular tumors in a murine model.
GM-CSF is a cytokine that induces myeloid precursor cells to proliferate and differentiate into neutrophils, monocytes, macrophages and eosinophils. GM-CSF also recruits and stimulates dendritic cells(15). In experiments studying tumor vaccines, GM-CSF was the most potent of a number of cytokines, adhesion molecules and other immunostimulatory molecules for the induction of specific and long-lasting antitumor immunity(16;17). Vaccination with irradiated B16 melanoma cells engineered to secrete murine GM-CSF stimulates potent, specific, and long-lasting antitumor immunity(7). Murine tumor cells infected ex vivo or in vivo with disabled infectious single-cycle herpes viral vectors expressing GM-CSF may function effectively both in preventing cancer formation as well as in treating established tumors(18;19).
MATERIALS AND METHODS
Cells and cell culture
The murine colorectal carcinoma cell line CT26, murine hepatoma cell line Hepa 1–6 and the murine melanoma cell line B16 were used in this study. All cells lines were obtained from American Type Culture Collection (Rockville, MD). CT26 cells were maintained in RPMI supplemented with 10% fetal calf serum (FCS), 100μg/ml penicillin, 100μg/ml streptomycin and 5mM non-essential amino acids. Hepa 1–6 and B16 cells were maintained in DME HG supplemented with 10% FCS, 100μg/ml penicillin, and 100μg/ml streptomycin.
Viruses
Structures of the two viruses used are shown in Figure 1. NV1023 is derived from R7020, a previously described attenuated, replication-competent virus based on the HSV-1 strain-F(20). R7020 has one copy of the γ134.5 neurovirulence gene deleted. Also deleted is the UL24, the UL56 genes and the L/S junction(20). The viral thymidine kinase gene has been moved and placed under the α4 promoter. NV1023 differs from R7020 in that the deleted endogenous thymidine kinase (TK) gene and the adjoining UL24 promoter region have been repaired. In addition the E. coli lacZ marker gene has been inserted into the ICP47 locus. NV1034 is a derivative of NV1023 in which a murine GM-CSF cDNA has been inserted. The synthesis of these viruses has previously been described(14). NV1023 and NV1034 were propagated on Vero cells and titered by standard plaque assay.
Figure 1.
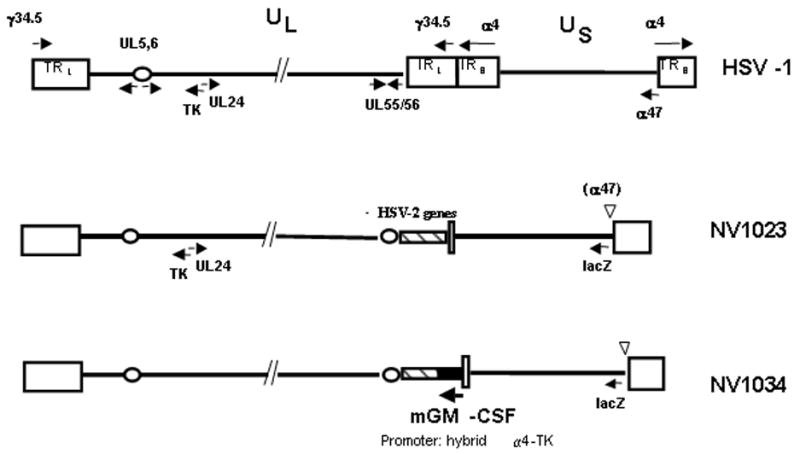
Structure of f-strain of wild-type HSV-1, NV1023, and NV1034. The wild-type HSV-1 genomic structure, shown here schematically (not to scale) in the prototype orientation, consists of long (L) and short (S) unique sequences (UL and US), each bound by inverted repeat regions (RL and RS). HSV-1 encodes over 80 genes. The locations of the following genes are shown for reference; Thymidine kinase (TK), UL24, UL5/6, γ34.5 (the major HSV-1 neurovirulence gene), and the immediate-early genes α4 and α47. The L/S junction of NV1023 contains 5.2 kb of HSV-2 DNA (hatched box), and carries the E.coli lacZ gene under control of the α47 promoter between nucleotides 144678 to 146007 of HSV-1 at the α47 locus (Genbank Accession No. X14112). NV1034 has a similar structure to NV1023 and carry the murine GM-CSF under control of a hybrid α4-TK promoter.
In vitro infectivity and Cytotoxicity
Ability of NV1034 to infect CT26 or Hepa 1–6 cells was assessed. Cells were plated at 5×104 cells/well in 12 well plates (Costar, Corning Inc., Corning, NY) and were infected with NV1034 at multiplicities of infection (MOI) ranging from 0.01 to 5.0. Control wells were treated with PBS. To assess infectivity, cells were fixed in 1% glutaraldehyde at 24 hour intervals for 6 days and stained with X-Gal (GibcoBRL, Grand Island, NY) to detect β-galactoside, the product of the lacZ marker gene. The number of stained (blue) cells/high power field (hpf) and the total number of cells/hpf were counted and the percentage of infected cells calculated. Cell viability was assessed by counting live cells at 24 hour intervals post-infection using trypan blue exclusion.
In vitro viral proliferation
Viral growth assays were performed to compare the ability of CT26 and Hepa 1–6 cells to support the replication of NV1034 or NV1023. Cells were infected with NV1034 or NV1023 at a MOI of 1.0. Cells and supernatants were harvested at 6 hours and daily post-infection, for 6 days. Three cycles of freeze-thaw lysis were performed and viral titers were determined by standard plaque assay on Vero cells.
In vitro GM-CSF production
The ability of the NV1034 to produce GM-CSF when it infects CT26 and Hepa 1–6 cells was assessed. Cells were then infected with NV1034 or NV1023 at multiplicities of infection (MOIs) of 0.1, 1.0, or 5.0. Supernatants were collected daily and concentration of GM-CSF determined by ELISA (R & D Systems, Minneapolis, MN).
