

Abstract
During an acute Plasmodium infection, uncontrolled pro-inflammatory responses can cause morbidity and mortality. Regulation of this response is required in order to prevent immunopathology. We therefore decided to investigate a recently characterized subset of regulatory dendritic cells (DCs) that expresses low levels of CD11c and high levels of CD45RB. During a P. yoelii infection, these regulatory CD11cloCD45RBhi DCs become the prevalent CD11c-expressing cells in the spleen, overtaking the conventional CD11chi DCs. Furthermore, the regulatory CD11cloCD45RBhi DCs induce IL-10-expressing CD4 T cells. A similar change in splenic DC subsets is seen when mice are injected with sublethal doses of lipopolysaccharide (LPS), suggesting that shifting the splenic DC subsets in favor of regulatory CD11cloCD45RBhi DCs can be triggered solely by a high inflammatory stimulus. This is the first time regulatory DCs have been observed in a natural immune response to an infectious disease or endotoxic shock.
Keywords: Parasitic-protozoan, Dendritic cells, Inflammation, Tolerance/Suppression/Anergy, Endotoxin shock
Introduction
The encounter of dendritic cells (DCs) with microbes or microbial products results in activation and maturation of the DCs, leading to optimal antigen-presenting function and activation of T cells. Part of this process involves the induction of a pro-inflammatory cytokine response that helps to activate T cells in appropriate ways to counter the pathogen. As the immune response progresses, there is a need to inhibit the inflammatory response in order to prevent immunopathology (1). While there has been a lot of research demonstrating the involvement of anti-inflammatory cytokines, such as IL-10, in the dampening of the inflammatory response, little is known about the host cells involved in this process.
Malaria is a devastating disease caused by the Plasmodium parasite, killing one person every 30 seconds and causing between 300 and 500 million clinical cases every year. Malaria creates a large inflammatory response during acute infection, inducing high levels of IFN-γ, TNF-α and IL-12 in the serum of infected individuals (2). Despite inducing high inflammatory responses, individuals do not gain immunity to the parasite. There is only partial protection to malaria associated with resistance to disease. This is the result of chronic exposure to the parasite, which can still be found in partially immune individuals at low levels in the blood (3). Partial immunity is not permanent, however, as it is lost soon after individuals leave the endemic region (4), suggesting that the immune system is not capable of creating suitable immune responses against malaria in order to maintain immunity. Moreover, there are also numerous studies that describe immune suppression as a result of Plasmodium infections (3).
DCs play an integral role in immunity by bridging the gap between the innate and adaptive immune systems. As the most potent antigen-presenting cell, DCs are the primary cell responsible for launching and coordinating appropriate immune responses to pathogens and priming memory so that the organism is protected against future infections (1). In playing such a central role in immunity, it is not surprising that the function of DCs is targeted by the parasite. Indeed, several studies in both humans (5, 6) and mice (7-13) have documented changes in DC function in response to Plasmodium. In contrast, other reports have found fully functional DCs when exposed to parasites (14-21). Therefore a consensus is lacking on how DCs are regulating T cell responses during an infection (22).
The splenic DC compartment is a heterogeneous population, with subsets of cells differing across a spectrum of functions and morphologies (23). However, malaria researchers have largely treated splenic DCs as a single, homogeneous population. Only recently, the possibilities of DC subsets playing different roles during the course of a murine Plasmodium infection have been explored (13, 14, 16, 18), yet these reports have only considered conventional DCs which are identified by their high expression of the integrin-α chain CD11c. In order to attempt to reconcile the different DC phenotypes as seen by different research groups, we chose the context of a non-lethal murine Plasmodium infection that we had previously characterized in detail (8-10) and decided to turn our attention to CD11clo-expressing DCs, a largely neglected subset of DCs.
Recently, several reports have discussed the potential regulatory function of a DC subset characterized by their particular CD11cloCD45RBhi surface marker expression (24-27). Though originally derived by the addition of IL-10 to in vitro differentiation cultures of bone marrow-derived DCs, CD11cloCD45RBhi DCs were also observed to be resident in the spleens of mice (24, 27). They display a characteristic cytokine profile, secreting high levels of IL-10 without IL-12 when stimulated. They also express lower levels of CD86 and class II major histocompatibility complex (MHC) than their CD11chi counterparts (24, 27). The CD11cloCD45RBhi DCs earned their regulatory label through their capacity to induce regulatory T cells in vitro and in vivo (24-26). Moreover, when regulatory CD11cloCD45RBhi DCs are differentiated in vitro and transferred into mice, they induce antigen-specific tolerance (24-26) and suppress LPS-induced host inflammatory responses (27). Based on these properties, regulatory CD11cloCD45RBhi DCs have been proposed as potential therapeutic tools for the treatment of inflammatory diseases (26, 27).
We have found that this regulatory CD11cloCD45RBhi DC subset becomes the predominant CD11c-expressing cell population in the spleen during an acute P. yoelii infection, overtaking the conventional CD11chi DCs. Furthermore, a similar rapid expansion of the regulatory DCs and contraction of the conventional DCs occurs after the administration of high doses of lipopolysaccharide (LPS) to mice. This is first time regulatory CD11cloCD45RBhi DCs have been observed over the course of a natural immune response to infection.
Materials and Methods
Mice
Female BALB/c, Swiss Webster and C57BL/6 mice were purchased from the National Insititutes of Health. Female DO11.10 TCR-transgenic mice were purchased from Jackson Laboratories. All mice were housed in the Medical Parasitology animal facility, and all experiments were approved by the Institutional Animal Care and Use Committee.
Parasites, Infections and Endotoxin Injection
Nonlethal strain Plasmodium yoelii 17XNL-infected erythrocytes were harvested by cardiac puncture of infected, anesthetized Swiss Webster mice before the peak in parasitemia. Erythrocytes were washed twice with PBS and separated from white blood cells by centrifugation at 2000 g for 3 minutes. Erythrocytes were then spun on an Accudenz (Accurate Chemical & Scientific Corporation) gradient to isolate schizontsand late trophozoite-stage infected erythrocytes. The collected infected erythrocytes were washed and resuspended in PBS. Infected erythrocytes of lethal strains P. berghei ANKA and P. yoelii YM were obtained by bleeding mice from the tail, and then diluting to the appropriate iRBC concentration based on parasitemia counts. To start blood-stage infections, BALB/c and C57BL/6 mice were injected intraperitoneally with infected erythrocytes (103, 104, 105 or 106 cells per mouse) resuspended in PBS. Uninfected erythrocytes as controls were obtained in a similar manner from naive Swiss Webster mice.
P. yoelii 17XNL sporozoites were obtained from the dissection of infected Anopheles stephensi mosquito salivary glands. To infect BALB/c mice, sporozoites (10, 100 or 1000 per mouse) resuspended in RPMI 1640 medium were injected intravenously in the tail vein. Injection of RPMI 1640 medium was used as controls respectively. In the group that received 10 sporozoites, only 4 of 6 mice became infected, indicating that we were infecting mice with a very low numbers of sporozoites.
To evaluate parasitemia, thin blood smears were made by bleeding mice from a nick in the tail. Smears were stained with KaryoMAX Giemsa (Gibco), and a minimum of 500 erythrocytes per smear was counted.
Sublethal endotoxic shock was induced in BALB/c mice by injecting 15 mg/kg of LPS (Sigma-Aldrich) intravenously in the tail vein. PBS was injected as a control.
Cell separation
Single cell suspensions of splenocytes were obtained by mechanical disruption through a cell strainer and then osmotically lysing erythrocytes by incubation in an ammonium chloride/potassium hydrogen carbonate buffer. All spleen cell preparations were resuspended in DMEM (Mediatech) supplemented with 10% fetal bovine serum (FBS; Gibco) and PSG antibiotic mix (100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine; Gibco), and kept on ice.
For DC subsets, single cell suspensions of splenocytes were enriched for the CD11c+ population by positive selection using anti-CD11c microbeads and MACS magnetic columns (Miltenyi Biotec) as instructed by the manufacturer. After blocking using anti-CD16/CD32, cells were stained with PE anti-CD45RB (16A) and APC anti-CD11c (HL3), then sorted on a MoFlo high-performance cell sorter (Dako). After the sort, cells were washed with DMEM and resuspended in complete DMEM medium (DMEM supplemented with 10% FBS, PSG antibiotic mix, 1 M HEPES, 10 mM non-essential amino acids, and 55 mM β-mercaptethanol; all Gibco).
CD4 T cells from DO11.10 mice were isolated from single cell suspensions of splenocytes by negative selection using the murine CD4+ T cell isolation kit and MACS magnetics columns (Miltenyi Biotec) as instructed by the manufacturer. After isolation, cells were washed and resuspended in complete DMEM medium.
DC-T Cell Co-cultures
Sorted DCs were cultured in various ratios with DO11.10 CD4 T cells (8 × 104) and OVA323−339 peptide (10 μg/ml; Peptides International) in 96-well U-bottom plates and incubated at 37°C, 5% CO2. After 80 hours, culture media were harvested and analyzed using the BD Cytometric Bead Array (BD Biosciences) as instructed by the manufacturer.
Flow Cytometry
All flow cytometry, outside of the cell sorting, was performed on a FACS Calibur (Becton Dickinson) and analyzed with either CellQuest (Becton Dickinson) or FlowJo (TreeStar). All antibodies for FACS were purchased from Biolegend or BD Biosciences unless otherwise indicated.
For analysis of spleen DCs, single cell suspensions of splenocytes were first blocked with anti-CD16/CD32 (Fcγ III/II receptor; 2.4G2), then stained using a combination of FITC anti-CD40 (3/23), FITC anti-CD80 (16−10A1), FITC anti-CD86 (GL1), PE anti-CD45RB (16A), and APC anti-CD11c (HL3 and N418). Isotype-matched antibodies were used as staining controls. mPDCA-1 staining was performed using biotinylated anti-mPDCA-1 (JF05−1C2.4.1, Miltenyi Biotec) followed by Cy5PE-conjugated streptavidin (Biolegend).
For T cell surface marker analysis, cells from 7-day DC-T cell co-cultures were harvested, blocked with anti-CD16/CD32, then stained using a combination of FITC anti-CD44 (IM7), FITC anti-CD25 (PC61), FITC anti-CD69 (H1.2F3), PE anti-LFA-1 (2D7), PE anti-CD62L (MEL-14), PE anti-CTLA-4 (UC10−4F10−11), Cy5PE anti-CD4 (GK1.5), and APC anti-CD3ε (145−2C11).
For intracellular cytokine staining of T cells, cells from 7-day DC-T cell co-cultures were restimulated with PMA (50 ng/ml) and ionomycin (1 μg/ml) alongside monensin (10 μg/ml) (all from Sigma-Aldrich) for 5 hours. After harvesting, cells were blocked with anti-CD16/CD32, then stained using Cy5PE anti-CD4, and APC anti-CD3ε. After fixation and permeabilization using FoxP3 staining buffer reagents (eBioscience) as instructed by the manufacturer, cells were incubated with FITC anti-IFN-γ (XMG1.2), PE anti-IL-10 (JES5−16E3), and Alexa Fluor 488 anti-IL-4 (11B11). Isotype-matched antibodies were used as staining controls.
Statistical analysis
Data were analyzed using Prism (GraphPad). Student's t-tests and one- or two-way ANOVA were performed as mentioned. All statistics were considered significant if P<0.05.
Results
Regulatory CD11cloCD45RBhi dendritic cells are the prevalent DC subpopulation in the spleen during an acute P. yoelii infection
Using markers previously described to phenotype DCs (24), we identified the two major subpopulations of CD11c-expressing cells in the spleens of BALB/c mice: conventional CD11chi (CD45RBint) cells (Figure 1A, gate R1) and regulatory CD11cloCD45RBhi DCs (Figure 1A, gate R2). During the course of an acute P. yoelii infection initiated by intraperitoneal injection of infected erythrocytes, there were dramatic changes in the composition of the CD11c+ splenic DC population. The proportion of regulatory CD11cloCD45RBhi cells increased dramatically over the course of infection while the conventional CD11chi DCs suffered a concurrent decrease (Figure 1B). These populations remained altered late into the acute infection. Moreover, the expansion of the regulatory CD11cloCD45RBhi DC subset was not unique to P. yoelii 17XNL as both lethal infections with P. berghei ANKA and P. yoelii YM demonstrated increases in this population (Figure 1C, D).
Figure 1. The regulatory CD11cloCD45RBhi subset of DCs becomes the predominant subset in the spleen during a P. yoelii infection.

(A-B, E-H) BALB/c mice were injected i.p. with 106 control (RBC, black circles) or P. yoelii-infected (iRBC, white squares) erythrocytes. Splenocytes from groups of 4 mice were stained with antibodies to CD11c and CD45RB. (A) Two subpopulations, R1 (CD11chi) and R2 (CD11cloCD45RBhi) were gated for analysis by flow cytometry. The FACS plots are representative of splenocytes from day 0 and day 10 post-infection. (B) At the indicated days post-injection, splenocytes were analyzed for the two subsets, expressed as the proportion of CD11c+ cells (DCs). (C-D) C57BL/6 mice were injected i.p. with 106 P. berghei ANKA-infected erythrocytes (C). BALB/c mice were injected i.p. with 106 P. yoelii YM-infected erythrocytes (D). Splenocytes were harvested on the days indicated and stained as in (A). The values on the plots represent the proportion of the CD11c+ gate that each subset comprises. (E) On the days indicated post-injection, two subpopulations, CD11chi and CD11cloCD45RBhi were gated, and analyzed for their expression of mPDCA-1 (heavy line) by flow cytometry. A staining control was also analyzed (grey line). (F-G) At the indicated days post-injection, the number of total spleen cells (F) and the absolute number of each DC subset (G) were quantified. (H) Parasitaemia were quantified by Giemsa-stained slides. The results are representative of three independent experiments. Error bars represent standard deviation within groups of 4 mice (*, P < 0.05; ***, P < 0.001 when comparing iRBC to RBC by two-way ANOVA).