Animal Studies
Four to 6 week old male Balb/c mice (Taconic; Germantown, NY) or C57 L/J mice (Jackson Laboratories; Ann Arbor, ME) were used. All animal studies were approved by the Memorial Sloan-Kettering Institutional Animal Care and Use Committee and performed under strict guidelines. Animals were anesthetized with an intraperitoneal (i.p) injection of ketamine (70mg/kg) and xylazine (10mg/kg) for experimental procedures.
Establishment of Subcutaneous Tumors
One ×105 CT26 cells or 5×105 Hepa 1–6 cells were injected subcutaneously into the flanks of Balb/c mice or C57L/J mice respectively. Palpable tumors, 3–5 mm in diameter, develop reliably in 14 days after tumor cell inoculation for CT26, and in 7 days for Hepa 1–6.
In vivo viral infection and GM-CSF production
This experiment was conducted to assess in vivo viral infection and GM-CSF production by NV1034. Subcutaneous CT26 tumors were induced in the flanks of Balb/c mice as described above. Tumors were injected with 5×107 plaque forming units (pfu) of NV1034 or NV1023 and were harvested on days 1, 2, 3, and 7 post-injection. Half the tumor was immediately frozen in liquid nitrogen and later homogenized in PBS for assessment for GM-CSF expression by ELISA (R & D Systems, Minneapolis, MN). The other half of the tumor was embedded in Tissue-Tek prior to cutting into 8μm frozen sections for subsequent histochemical staining with Xgal (GibcoBRL, Grand Island, NY) to detect marker gene β-galactosidase (lacZ) expression as an indicator of viral infection.
Treatment of subcutaneous flank tumors with virus
The efficacy of NV1034 to treat CT26 tumors in immune competent mice was compared to that of NV1023. Palpable CT26 flank tumors (3–5mm diameter) were treated with intratumoral injection of 1×106 pfu(low dose), 1×107 pfu (medium dose), or 5×107 pfu (high dose) of NV1034, NV1023, or PBS (n=6–7/group). These experiments were then repeated to ensure consistency. Hepa 1–6 tumors were treated with 1×105 pfu, 1×106 pfu, or 5×106 pfu of these viruses in similar experiments. The longest and shortest diameter of the tumors were measured over 14 days after treatment, and the tumor volume in each case was calculated using the following formula:
where DL is the longest diameter and DS is the shortest diameter. Mean tumor volume in each group was compared using Student’s t-test.
Tumor cell rechallenge after excision of flank tumor
This experiment was conducted to assess the ability of viral therapy to induce antitumor immunity and to protect the mice against future growth of tumor of the same type. CT26 flank tumors treated as stated above, were excised after three weeks. The opposite flank was subsequently subcutaneously injected with 1×106 CT26 cells. In addition, 8 naïve mice were also injected subcutaneously with 1×106 CT26 cells in the flank as controls. The mice were followed for 40 days for the development of any flank tumor. A similar experiment was also performed in animals with Hepa 1–6 flank tumors. Those mice that did not develop tumor after the second tumor cell injection, were further injected with 1×106 B16 melanoma cells subcutaneously into the same flank and followed to determine if the protection against tumor development was tumor type specific. Results were compared by Fisher’s Exact.
Effect of CD4+ and CD8+ T lymphocyte depletion on antitumor efficacy of virus
Both CD4+ and CD8+ T-lymphocytes were depleted simultaneously by i.p injection of monoclonal antibodies (0.2mg of each antibody was injected i.p. on days 0, 1, and 2) derived from the hybridomas GK1.5 and 53–6.72. Depletion was confirmed by FACS analysis of spleens on day 6. The depletion was maintained with i.p injection of 0.5mg of each antibody once a week thereafter. CT26 flank tumors were induced in the depleted mice (n=49) as described above. Once the flank tumors were palpable (3–5mm diameter), they were treated with intratumoral injection of 1×107 pfu of NV1034, NV1023, or PBS (n=7/group). PBS-treated animals were also depleted of CD4+ and CD8+ T-lymphocytes. Tumor volumes were measured, and the mean tumor volumes were compared using the Student’s t-test. After excision of the primary tumor, the opposite flank was re-inoculated with 1×106 CT26 cells. The mice were followed for development of tumor in the opposite flank. A similar experiment was also conducted using syngeneic Hepa1–6 cells in C57 L/J mice.
Statistical Analysis
All continuous variables are presented as mean ± standard error of mean (SEM). Statistical test used in each experiment is mentioned in the section.
RESULTS
In vitro infectivity assay
The first objective was to determine the infectivity of NV1034 for colorectal or hepatocellular adenocarcinomas. None of the control wells treated with PBS showed any blue staining with Xgal. The percentage of CT26 cells expressing the lacZ gene as a marker of NV1034 infection at the various time points is shown in Table 1. It was observed that NV1034 at an MOI of 1.0 infected 100% cells by 5 days. The secretion of GM-CSF by the tumor cells did not alter the infectivity of the virus.
Table 1.
Infection of CT26 cells in vitro with various MOIs of NV1034.
| Days post-infection | Control | MOI 0.01 | MOI 0.1 | MOI 1.0 |
|---|---|---|---|---|
| 0 | 0 | 0 | 0 | 0 |
| 1 | 0 | 0 | 8 | 17 |
| 2 | 0 | 0 | 10 | 23 |
| 3 | 0 | 0 | 27 | 46 |
| 4 | 0 | 0 | 53 | 89 |
| 5 | 0 | 0 | 79 | 100 |
| 6 | 0 | 29 | 100 | 100 |
CT26 cells were exposed to various MOIs of NV1034 and X-Gal staining at various time points determined the percentage of lacZ expressing cells. MOIs of 0.1 and 1.0 infected all the cells by 6 days but an MOI of 0.01 infected only about 30% of the cells in the same time period.
In vitro cytotoxicity
These studies were performed to determine the oncolytic efficacy of NV1034 compared to its parent virus NV1023. It demonstrated a 100% CT26 cell kill by NV1034 and NV1023 with an MOI of 1.0 at 7 days post-injection. At an MOI of 0.1, NV1034 killed 92% of the CT26 cells, while NV1023 killed 89%. The viable cells remaining at various time points (as a percent of control) are demonstrated in Figure 2 (A1 and B1). Hepa 1–6 cells demonstrated a similar sensitivity to these viral agents (Figure 2- A2 and B2). The additional secretion of GM-CSF by NV1034 did not decrease the in vitro cytotoxicity of the virus.
Figure 2.
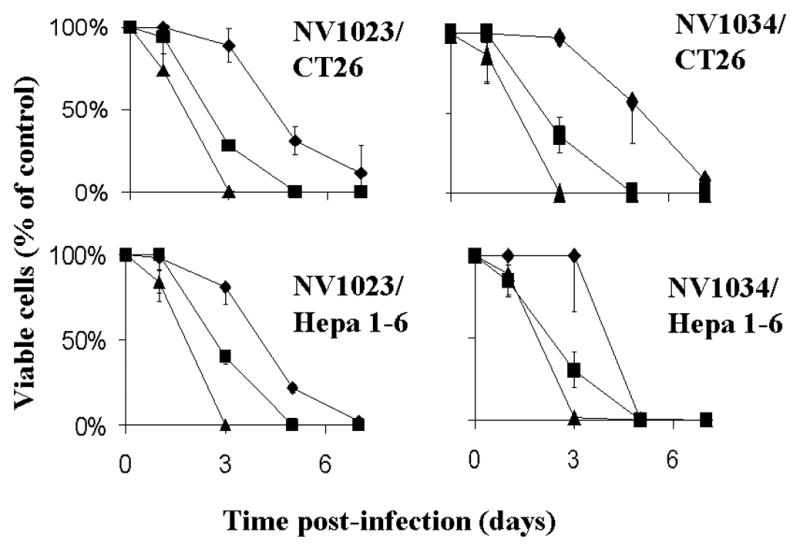
In vitro cytotoxicity of A. NV1023, and B. NV1034 in 1) CT 26 (TOP) and 2) Hepa1-6 (BOTTOM) cells. Tumor cells were infected with each virus at varying MOI’s (Diamond-0.1; Rectangle-1.0; Triangle-5.0) and the proportion of viable cells at different time points was determined by counting using trypan blue exclusion (mean ± SD). X axis-Time post-infection (days), Y axis-Viable cells (% of control). MOI: Multiplicity of infection
In vitro viral proliferation
Plaque assays were performed on cells and supernatants to determine the ability of CT26 and Hepa 1–6 cells to support the replication of these viruses. Viral titers were determined over 6 days for CT26 cells and 7 days for Hepa 1–6 cells. Both cell lines supported the replication of all viruses tested (Figure 3).
Figure 3.
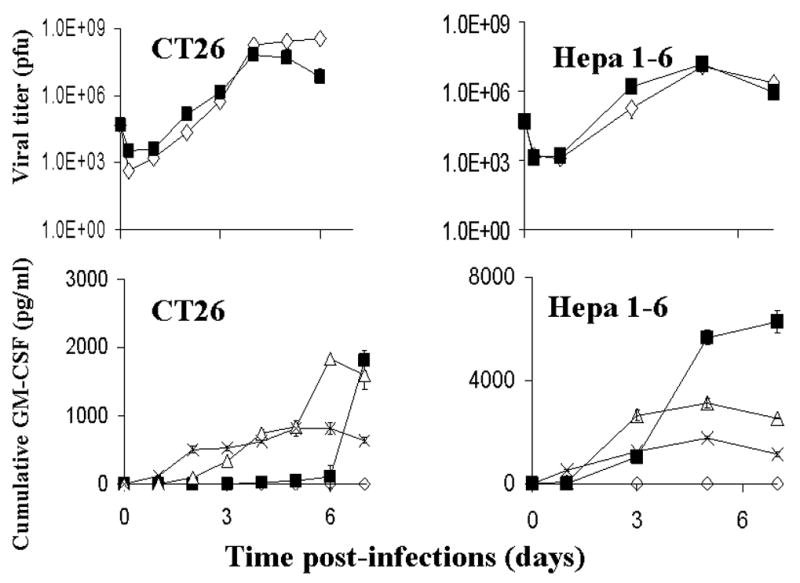
Viral replication (A) and GMCSF production (B) in CT26 or Hepa 1–6. For viral replication experiments, tumor cells were infected with NV1023 (square) or NV1034 (diamond) at an MOI of 1. Cells and supernatants were collected periodically and titered by a standard plaque assay (mean ± SD). X axis- Time post-infection (days), Y axis- Viral titer in pfu. For GM-CSF production, tumor cells were infected with NV1034 at MOIs of 5.0 (cross), MOI of 1.0 (triangle), or MOI 0.1 (square). Treatment with media only (diamonds) produced no GM-CSF. X axis- Time post infection (days), Y axis- Cumulative GM-CSF (picograms/ml). MOI: Multiplicity of infection, PFU: plaque forming units
In vitro GM-CSF production
The ability of NV1034 to produce GM-CSF when it infects tumor cells was then tested by performing ELISA on supernatants collected from tumor cells infected with either virus. NV1034 produced picogram amounts of GM-CSF in both the cell lines tested (Figure 3C). Treatment of tumor cells by NV1023 did not result in production of any GM-CSF. In the CT26 cell line, infection of the cells with an MOI of 5.0 resulted in a cumulative GM-CSF plateau of only 819 pg/ml media by 5 days post-infection as most of the tumor cells were lysed by the oncolytic effect of the virus early in the assay. Infection at an MOI of 0.1 on the other hand produced 1810±150 pg/ml of GM-CSF by 7 days and had not peaked during the time period assayed. Similar results were seen for the Hepa 1–6 cells as shown in the Figure 3D.
In vivo Infectivity and GM-CSF Production
Frozen sections of tumor injected with NV1034 and stained with Xgal demonstrated viral lacZ gene expression throughout the first seven days assayed (Figure 4). Maximum expression was seen on day 2–3. ELISA performed on tumors for in vivo murine GM-CSF production detected no GM-CSF in the tumors treated with NV1023. For tumors treated with NV1034, the GM-CSF production (in pg/gm tumor tissue) on days 1, 2, 3, and 7 was 2400±700, 2000±600, 1000±200, and 440±370 respectively.