As plasmacytoid DCs also express low levels of CD11c, we stained splenocytes from mice using the plasmacytoid DC marker, mPDCA-1 (28). Regulatory CD11cloCD45RBhi DCs isolated from mice on day 10 of blood stage infection did not express the marker, though it is indeed present on 10% of the CD11cloCD45RBhi cells from naive mice (Figure 1E).
Because of the splenomegaly induced by P. yoelii infections, which results in an increase of 10- to 15-fold in the number of total cells in the spleen (Figure 1F), we calculated the absolute number of these two subpopulations in the spleen. The number of regulatory CD11cloCD45RBhi DCs increased over 30-fold during the 14 days of acute infection. On the other hand, the number of conventional CD11chi DCs only increased 3-fold by day 10 (Figure 1G). This indicates that the ratio of regulatory versus conventional DCs has changed from 3:1 in control mice to 28:1 at day 6 after infection, before the peak of parasitemia at day 10 (Figure 1H).
Regulatory DCs do not upregulate surface expression of CD40 over the course of an acute P. yoelii infection
We also determined the maturation phenotype of the splenic DCs as a whole or analyzed separately in the subpopulations described above in Figure 1A. Cells were assayed for their expression of costimulatory molecules CD40, CD80 and CD86 since upregulation of these molecules is considered an indicator of DC maturation (1). CD11chi DCs expressed increased levels of all three costimulatory markers over the course of infection (Figure 2A, left panels), confirming that this subset correlates with the conventional DC population (24). Regulatory CD11cloCD45RBhi DCs, though increasing their CD80 and CD86 early in the acute infection, only had CD80 upregulated on day 10. More striking was that regulatory DCs did not upregulate CD40 at any point during the acute infection. In fact, expression of CD40 on this subset dropped below the levels found in the uninfected control mice late in infection (Figure 2A, center panels; Figure 2B).
Figure 2. Regulatory CD11cloCD45RBhi DCs upregulate CD80 and CD86, but not CD40.

Mice were injected i.p. with 106 control or infected erythrocytes. At the indicated days post-injection, spleen CD11c+ DC subsets were analyzed for their expression of the costimulatory molecules. (A) CD40, CD80 and CD86 expression on DCs from control (RBC, black circles) or infected (iRBC, white squares) mice, expressed as mean fluorescence intensity (MFI). Error bars represent standard deviation within groups of 5 mice (*, P < 0.05; **, P<0.01; ***, P < 0.001 when comparing iRBC to RBC by twoway ANOVA). (B) FACS plots showing expression of CD40 on DCs from control (grey shaded histogram) and infected (empty histogram with heavy line) mice. An isotype-matched control was also analyzed (thin grey line). Parasitaemia were similar to those indicated in Figure 1. The results are representative of two independent experiments.
When analyzing the entire DC population (CD11c+ cells), their expression of costimulatory molecules appeared to mirror that of the regulatory CD11cloCD45RBhi subpopulation, with the reduction of CD86 to background levels by day 10, and with very poor upregulation of CD40 at any point during the infection (Figure 2A, right panels). This is a consequence of the fact that the regulatory subset is the dominant DC population in the spleen (Figure 1B).
Regulatory CD11cloCD45RBhi DCs induce proliferation and IL-10 expression in antigen-specific CD4 T cells
To functionally characterize the subsets of DCs analyzed, we determined the capacity of each subset to activate antigen-specific CD4 T cells in vitro. The two subpopulations of DCs as described above in Figure 1A were FACS-sorted from the spleens of mice at day 10 post-infection and co-cultured with ovalbumin (OVA)-specific CD4 T cells. These CD4 T cells, purified from the spleens of DO11.10 mice, express a specific T cell receptor that recognizes the CD4 OVA323−339 epitope. We also sorted the two DC subpopulations from naive littermates to determine whether they acted differently within the context of a Plasmodium infection. When assayed for proliferation after 4 days of peptide-induced stimulation, both DC subsets from naive and infected mice were equally capable of inducing proliferation from the OVA-specific T cells (Figure 3A). Indeed, similar levels of T cell proliferation are induced by conventional and regulatory DCs in response to peptide (24).
Figure 3. Regulatory CD11cloCD45RBhi DCs induce proliferation and IL-10 expression in antigen-specific CD4 T cells.

Mice were injected i.p. with 106 infected erythrocytes. At days 0 (naive mice) and 10 post-injection, DC subsets were sorted by FACS. DC subsets were then co-cultured with naive CD4 T cells isolated from DO11.10 mice in the presence of OVA peptide. (A) After 4 days of co-culture, T cell proliferation was determined using the incorporation of [H3]-thymidine for the final 16 hours of culture as a readout. Error bars represent standard deviation within co-cultures of cells sorted from 3 individual mice. (B-C) After 24 hours or 7 days of co-culture with DCs sorted from infected (B) or naive (C) mice, T cells were collected and assayed via flow cytometry for markers of T cell activation. FACS histograms show the expression of the surface marker when T cells are co-cultured with CD11chi DCs (thick line) or CD11cloCD45RBhi DCs (thin line). Stainings with isotype control antibodies are shown in the grey shaded histograms. Each FACS plot is representative of one out of three individual mice. (D) After 7 days of co-culture with sorted DCs from day 0 (naïve) or day 10-infected mice, cells were restimulated with PMA and ionomycin for 5 hours. CD4 T cells were stained intracellularly for cytokine expression. Isotype controls for the cytokine-specific antibodies are shown as FACS histograms, as detailed in B. Each FACS plot is representative of one out of three DC-T cell co-cultures, and are representative of two independent experiments.
T cells were also assayed for their cell surface expression of T cell activation molecules after co-incubation with either the conventional CD11chi or the regulatory CD11cloCD45RBhi DC subsets in the presence of the OVA epitope. T cells demonstrated similar activation phenotypes when looking at the activation markers CD25, CD44, CD69, and LFA-1 after 24 hours or 7 days of co-incubation with DC subsets (Figure 3B). The notable differences were in their day 7 expression of CD62L and CTLA-4. T cells incubated with conventional DCs had downregulated their expression of CD62L by day 7 of the co-culture. In contrast, T cells incubated with regulatory DCs retained their high expression of CD62L (Figure 3B). Similarly, though CTLA-4 expression was induced on T cells by both DC subsets after 24 hours of co-culture, by day 7 CTLA-4 had been downregulated on the T cells co-cultured with the conventional CD11chi DCs while it remained high on T cells co-cultured with regulatory CD11cloCD45RBhi DCs (Figure 3B). Importantly, similar trends were observed in the T cell surface marker phenotype if they were co-cultured with DC subpopulations taken from naive mice (Figure 3C).