Figure 4.
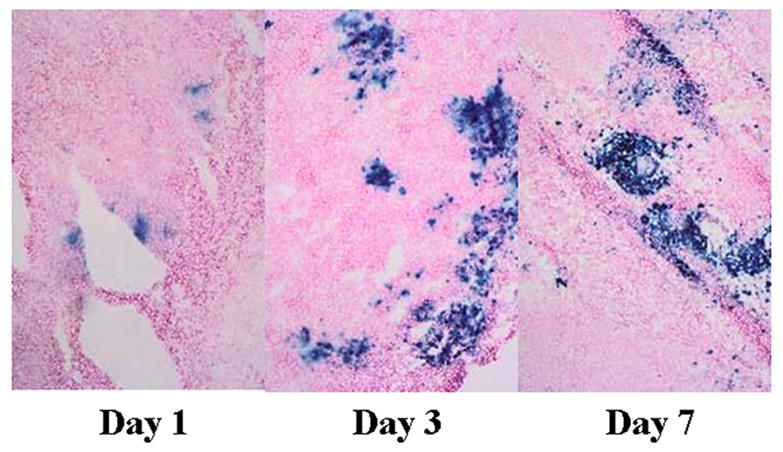
In vivo infection of CT26 tumors with NV1034. CT26 flank tumors were injected with 1×107 pfu of NV1034. Tumors were harvested on days 1, 2, 3, and 7 and 8μm frozen sections stained with Xgal solution. LacZ expression (blue cells) could be detected as early as day1, peaked on day 2 and 3, but still could be detected on day 7. PFU: plaque forming units
Treatment of subcutaneous flank tumors with virus
Subcutaneous injection of 1×105 CT26 cell in the flank of 4–6 week old Balb/c mice reliably produced tumors 3–5mm in diameter in 2 weeks. When treated with a single intratumoral injection of 1×106 pfu of virus, the tumor volume on day 14 post treatment was 150±70 mm3 in the NV1034 group (p=0.01 vs. control), 270±120 mm3 in the NV1023 group (p<0.05 vs. control), and 700±200 mm3 in the control group (Figure 5B). When treated with a single intratumoral injection of 1×107 pfu of virus, the tumor volume on day 14 post-treatment was 37±23 mm3 in the NV1034 group (p=0.01 vs. control), 210±70 mm3 in the NV1023 group (p<0.05 vs. control), and 710±203 mm3 in the control group (Figure 5A). The antitumor effect of NV1034 was significantly better than that of NV1023 (p=0.05)(Figure 5A and B). Similar results were observed when subcutaneous Hepa 1–6 flank tumors were treated with a single intratumoral injection. When treated with a single intratumoral injection of 1×106 pfu of virus, the tumor volume on day 14 post-treatment was 0.5±0.5 mm3 in the NV1034 group (p=0.01 vs. control), 12±3 mm3 in the NV1023 group (p<0.05 vs. control), and 290±90 mm3 in the control group (Figure 5C). When treated with a single intratumoral injection of 1×105 pfu of virus, the tumor volume on day 14 post-treatment was 2±2 mm3 in the NV1034 group (p=0.01 vs. control), 72±38 mm3 in the NV1023 group (p<0.05 vs. control), and 280±80 mm3 in the control group (Figure 5C). At highest doses (5×107 for CT-26, and 5×106 for Hepa 1–6), the effects of NV1034 were indistinguishable from those of NV1023, likely due to the dominant oncolytic effects at high doses.
Figure 5.
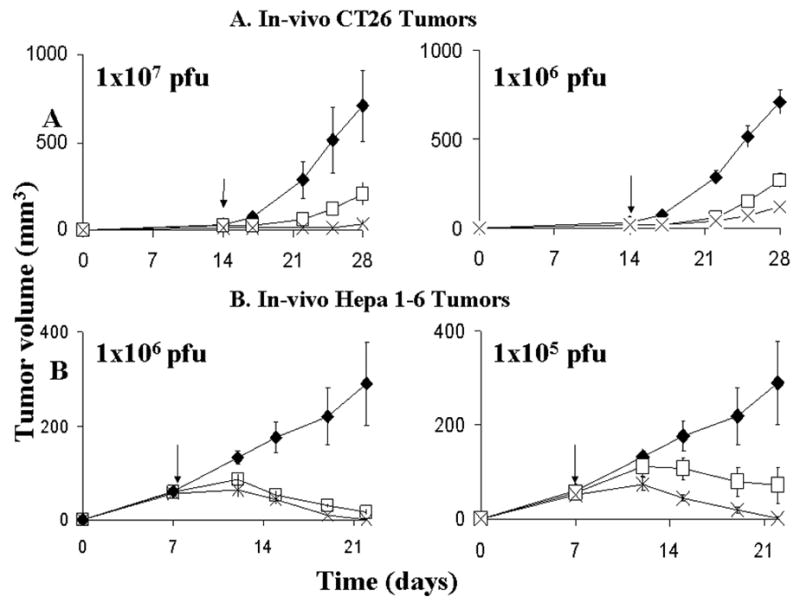
In vivo effects of tumor treatment with NV1023 (square), NV1034 (cross), or PBS (diamond). Single dose intratumoral viral therapy of CT26 (A) and Hepa 1–6 (B) flank tumors in immunocompetent Balb/c and C57 L/J mice respectively. In CT26 tumors, a single intratumoral injection of NV1023, or NV1034 at 1×107 pfu or 1×106 significantly reduced tumor volume (mean ± SEM) compared to PBS injected controls (p<0.05; two-tailed T test). In addition, the NV1034 treated group had a significant reduction in tumor volume compared to those treated with the non-cytokine expressing virus NV1023 (p<0.05; paired, two-tailed T test, day28). In Hepa 1–6 tumors (B), a single intratumoral injection of NV1023 or NV1034 at 1×106 pfu or 1×105 significantly reduced tumor volume (mean ± SEM) compared to PBS injected controls (p<0.05; two-tailed T test). In addition, the NV1034 treated group had a significant reduction in tumor volume compared to those treated with the non-cytokine expressing virus NV1023 (p<0.04; two-tailed T test). X axis- Time (days), Y axis- Tumor volume (mm3). PFU: plaque forming units
Thus we observed that all oncolytic viruses reduced tumor burden compared to PBS treated controls in both tumor models. In addition, the cytokine-secreting NV1034 significantly reduced tumor burden compared to the non-cytokine-secreting NV1023 virus in both the murine colorectal and the hepatoma tumor models.
Effect of CD4+ and CD8+ T lymphocyte depletion on antitumor effects of virus
This experiment was conducted to demonstrate the effect of simultaneous depletion of both CD4+ and CD8+ T lymphocyte depletion on the antitumor effects of NV1034 and NV1023. When 3–5mm CT26 flank tumors were treated with 1×107 pfu of virus in the setting of T-cell depletion, the tumor volume (in mm3) on day 14 post-treatment was as follows: Controls, 1010±110; NV1034, 680±100; and NV1023, 730±120 (Figure 6A). Only NV1034 reduced tumor burden significantly (p=0.05). However, the antitumor effects of NV1023 and NV1034 were similar.
Figure 6.
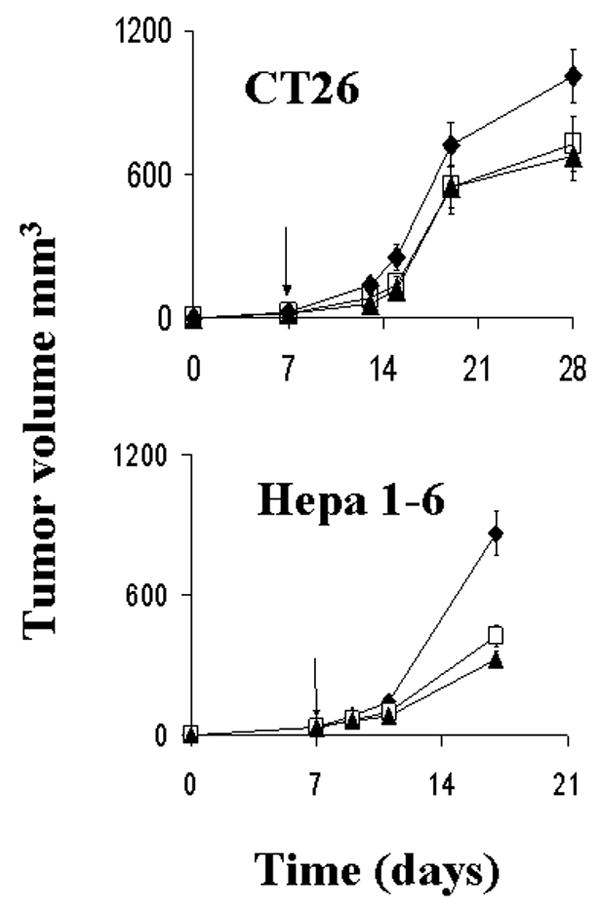
Intratumoral herpes viral therapy for CT26 and Hepa 1–6 flank tumors in mice depleted of CD4+ and CD8+ T-lymphocytes. Established flank tumors in depleted animals (n=6–8/group) were treated with a single intratumoral dose of NV1023 (square), NV1034 (triangle), or PBS (diamond). In both models, both viruses demonstrated similar efficacy in reducing tumor burden (mean tumor volume ± SEM) compared to controls (p≤0.05; two-tailed T test). There was no difference in the antitumor efficacy between the two viruses (p=NS; two-tailed T test). X axis- Time (days), Y axis- Tumor volume (mm3). PBS: Phosphate buffered saline
A similar experiment using 1×106 pfu of virus was also conducted with Hepa 1–6 flank tumors in C57 L/J mice. The tumor volume (mm3) on day 10 post-treatment was as follows: Control, 860±97; NV1023, 420±40; and NV1034, 324±39 (Figure 6B). Both NV1023 and NV1034 reduced tumor burden significantly compared to control (p<0.05). However there was no difference between the effects of NV1023 and NV1034 (p=not significant; NS). Thus the cytokine-secreting virus NV1034 has superior antitumor effects only in mice with an intact cellular immune system and is mediated via CD4+ or CD8+ T lymphocytes.
Tumor cell rechallenge
An experiment was performed to determine whether treatment of primary tumors provided any protection against a tumor rechallenge of the same tumor type. CT26 flank tumors treated with NV1023, NV1034, or PBS as in the previous experiments were resected and the opposite flank challenged with 1×106 CT26. Eight naïve animals were also injected with tumor cells. It was observed that all the naïve mice developed flank tumors. Eighty percent (4/5) in the control group (treated with PBS) developed flank tumors, whereas only 20% (1/5) in the groups treated with NV1023 or NV1034 developed tumors when followed for 40 days. All mice depleted of CD4+ and CD8+ T lymphocytes developed tumors when rechallenged after resection of the primary treated tumor.
This was repeated for Hepa 1–6- bearing animals. When C57 L/J mice with Hepa 1–6 tumors were rechallenged in similar fashion, all the naïve, as well as PBS control mice developed tumors within 7 days. Fifty percent (4/8) of mice treated with NV1023 (p=NS) and 25% (2/8) treated with NV1034 (p<0.01) also developed tumors within 7 days. To determine whether the protection against tumor rechallenge was specific for the tumor type, the animals that did not develop tumors after rechallenge with Hepa 1–6 cells were injected with syngeneic B16 melanoma cells subcutaneously into the flank. All animals inoculated with B16 developed tumors. Thus, not only does treatment with the cytokine-producing virus protect against future tumor challenge, but this protection is tumor specific.