In order to get a better functional phenotype of the T cells being activated by conventional and regulatory DCs, we looked at the intracellular cytokine expression of the T cells after 7 days co-culture with the separate DC subsets from infected and naive mice. When the OVA-specific CD4 T cells were stained for expression of intracellular cytokines after 7 days incubation, a large proportion of T cells co-cultured with conventional DCs from day 10-infected mice expressed high amounts of IFN-γ without IL-10 (33.1 ± 2.4% of CD4 T cells). If instead the CD4 T cells were co-cultured with regulatory DCs, the proportion of T cells expressing IFN-γ without IL-10 was decreased by half (15.3 ± 0.1%, P<0.0005). On the other hand, regulatory DCs from day 10-infected mice induced four times as many T cells to express IL-10 without IFN-γ (35.1 ± 1.3% of CD4 T cells by regulatory DCs versus 8.1 ± 2.4% by conventional DCs, P<0.00001) (Figure 3D, day 10 top panels). Moreover, co-culture with regulatory DCs induced twice the number of CD4 T cells expressing IL-10 without IL-4 (52.0 ± 0.9% by regulatory DCs versus 21.9 ± 3.0% by conventional DCs, P<0.0001) (Figure 3D, day 10 bottom panels). Co-cultures with DCs isolated from naive mice (day 0) demonstrated similar trends, however in a much more polarized fashion. Conventional DCs induce 15 times as many CD4 T cells to express IFN-γ without IL-10, whereas regulatory DCs induce IL-10 without IFN-γ from nearly 70% of the T cells (Figure 3D, day 0 top panels). Notably, naive regulatory DCs induced co-expression of IL-4 and IL-10 from a good proportion of CD4 T cells (15.3 ± 1.3% of CD4 T cells) which was not the case when the T cells were co-cultured with regulatory DCs from infected mice (2.6 ± 0.3%) (Figure 3D, day 0 bottom panels), suggesting that regulatory DCs, particularly those from infected mice, may induce a regulatory phenotype in T cells as characterized by expression of IL-10 but not IL-4 or IFN-γ (29). In summary, these results appear to indicate that although both DC subsets can induce CD T cells to proliferate, conventional CD11chi DCs induce classical activation of IFN-γ-expressing T cells, while the regulatory CD11cloCD45RBhi DCs preferentially induce CD62LhiCTLA-4+ T cells that express IL-10 without IL-4. Interestingly, prolonged CD62L and CTLA-4 expression has been correlated to a regulatory T cell phenotype (30-32).
Conventional DCs induce an inflammatory cytokine milieu
We looked at the cytokine milieu induced by the co-culture of each DC subset with OVA-specific CD4 T cells. After sorting DCs from naive or day 10-infected mice, DCs were co-cultured with OVA-specific CD4 T cells in the presence of OVA peptide for 80 hours. The culture media from the wells were then assayed for their cytokine contents. Culture medium from the co-cultures with conventional DCs from either naive or infected mice contained high levels of the inflammatory cytokines IL-6, TNF-α and IL-12p40. Culture media from co-cultures with regulatory DCs contained 50−75% less IL-6, 25−60% less TNF-α, and virtually no IL-12p40 (Figure 4). IFN-γ was also elevated in the co-culture of CD4 T cells with conventional DCs from naive mice when compared to regulatory DCs, however this difference is not as evident in the co-cultures with DCs from infected mice. We had found comparable results when analyzing intracellular cytokine staining of T cells from similar co-cultures (Figure 3D). Regulatory DCs from naive mice induced more IL-10 in the co-cultures when compared to their conventional DC counterparts; however, the trend is less pronounced when considering the co-cultures of DCs from infected mice (Figure 4). It is likely that both DCs and T cells contribute to the cytokines detected in this assay, and in particular IL-10 since both the regulatory DC and the malaria literature document the expression of IL-10 by DCs (9, 18, 24, 25).
Figure 4. Conventional DCs induce a stronger inflammatory cytokine environment.
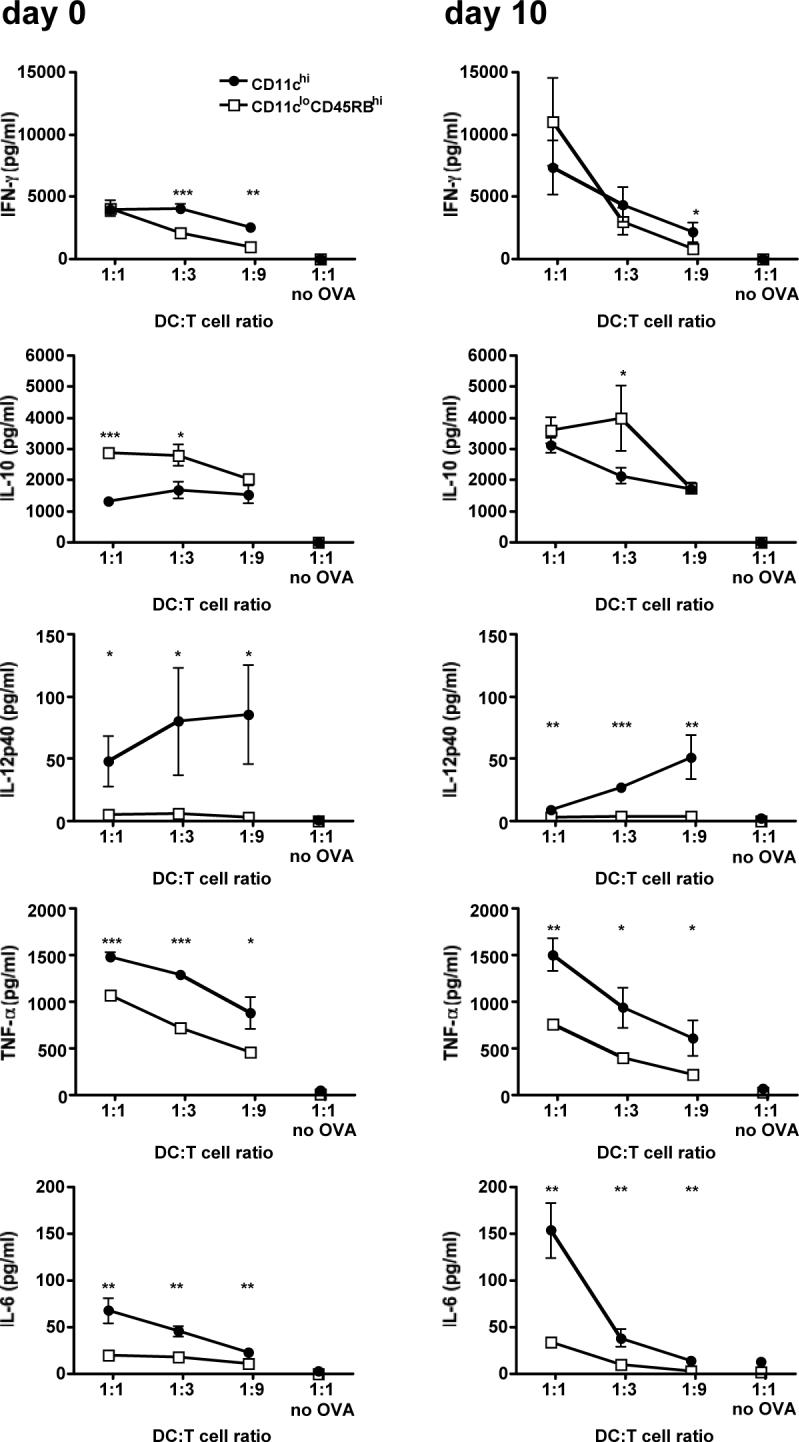
Mice were injected i.p. with 106 infected erythrocytes. At days 0 (naive mice) and 10 post-injection, DC subsets were sorted by FACS. DC subsets were then co-cultured at various ratios with naive CD4 T cells isolated from DO11.10 mice, with or without OVA peptide. After 80 hours of co-culture, the culture media was assayed for cytokine content using the BD Cytometric Bead Array. Error bars represent standard deviation within co-cultures of cells sorted from 3 individual mice (*, P < 0.05; **, P<0.01; ***, P < 0.001 when comparing CD11chi co-cultures to CD11cloCD45RBhi co-cultures by Student's t-test).