DISCUSSION
In this study we compared the antitumor efficacy of a GM-CSF producing oncolytic virus NV1034 to that of the oncolytic virus NV1023 in a murine model of colorectal carcinoma and hepatoma. NV1023 is an attenuated, replication-competent HSV-1 derived from R7020, a virus originally designed as a vaccine against both HSV-1 and HSV-2 infections(20). R7020 is an oncolytic virus that has demonstrated efficacy in the treatment of a wide variety of tumors in murine models(4;21;22). It is currently undergoing phase I clinical trials for metastatic liver tumors. Compared to the parent R7020 virus, NV1023 and NV1034 have had their endogenous HSV thymidine kinase and UL24 genes restored and contain the lacZ marker gene within the ICP47 locus. ICP47 is involved in the virus’s ability to inhibit MHC class I peptide expression in humans by the infected cell(23). The interruption of ICP47 by the lacZ gene may therefore potentially increase MHC class I antigen expression and theoretically make the tumor cell more recognizable by circulating immune cells. NV1034 differs from NV1023 by the insertion of the murine GM-CSF gene adjacent to the HSV-2 fragment. These studies demonstrate NV1023 and NV1034 to be potent oncolytic viruses. Furthermore, the addition of the GM-CSF gene did not alter the in vitro infectivity, replication, or direct cytotoxicity of NV1034 compared to the non-cytokine-secreting control NV1023. The GM-CSF gene is under the control of a hybrid α4 enhancer-TK promoter. The current studies demonstrate that both NV1023 and NV1034 have similar infective and replicative abilities in CT26 and Hepa 1–6 cells in vitro. When administered in vivo by intratumoral injection, both viruses decreased tumor volume significantly compared to saline treated controls. In addition, GM-CSF secreting NV1034 demonstrated an enhanced antitumor efficacy in vivo, likely through CD4+ and CD8+ T-cell mediated immune mechanisms.
GM-CSF is a cytokine that induces myeloid precursor cells to proliferate and differentiate into neutrophils, monocytes, macrophages and eosinophils. GM-CSF also recruits and stimulates dendritic cells(24). In experiments studying tumor vaccines, GM-CSF was the most potent of a number of cytokines, adhesion molecules and other immunostimulatory molecules for the induction of specific and long-lasting antitumor immunity(16;17). Vaccination with irradiated B16 melanoma cells engineered to secrete murine GM-CSF stimulates potent, specific, and long-lasting antitumor immunity(7). Murine tumor cells infected ex vivo or in vivo with disabled infectious single-cycle herpes viral vectors expressing GM-CSF may function effectively both in preventing cancer formation as well as in treating established tumors(18;19). In the current study, the GM-CSF producing NV1034 produces a significant reduction in tumor burden compared to the purely oncolytic virus NV1023. It is a demonstration that local cytokine production can add to the already potent antitumor efficacy of oncolytic viruses.
Previous studies have demonstrated that even non-cytokine expressing oncolytic HSV (G207) can induce antitumor immune responses(24). Results from the current studies confirm previous observations that oncolytic viral therapy alone induces some immunity against tumor. Further enhancing this anticancer immune response during oncolytic therapy has great appeal, because during the time required to generate an immune system, the oncolytic properties of the HSV provide a direct and immediate means of controlling tumor growth. Some previous attempts to combine use of oncolytic viruses and cytokine immunotherapy had used combined delivery of oncolytic viruses along with delivery of separate replication incompetent amplicon vectors for cytokine gene transfer(13;25). The use of a single vector for oncolysis as well as immune stimulation greatly simplifies application. The current study demonstrates that such single vector multimodality therapy is feasible, safe, and efficacious.
The ability of NV1034 to induce a potent, specific, and lasting immune response against the CT26 and Hepa 1–6 tumors was tested by reinoculating the animals with tumor cells after surgically excising the residual tumor after viral therapy. Indeed, treatment with NV1023 protected some of the mice against tumor growth in the CT-26 model. CT26 is a relatively immunogenic tumor, and treatment of tumors with saline alone protected 40% of the mice against rechallenge. Thus, experiments were repeated in the Hepa 1–6 tumor model that is much less immunogenic. The results indicate that addition of GM-CSF enhances the antitumor immunity of oncolytic therapy. Furthermore, this protection was tumor specific since all the mice that did not develop tumor with Hepa 1–6 rechallenge developed tumors when inoculated with B16 melanoma cells. When virus (NV1023 or NV1034) is injected into the tumor in high doses, the oncolytic effect is so strong that the effect of immunostimulation by the GM-CSF is not noticeable. However, when the tumor burden is high, host toxicity of the virus will limit the dose that can be administered.
Immunity may also play a detrimental role in oncolytic viral therapy. Since 90% of adults carry antibodies to HSV type 1(26), the host immune response can theoretically decrease efficacy of such viral therapy. However, experimental data indicate that treatment using HSV in mice pre-immunized to produce cellular and humoral immunity against HSV can still have biologic and antitumor effects(22;27). What is not known is whether use of additional immunostimulatory agents such as GM-CSF will further enhance the antiviral response and decrease viral antitumor efficacy.
Five-year survival after surgical resection of colorectal metastases to the liver or hepatocellular carcinoma is only 23–38%(28;29) and 40–50% respectively(30). Local and distant recurrences after surgical therapy for colorectal as well as hepatocellular cancer, occur as a result of microscopic disease that was not detectable at the time of resection(31). A number of conventional strategies including chemotherapy and radiation therapy have had little or no impact on improving recurrence rates and survival(32–38). Indeed, most patients have distribution of disease that is not surgically treatable, and survival for these patients even with our best palliative chemotherapy regimens, is usually measured in months(39;40). There is a need to develop novel forms of therapy that can minimize these recurrences after resection and to improve results of palliative treatments. The current data are encouraging for investigation of combined oncolytic and immunotherapy in the therapy of these tumors.
Acknowledgments
The authors thank Brian Horsburgh, Ph.D. and Medigene, Inc. for constructing and providing us with the NV1034 virus.
Footnotes
Supported in part by grants RO1 CA 75416, RO1 CA 72632, and RO1 RO1CA/DK80982 (Y.F.) from the National Institutes of Health and Grant MBC-99366 (Y.F.) from the American Cancer Society, and a grant from the Lustgarten Foundation
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Mineta T, Rabkin SD, Yazaki T, Hunter WD, Martuza RL. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1(9):938–43. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 2.Yazaki T, Manz HJ, Rabkin SD, Martuza RL. Treatment of human malignant meningiomas by G207, a replication-competent multimutated herpes simplex virus 1. Cancer Research. 1995;55(21):4752–6. [PubMed] [Google Scholar]
- 3.Walker JR, McGeagh KG, Sundaresan P, Jorgensen TJ, Rabkin SD, Martuza RL. Local and systemic therapy of human prostate adenocarcinoma with the conditionally replicating herpes simplex virus vector G207. Hum Gene Ther. 1999;10(13):2237–43. doi: 10.1089/10430349950017211. [DOI] [PubMed] [Google Scholar]
- 4.Bennett JJ, Kooby DA, Delman K, McAuliffe P, Halterman MW, Federoff H, et al. Antitumor efficacy of regional oncolytic viral therapy for peritoneally disseminated cancer. Journal of Molecular Medicine. 2000;78(3):166–74. doi: 10.1007/s001090000092. [DOI] [PubMed] [Google Scholar]
- 5.Kooby DA, Carew JF, Halterman MW, Mack JE, Bertino JR, Blumgart LH, et al. Oncolytic viral therapy for human colorectal cancer and liver metastases using a multi-mutated herpes simplex virus type-1 (G207) FASEB Journal. 1999;13(11):1325–34. doi: 10.1096/fasebj.13.11.1325. [DOI] [PubMed] [Google Scholar]
- 6.Carew JF, Kooby DA, Halterman MW, Federoff HJ, Fong Y. Selective infection and cytolysis of human head and neck squamous cell carcinoma with sparing of normal mucosa by a cytotoxic herpes simplex virus type 1 (G207) Human Gene Therapy. 1999;10(10):1599–606. doi: 10.1089/10430349950017608. [DOI] [PubMed] [Google Scholar]
- 7.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993 April;90:3539–43. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kemeny N, Gonen M, Sullivan D, Schwartz L, Benedetti F, Saltz L, et al. Phase I study of hepatic arterial infusion of floxuridine and dexamethasone with systemic irinotecan for unresectable hepatic metastases from colorectal cancer. J Clin Oncol. 2001;19(10):2687–95. doi: 10.1200/JCO.2001.19.10.2687. [DOI] [PubMed] [Google Scholar]
- 9.Andreansky S, He B, van Cott J, McGhee J, Markert JM, Gillespie GY, et al. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Therapy. 1998;5(1):121–30. doi: 10.1038/sj.gt.3300550. [DOI] [PubMed] [Google Scholar]
- 10.Bramson JL, Hitt M, Addison CL, Muller WJ, Gauldie J, Graham FL. Direct intratumoral injection of an adenovirus expressing interleukin- 12 induces regression and long-lasting immunity that is associated with highly localized expression of interleukin-12. Hum Gene Ther. 1996;7(16):1995–2002. doi: 10.1089/hum.1996.7.16-1995. [DOI] [PubMed] [Google Scholar]
- 11.D’Angelica M, Karpoff H, Halterman M, Ellis J, Klimstra D, Edelstein D, et al. In vivo interleukin-2 gene therapy of established tumors with herpes simplex amplicon vectors. Cancer Immunology & Immunotherapy. 1999;47(5):265–71. doi: 10.1007/s002620050530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Angelica M, Tung C, Allen P, Halterman M, Delman K, Delohery T, et al. Herpes simplex virus (HSV)-mediated ICAM-1 gene transfer abrogates tumorigenicity and induces anti-tumor immunity. Molecular Medicine. 1999;5(9):606–16. [PMC free article] [PubMed] [Google Scholar]
- 13.Toda M, Martuza RL, Kojima H, Rabkin SD. In situ cancer vaccination: an IL-12 defective vector/replication-competent herpes simplex virus combination induces local and systemic antitumor activity. Journal Of Immunology. 1998;160(9):4457–64. [PubMed] [Google Scholar]
- 14.Wong RJ, Patel SG, Kim S, DeMatteo RP, Malhotra S, Bennett JJ, et al. Cytokine gene transfer enhances herpes oncolytic therapy in murine squamous cell carcinoma. Human Gene Therapy. 2001;12(3):253–65. doi: 10.1089/10430340150218396. [DOI] [PubMed] [Google Scholar]
- 15.Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256(12):1550–2. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 16.Dranoff G, Mulligan RC. Activities of granulocyte-macrophage colony-stimulating factor revealed by gene transfer and gene knockout studies. Stem Cells. 1994;12 (Suppl 1):173–82. [PubMed] [Google Scholar]
- 17.Kim SH, Carew JF, Kooby DA, Shields J, Entwisle C, Patel S, et al. Combination gene therapy using multiple immunomodulatory genes transferred by a defective infectious single-cycle herpes virus in squamous cell cancer. Cancer Gene Therapy. 2000;7(9):1279–85. doi: 10.1038/sj.cgt.7700231. [DOI] [PubMed] [Google Scholar]
- 18.Todryk S, McLean C, Ali S, Entwistle C, Boursnell M, Rees R, et al. Disabled infectious single-cycle herpes simplex virus as an oncolytic vector for immunotherapy of colorectal cancer. Hum Gene Ther. 1999;10(17):2757–68. doi: 10.1089/10430349950016492. [DOI] [PubMed] [Google Scholar]
- 19.Ali SA, McLean CS, Boursnell MEG, Martin G, Holmes CL, Reeder S, et al. Preclinical Evaluation of “Whole” Cell Vaccines for Prophylaxis and Therapy Using a Disabled Infectious Single Cycle-Herpes Simplex Virus Vector to Transduce Cytokine Genes. Cancer Research. 2000;60:1663–70. [PubMed] [Google Scholar]