Higher infection doses result in a faster progression towards a regulatory DC phenotype in the spleen
Since infection of humans with low, subphysiological doses of blood-stage parasites result in effective immunity against the disease (33), we were curious as to whether the shifting of the DC subsets could be dependent on the dose of parasite inoculum.
We infected mice with 106 infected erythrocytes, similar to our previous experiments, and 10-fold dilutions to the subphysiological amount of 1000 infected erythrocytes. Splenic DC subsets (Figure 5A) and parasitemia (Figure 5B) were assayed at different points up until day 10 post-injection.
Figure 5. The dose of infected erythrocytes dictates the balance of DC subsets observed during an acute P. yoelii infection.

(A-C) Mice were injected i.p. with 106 control (RBC, black circles) or 106, 105, 104, or 103 infected (white symbols) erythrocytes. (A) At the indicated days post-injection, splenocytes from groups of 3 mice were analyzed for the two subpopulations, expressed as the proportion of CD11c+ cells (DCs). (B) Parasitaemia were quantified by Giemsa-stained slides. (C) The expression of costimulatory molecules for the splenic DC subsets from (A), expressed as mean fluorescence intensity (MFI). The results are representative of two similar independent experiments. (D-E) Mice were injected i.p. with 106 control or infected erythrocytes. At days 0, 1 or 2 post-injection, splenocytes from groups of 3 mice were analyzed for the two subpopulations. (D) Subpopulations from control (RBC, black circles) or infected (iRBC, white squares) mice are expressed as the proportion of CD11c+ cells (DCs). Error bars represent standard deviation within groups of at least 3 mice (no significant difference when comparing iRBC to RBC by two-way ANOVA). (E) FACS plots showing expression of CD40 on DCs from control (grey shaded histogram) and infected (empty histogram with heavy line) mice. An isotype-matched control was also analyzed (thin grey line).
When mice were infected with the high dose of 106 infected erythrocytes, the regulatory CD11cloCD45RBhi DCs had already begun to expand by day 3. By day 6, more than 85% of the splenic DCs were comprised of this population. In contrast, expansion of the regulatory DC subset was delayed to day 10 in mice that received the lower doses of 104 or 103 infected erythrocytes (Figure 5A, right panel). The levels of CD86 mirror this temporal delay between high and low parasite doses, with upregulation seen earlier on day 3 on the DCs from mice that had received 106 infected erythrocytes, and later (day 6) on the DCs from mice that had received 104 and 103 infected erythrocytes (Figure 5C, bottom right panel).
The conventional CD11chi subset had declined to its minimum proportion by day 10 in all groups of mice. Interestingly, on day 3 the three lower doses had induced an expansion of this population. This is in contrast to mice infected with the highest dose of 106 infected erythrocytes, which on day 3 already showed lower proportions of this subset than the uninfected controls (Figure 5A, left panel). We therefore questioned whether there had been an earlier expansion of the conventional DC subpopulation in mice that had received 106 infected erythrocytes. We analyzed the splenic DC subsets at 24 and 48 hours post-infection. Neither of the early time-points demonstrated any significant changes in the proportions (Figure 5D) or absolute numbers (data not shown) of either of the two DC subpopulations analyzed. Moreover, regulatory DCs maintained low levels of CD40 even at these early time points (Figure 5E).
Malaria starts with an average of 15−123 sporozoites inoculated into the skin by an infected mosquito. A fraction of these sporozoites will reach the liver where they infect hepatocytes. After vigorous asexual replication, each infected hepatocyte produces thousands of merozoites (34). P. yoelii can produce more than 8,000 merozoites per infected hepatocyte (35), compared with 40,000 merozoites produced by P. falciparum (36). This initial step guarantees that blood-stage infection starts with a high dose of merozoites delivered to the blood stream. We also investigated if infection started by sporozoites would change the proportions of splenic DC subsets. We therefore injected 10, 100 and 1000 sporozoites intravenously into mice and assayed their splenic DC subsets on day 12 post-injection. As P. yoelii sporozoites take 2 days to develop in the liver and start a blood-stage infection (35), day 12 post-injection of sporozoites correlates to day 10 of a blood-stage infection. By day 10 of the blood-stage infection, even the mice that had received only 10 sporozoites demonstrated the maximum regulatory DC phenotype in their spleens (Figure 6A). Parasitemia curves from sporozoite-injected mice paralleled those of the dose of 106 infected erythrocytes (Figure 6B). Notably, on day 3 after the start of blood-stage infection, mice that had received 10 sporozoites had parasitemias similar to those mice injected with 106 infected erythrocytes, while mice that had received 105 infected erythrocytes still had parasite-negative slides at this point of blood-stage infection. This indicates that a dose of 106 infected erythrocytes is the best physiological correlate to infection with low numbers of sporozoites.
Figure 6. Blood-stage infections started sporozoites are similar to those started with 106 infected erythrocytes.

Mice were injected i.v. with 1000, 100 or 10 sporozoites (spz, black and grey bars) or the vehicle RPMI (white bars). (A) At the indicated days post-injection, splenocytes from groups of 3 mice were analyzed for the two subpopulations, expressed as the proportion of CD11c+ cells (DCs). Error bars represent standard deviation within groups of 3 mice (***, P < 0.001 when comparing iRBC to RBC by one-way ANOVA). (B) Parasitaemia were counted via Giemsa-stained slides (white symbols) and are overlaid here with the parasitemia curves from infections started with 106 and 105 infected erythrocytes from Figure 5B for comparison (grey symbols). The differences between the doses of 106 and 105 infected erythrocytes on day 3 become apparent because of the logarithmic scale used. The graph is normalized to account for the 2 days of development required by sporozoites before starting blood-stage infections. Parasitemia counts below the level of detection are plotted below the black dashed line.