- 20.Meignier B, Longnecker R, Roizman B. In vivo behavior of genetically engineered herpes simplex viruses R7017 and R7020: Construction and evaluation in rodents. Journal Of Infectious Diseases. 1999;158:602–14. doi: 10.1093/infdis/158.3.602. [DOI] [PubMed] [Google Scholar]
- 21.Bradley JD, Kataoka Y, Advani S, Chung SM, Arani RB, Gillespie GY, et al. Ionizing radiation improves survival in mice bearing intracranial high- grade gliomas injected with genetically modified herpes simplex virus. Clin Cancer Res. 1999;5(6):1517–22. [PubMed] [Google Scholar]
- 22.Delman KA, Bennett JJ, Zager JS, Burt BM, McAuliffe PF, Petrowsky H, et al. Effects of preexisting immunity on the response to herpes simplex-based oncolytic viral therapy. Human Gene Therapy. 2000;11(18):2465–72. doi: 10.1089/10430340050207957. [DOI] [PubMed] [Google Scholar]
- 23.Jugovic P, Hill AM, Tomazin R, Ploegh H, Johnson DC. Inhibition of major histocompatibility complex class I antigen presentation in pig and primate cells by herpes simplex virus type 1 and 2 ICP47. Journal of Virology. 1998;72(6):5076–84. doi: 10.1128/jvi.72.6.5076-5084.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Todo T, Rabkin SD, Sundaresan P, Wu A, Meehan KR, Herscowitz HB, et al. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum Gene Ther. 1999;10(17):2741–55. doi: 10.1089/10430349950016483. [DOI] [PubMed] [Google Scholar]
- 25.Carew JF, Kooby DA, Halterman MW, Kim SH, Federoff HJ, Fong YM. A novel approach to cancer therapy using an oncolytic herpes virus to package amplicons containing cytokine genes. Molecular Therapy. 2001;4(3):250–6. doi: 10.1006/mthe.2001.0448. [DOI] [PubMed] [Google Scholar]
- 26.Whitley RJ, Kimberlin DW, Roizman B. Herpes simplex viruses. Clinical Infectious Diseases. 1998;26(3):541–53. doi: 10.1086/514600. [DOI] [PubMed] [Google Scholar]
- 27.Herrlinger U, Kramm CM, Aboody-Guterman KS, Silver JS, Ikeda K, Johnston, et al. Pre-existing herpes simplex virus 1 (HSV-1) immunity decreases, but does not abolish, gene transfer to experimental brain tumors by a HSV-1 vector. Gene Therapy. 1998;5(6):809–19. doi: 10.1038/sj.gt.3300643. [DOI] [PubMed] [Google Scholar]
- 28.Fong Y, Cohen AM, Fortner JG, Enker WE, Turnbull AD, Coit DG, et al. Liver resection for colorectal metastases. J Clin Oncol. 1997;15(3):938–46. doi: 10.1200/JCO.1997.15.3.938. [DOI] [PubMed] [Google Scholar]
- 29.Weber SM, Jarnagin WR, DeMatteo RP, Blumgart LH, Fong Y. Survival after resection of multiple hepatic colorectal metastases. Annals of Surgical Oncology. 2000;7(9):643–50. doi: 10.1007/s10434-000-0643-3. [DOI] [PubMed] [Google Scholar]
- 30.Fong Y, Sun RL, Jarnagin W, Blumgart LH. An analysis of 412 cases of hepatocellular carcinoma at a Western center. Annals Of Surgery. 1999;229(6):790–9. doi: 10.1097/00000658-199906000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Panis Y, Ribeiro J, Chretien Y, Nordlinger B. Dormant liver metastases: an experimental study. British Journal Of Surgery. 1992;79(3):221–3. doi: 10.1002/bjs.1800790309. [DOI] [PubMed] [Google Scholar]
- 32.Ono T, Nagasue N, Kohno H, Hayashi T, Uchida M, Yukaya H, et al. Adjuvant chemotherapy with epirubicin and carmofur after radical resection of hepatocellular carcinoma: a prospective randomized study. Seminars in Oncology. 1997;24(2 Suppl 6):S6. [PubMed] [Google Scholar]
- 33.Kohno H, Nagasue N, Hayashi T, Yamanoi A, Uchida M, Ono T, et al. Postoperative adjuvant chemotherapy after radical hepatic resection for hepatocellular carcinoma (HCC) Hepato-Gastroenterology. 1996;43(12):1405–9. [PubMed] [Google Scholar]
- 34.Izumi R, Shimizu K, Iyobe T, Ii T, Yagi M, Matsui O, et al. Postoperative adjuvant arterial infusion of lipiodol containing anticancer drugs in patients with hepatocellular carcinoma. Hepatology. 1994;20:295–301. [PubMed] [Google Scholar]
- 35.Wu CC, Ho YZ, Ho WL, Wu TC, Liu TJ, P’eng FK. Preoperative transcatheter arterial chemoembolization for resectable large hepatocellular carcinoma. A reappraisal Br J Surg. 1995;82:122–6. doi: 10.1002/bjs.1800820141. [DOI] [PubMed] [Google Scholar]
- 36.Whalen GF, Bird I, Tanski W, Russell JC, Clive J. Laparoscopic cholecystectomy does not demonstrably decrease survival of patients with serendipitously treated gallbladder cancer. Journal of the American College of Surgeons. 2001 Feb;192(2):189–95. doi: 10.1016/s1072-7515(00)00794-8. [DOI] [PubMed] [Google Scholar]
- 37.Lai EC, Lo CM, Fan ST, Liu CL, Wong J. Postoperative adjuvant chemotherapy after curative resection of hepatocellular carcinoma: a randomized controlled trial. Arch Surg. 1998;133(2):183–8. doi: 10.1001/archsurg.133.2.183. [DOI] [PubMed] [Google Scholar]
- 38.Fong Y, Jarnagin W, Blumgart LH. Gallbladder cancer: comparison of patients presenting initially for definitive operation with those presenting after prior noncurative intervention. Annals Of Surgery. 2000;232(4):557–69. doi: 10.1097/00000658-200010000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kemeny N, Fong Y. Treatment of liver metastases. In: Holland JF, Frei E, Bast RC, Kufe DW, Morton DL, Weichselbaum RR, editors. Cancer Medicine. 4. Baltimore: Williams & Wilkins; 1997. pp. 1939–54. [Google Scholar]
- 40.Fong Y, Kemeny N, Lawrence TS. Cancer of the Liver and Biliary Tree. In: DeVita VT, Hellman S, Rosenberg SA, editors. Cancer: principles and practice of oncology. 6. Philadelphia: Lippincott Williams and Wilkins; 2001. pp. 1162–203. [Google Scholar]