Taken altogether, these results indicate that a P. yoelii blood-stage infection, even if started with subphysiological doses (103, 104 and 105) of infected erythrocytes, will result with the emergence of the regulatory CD11cloCD45RBhi DCs in the spleen. The results also demonstrate that subphysiological doses of infected erythrocytes induce an early expansion of the conventional DC population that is not observed at the higher physiological doses of parasites.
Injection of LPS also induces the expansion of the regulatory CD11cloCD45RBhi DCs
We were curious whether the predominance of regulatory CD11cloCD45RBhi DCs observed during infection was unique to malaria. Indeed, the loss of conventional CD11chi DCs in the spleen has been documented as part of the tolerance response to endotoxin (37). We therefore used a high, but sublethal, dose of LPS to investigate changes in splenic DC subsets after an acute inflammatory response.
Mice were injected intravenously with a sublethal dose of LPS (15 mg/kg) and their splenic DC subsets were assayed at different points up until day 10 post-injection. Surprisingly, as early as 24 hours after LPS administration the regulatory DC subset had rapidly expanded to represent over 75% of the splenic DCs (Figure 7A, left panel). Over the same period, the conventional DCs had reduced to half their starting proportion (Figure 7A, right panel). Both of these trends continued and peaked at 48 hours post-injection when the conventional DCs are reduced to less than 1% of splenic DCs. On day 3, the regulatory DC shift began to resolve, and was back to control levels by day 6. Notably, unlike during a Plasmodium infection (Figure 2), regulatory CD11cloCD45RBhi DCs had upregulated their CD40 at 24 hours after LPS administration, similarly to the conventional DCs (Figure 7B, C). In conclusion, it appears that the expansion of the regulatory DC population also occurs in response to LPS and is not distinctive of the immune response to malaria.
Figure 7. A sublethal dose of LPS induces expansion of regulatory CD11cloCD45RBhi DCs.

Mice were injected i.v. with 15μg LPS (white squares) or vehicle (PBS, black circles). (A) At the indicated days post-injection, splenocytes from groups of 3 mice were analyzed for the two subpopulations, expressed as the proportion of CD11c+ cells (DCs). (B) Each subset was also assayed for expression of costimulatory molecules, expressed as mean fluorescence intensity (MFI). Error bars represent standard deviation within groups of at least 3 mice (*, P < 0.05; ***, P < 0.001 when comparing LPS to PBS by two-way ANOVA). (C) FACS plots showing expression of CD40 on DCs from mice that had received vehicle (grey shaded histogram) or LPS (empty histogram with heavy line). An isotype-matched control was also analyzed (thin grey line).
Discussion
We have analyzed the prevalence and function of regulatory DCs, a recently characterized subset, identified by their low expression of CD11c and the high expression of CD45RB. These CD11cloCD45RBhi DCs have been characterized primarily in vitro, where their regulatory characteristics were initially observed (24). Their regulatory function has also been shown in vivo, where regulatory CD11cloCD45RBhi DCs were transferred into mice in order induce tolerance by inducing regulatory T cells (24-26) or to suppress inflammatory immune responses (27). Though it has been demonstrated that this regulatory DC subset exists naturally in vivo (24, 27), we are the first to document their expansion during an inflammatory immune response, either in response to endotoxic shock or malaria. During an acute P. yoelii infection, we found that the regulatory CD11cloCD45RBhi DCs increase in proportion, making up over 85% of the splenic CD11c+ DC population. This is accompanied by a concurrent decrease in proportion of the conventional CD11chi DC subset. This change in the composition of the splenic DC population is maintained during the course of the disease. Moreover, these changes are seen irrespective of whether the blood-stage infection was started directly with the injection of infected erythrocytes or by first going through the liver stage via the injection of sporozoites. As the same switch is seen in both non-lethal (P. yoelii 17XNL) and lethal malaria infections (P. berghei ANKA and P. yoelii YM), it seems that the capacity to expand the regulatory DC population is not related to the lethality of the parasite strain. However, it is likely that different parasite strains may induce differential cytokine and T cell responses from the regulatory DCs population leading to differences in parasite clearance mechanisms.
Regulatory CD11cloCD45RBhi DCs differentiate as a result of a high IL-10 environment (24, 27). Since malaria is known to induce high amounts of IL-10 in mice (9, 38) as well as in humans (39), such an environment could foster the development and expansion of regulatory CD11cloCD45RBhi DCs. We have observed the increase in proportion of the regulatory DC subset in response to LPS and P. yoelii infection, suggesting that this expansion could be the result of the inflammation induced by these two stimuli, however further study would be required to substantiate this hypothesis. Indeed, early in Plasmodium infections there are large amounts of IFN-γ, TNF-α and IL-12 found in the serum (2). While an early pro-inflammatory response appears to be essential for the survival of a lethal infection (40-42), the complications associated with inflammation cause most of the pathology associated with malaria (43). IL-10 is an important anti-inflammatory mediator that can prevent death due to LPS- or malaria-induced inflammation (44-46). Notably, regulatory CD11cloCD45RBhi DCs induce IL-10-expressing T cells (24-26). We too have seen a similar functional role for the regulatory CD11cloCD45RBhi DCs isolated from P. yoelii-infected mice since they do not induce T cells to express high amounts of IFN-γ. Instead, the regulatory DCs induce IL-10-expressing T cells and reduce the level of inflammatory cytokines in the medium. Thus by eliciting the regulatory CD11cloCD45RBhi DC subset, the immune response may be seeking to stem the inflammation, saving the organism from an uncontrolled inflammatory response. It must be emphasized that the mechanisms by which regulatory DCs are induced, either in response to LPS or P. yoelii, are not yet known and could differ between the two systems.
The similarities between bacterial endotoxin tolerance and malaria have been discussed in detail (47). As in malaria, the regulatory CD11cloCD45RBhi DC subset expands rapidly in response to LPS injection. This is matched by a quick contraction of the conventional CD11chi subset that is a contributor of pro-inflammatory mediators during an immune response (1). Indeed, a decrease in the conventional CD11chi DCs has already been documented as part of the endotoxin tolerance response (37). Moreover, conventional DCs have recently been shown to be key mediators of inflammation in the experimental murine model for cerebral malaria (48), and our results indicate that this subset in particular induces high amounts of inflammatory cytokines when co-cultured with T cells. Considering the regulatory effects of the CD11cloCD45RBhi DCs on T cells, the changes in relative abundance of regulatory DCs will likely influence the course of the adaptive immune response during high inflammatory conditions. It is interesting to point out that in malaria, it seems to be the balance between the regulatory and conventional DCs that provides the tolerogenic response rather than the induction of a refractory DC phenotype. This is suggested by the fact that both conventional and regulatory DCs extracted from infected appear to act in a similar way to those taken from naive mice. Moreover, conventional DCs during malaria do not appear to be refractory, since even in late infection they are found in a mature state and are able to stimulate T cells to produce IFN-γ.
There is a notable difference between the regulatory CD11cloCD45RBhi DCs induced by a Plasmodium infection and those induced by endotoxic shock: while LPS administration causes regulatory DCs to upregulate all their costimulatory molecules including CD40, at no point during a P. yoelii infection is CD40 upregulated on this DC subset. CD40-CD40L interactions are very important both in their involvement in priming T cells as well as in their role in fully licensing DCs to become professional antigen-presenting cells (49). Failure to provide this signal inhibits CD4 T cell activation (50), confers tolerance(49), and induces regulatory T cells (51). Indeed, our results demonstrate that regulatory DCs do not activate T cells as well as their conventional DC counterparts. Recently, it has been shown that induction of CD4 memory T cells requires CD40; moreover, CD40 is also necessary to elicit effector functions (IFN-γ secretion) from CD4 memory T cells (52). Thus by retaining low surface expression of CD40 on the regulatory DCs, the parasite may be interfering with the induction of effector and memory T cell responses.
There has been much debate in trying to understand how DCs function during a Plasmodium infection. On one side, several groups have demonstrated that DCs mature in response to the malaria parasite, upregulating costimulatory and MHC molecules, secreting Th1-inducing and pro-inflammatory cytokines, and initiating antigen-specific T cell responses (14-21). In contrast, other groups have also documented seemingly opposite results, where the DC maturation response is modulated by Plasmodium, resulting in the inability to upregulate costimulatory and histocompatibility molecules, secretion of anti-inflammatory mediators, and inhibition of T cell responses (5-13). More recently, there has been a renewed attempt to understand these differences, looking at the time of infection (14, 18), CD11chi DC subpopulations (13, 16, 18), and the different Plasmodium species being used as model infections (13, 21).
In this study, we show that there are several factors that will dictate the phenotype of the DC population during a Plasmodium infection: the definition of the DC population based on CD11c expression, the time during infection at which the cells are assayed, and the dose of infected erythrocytes used to begin the blood-stage infection in the murine malaria model.
CD11c is an integrin receptor used as a marker to define DCs in mice (23). Classically, the analysis of DCs isolated from infected mice or humans typically involves the use of flow cytometry that will lead to different results depending on whether only CD11chi (14-16) or all CD11c+ cells are considered for the analysis (8-10, 18, 19). We found that when considering all the CD11c+ cells (and not CD11chi cells alone), the low costimulatory molecule expression pattern resembled that of the CD11clo cells because of the dominance of this subset in the spleen, especially late in infection. Considering exclusively the CD11chi cells shows DCs with upregulated levels of costimulatory molecules. And so it is likely that the discrepancy in the maturation state of DCs during malaria, as reported by different groups, may have simply been the result of the differential gating of the CD11c+ population. Moreover, consideration must be given to the method of ex vivo enrichment of DCs, as certain magnetic bead protocols can preferentially select for the CD11chi population.
We have also analyzed costimulatory molecules surface expression changes over the course of the acute blood-stage infection. We observed that the day post-infection at which cells are harvested also determines the level of maturation seen on DCs. This is not so much a result of temporal changes in DC maturation as it is a shift in the balance towards the regulatory DC subset later in infection.
Finally, the dose of infected erythrocytes used to start a blood-stage infection can influence the phenotype of the immune response, especially early in infection. We have shown that subphysiological doses of infected erythrocytes appear to induce a different immune response, particularly within the first 3 days of infection. There is an expansion of the conventional DCs on day 3 that is not seen with the higher physiological dose of infected erythrocytes, and which could be relevant for the downstream immune effector functions. This is perhaps not surprising as there have been studies in both humans and mice illustrating that subpatent infections (infections below the limit of detection by blood smears) can elicit protective responses (33, 53). Indeed, our results suggest that the parasite may use the liver stage replication to provide a large inoculum into the blood stream that would avoid the expansion of conventional DCs induced by the subphysiological doses of infected erythrocytes.
In conclusion, we have described the emergence of a regulatory DC subset that is induced in both a murine model of malaria as well as in sublethal endotoxin shock. The regulatory DCs appear to influence the immune response at least in part by modulating T cell activity, inducing a more regulatory over an effector T cell phenotype. The downstream effects of this anti-inflammatory response, both the involvement in parasite clearance and development of memory, needs to be addressed. Similarly, understanding the mechanism by which these DCs emerge could provide an opportunity for manipulation of the immune response to allow for effective clearance while avoiding pathologies associated with excessive inflammation.
Acknowledgements
We thank Dabeiba Bernal-Rubio and Sandra Gonzalez for help with murine infections and Jamie Orengo for her critical reading of the manuscript.
KW was partially funded by the Natural Sciences and Engineering Research Council of Canada. AR was supported by NIH grant AI 053698.
Abbreviations used in this paper
- DC
dendritic cell
- LPS
lipopolysaccharide
- OVA
ovalbumin
- iRBC
infected erythrocyte
- MFI
mean fluorescence intensity
Footnotes
Disclosures The authors have no financial conflict of interest.
References
- 1.Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 2.Stevenson MM, Riley EM. Innate immunity to malaria. Nat. Rev. Immunol. 2004;4:169–180. doi: 10.1038/nri1311. [DOI] [PubMed] [Google Scholar]
- 3.Plebanski M, Hill AV. The immunology of malaria infection. Curr. Opin. Immunol. 2000;12:437–441. doi: 10.1016/s0952-7915(00)00117-5. [DOI] [PubMed] [Google Scholar]
- 4.Marsh K. Malaria--a neglected disease? Parasitology. 1992;104(Suppl):S53–69. doi: 10.1017/s0031182000075247. [DOI] [PubMed] [Google Scholar]
- 5.Urban BC, Ferguson DJ, Pain A, Willcox N, Plebanski M, Austyn JM, Roberts DJ. Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature. 1999;400:73–77. doi: 10.1038/21900. [DOI] [PubMed] [Google Scholar]
- 6.Urban BC, Mwangi T, Ross A, Kinyanjui S, Mosobo M, Kai O, Lowe B, Marsh K, Roberts DJ. Peripheral blood dendritic cells in children with acute Plasmodium falciparum malaria. Blood. 2001;98:2859–2861. doi: 10.1182/blood.v98.9.2859. [DOI] [PubMed] [Google Scholar]
- 7.Millington OR, Di Lorenzo C, Phillips RS, Garside P, Brewer JM. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J. Biol. 2006;5:5. doi: 10.1186/jbiol34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carapau D, Kruhofer M, Chatalbash A, Orengo JM, Mota MM, Rodriguez A. Transcriptome profile of dendritic cells during malaria: cAMP regulation of IL-6. Cell. Microbiol. 2007;9:1738–1752. doi: 10.1111/j.1462-5822.2007.00910.x. [DOI] [PubMed] [Google Scholar]
- 9.Ocaña-Morgner C, Mota M, Rodriguez A. Malaria blood-stage suppression of liver-stage immunity by dendritic cells. J. Exp. Med. 2003;197:143–151. doi: 10.1084/jem.20021072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ocaña-Morgner C, Wong KA, Lega F, Dotor J, Borras-Cuesta F, Rodriguez A. Role of TGF-beta and PGE(2 )in T cell responses during Plasmodium yoelii infection. Eur. J. Immunol. 2007;37:1562–1574. doi: 10.1002/eji.200737068. [DOI] [PubMed] [Google Scholar]
- 11.Pouniotis DS, Proudfoot O, Bogdanoska V, Apostolopoulos V, Fifis T, Plebanski M. Dendritic cells induce immunity and long-lasting protection against blood-stage malaria despite an in vitro parasite-induced maturation defect. Infect. Immun. 2004;72:5331–5339. doi: 10.1128/IAI.72.9.5331-5339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pouniotis DS, Proudfoot O, Bogdanoska V, Scalzo K, Kovacevic S, Coppel RL, Plebanski M. Selectively impaired CD8+ but not CD4+ T cell cycle arrest during priming as a consequence of dendritic cell interaction with plasmodium-infected red cells. J. Immunol. 2005;175:3525–3533. doi: 10.4049/jimmunol.175.6.3525. [DOI] [PubMed] [Google Scholar]
- 13.Wykes MN, Liu XQ, Beattie L, Stanisic DI, Stacey KJ, Smyth MJ, Thomas R, Good MF. Plasmodium Strain Determines Dendritic Cell Function Essential for Survival from Malaria. PLoS Pathog. 2007;3:e96. doi: 10.1371/journal.ppat.0030096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ing R, Segura M, Thawani N, Tam M, Stevenson MM. Interaction of mouse dendritic cells and malaria-infected erythrocytes: uptake, maturation, and antigen presentation. J. Immunol. 2006;176:441–450. doi: 10.4049/jimmunol.176.1.441. [DOI] [PubMed] [Google Scholar]
- 15.Leisewitz AL, Rockett KA, Gumede B, Jones M, Urban B, Kwiatkowski DP. Response of the splenic dendritic cell population to malaria infection. Infect. Immun. 2004;72:4233–4239. doi: 10.1128/IAI.72.7.4233-4239.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sponaas AM, Cadman ET, Voisine C, Harrison V, Boonstra A, O'Garra A, Langhorne J. Malaria infection changes the ability of splenic dendritic cell populations to stimulate antigen-specific T cells. J. Exp. Med. 2006;203:1427–1433. doi: 10.1084/jem.20052450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luyendyk J, Olivas OR, Ginger LA, Avery AC. Antigen-presenting cell function during Plasmodium yoelii infection. Infect. Immun. 2002;70:2941–2949. doi: 10.1128/IAI.70.6.2941-2949.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perry JA, Olver CS, Burnett RC, Avery AC. Cutting edge: the acquisition of TLR tolerance during malaria infection impacts T cell activation. J. Immunol. 2005;174:5921–5925. doi: 10.4049/jimmunol.174.10.5921. [DOI] [PubMed] [Google Scholar]
- 19.Perry JA, Rush A, Wilson RJ, Olver CS, Avery AC. Dendritic cells from malaria-infected mice are fully functional APC. J. Immunol. 2004;172:475–482. doi: 10.4049/jimmunol.172.1.475. [DOI] [PubMed] [Google Scholar]
- 20.Seixas E, Cross C, Quin S, Langhorne J. Direct activation of dendritic cells by the malaria parasite, Plasmodium chabaudi chabaudi. Eur. J. Immunol. 2001;31:2970–2978. doi: 10.1002/1521-4141(2001010)31:10<2970::aid-immu2970>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 21.Wykes MN, Liu XQ, Jiang S, Hirunpetcharat C, Good MF. Systemic tumor necrosis factor generated during lethal Plasmodium infections impairs dendritic cell function. J. Immunol. 2007;179:3982–3987. doi: 10.4049/jimmunol.179.6.3982. [DOI] [PubMed] [Google Scholar]
- 22.Wykes M, Keighley C, Pinzon-Charry A, Good MF. Dendritic cell biology during malaria. Cell. Microbiol. 2007;9:300–305. doi: 10.1111/j.1462-5822.2006.00865.x. [DOI] [PubMed] [Google Scholar]
- 23.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 24.Wakkach A, Fournier N, Brun V, Breittmayer JP, Cottrez F, Groux H. Characterization of dendritic cells that induce tolerance and T regulatory 1 cell differentiation in vivo. Immunity. 2003;18:605–617. doi: 10.1016/s1074-7613(03)00113-4. [DOI] [PubMed] [Google Scholar]
- 25.Svensson M, Maroof A, Ato M, Kaye PM. Stromal cells direct local differentiation of regulatory dendritic cells. Immunity. 2004;21:805–816. doi: 10.1016/j.immuni.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 26.Delgado M, Gonzalez-Rey E, Ganea D. The neuropeptide vasoactive intestinal peptide generates tolerogenic dendritic cells. J. Immunol. 2005;175:7311–7324. doi: 10.4049/jimmunol.175.11.7311. [DOI] [PubMed] [Google Scholar]
- 27.Fujita S, Seino K, Sato K, Sato Y, Eizumi K, Yamashita N, Taniguchi M, Sato K. Regulatory dendritic cells act as regulators of acute lethal systemic inflammatory response. Blood. 2006;107:3656–3664. doi: 10.1182/blood-2005-10-4190. [DOI] [PubMed] [Google Scholar]
- 28.Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol. Rev. 2006;212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez I, Zeiser R, Karsunky H, Kambham N, Beilhack A, Soderstrom K, Negrin RS, Engleman E. CD101 surface expression discriminates potency among murine FoxP3+ regulatory T cells. J. Immunol. 2007;179:2808–2814. doi: 10.4049/jimmunol.179.5.2808. [DOI] [PubMed] [Google Scholar]
- 31.Lepault F, Gagnerault MC. Characterization of peripheral regulatory CD4+ T cells that prevent diabetes onset in nonobese diabetic mice. J. Immunol. 2000;164:240–247. doi: 10.4049/jimmunol.164.1.240. [DOI] [PubMed] [Google Scholar]
- 32.Sansom DM, Walker LS. The role of CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4) in regulatory T-cell biology. Immunol. Rev. 2006;212:131–148. doi: 10.1111/j.0105-2896.2006.00419.x. [DOI] [PubMed] [Google Scholar]
- 33.Pombo DJ, Lawrence G, Hirunpetcharat C, Rzepczyk C, Bryden M, Cloonan N, Anderson K, Mahakunkijcharoen Y, Martin LB, Wilson D, Elliott S, Elliott S, Eisen DP, Weinberg JB, Saul A, Good MF. Immunity to malaria after administration of ultra-low doses of red cells infected with Plasmodium falciparum. Lancet. 2002;360:610–617. doi: 10.1016/S0140-6736(02)09784-2. [DOI] [PubMed] [Google Scholar]
- 34.Prudencio M, Rodriguez A, Mota MM. The silent path to thousands of merozoites: the Plasmodium liver stage. Nat. Rev. Microbiol. 2006;4:849–856. doi: 10.1038/nrmicro1529. [DOI] [PubMed] [Google Scholar]
- 35.Landau I, Killick-Kendrick R. Rodent plasmodia of the Republique Centrafricaine: the sporogony and tissue stages of Plasmodium chabaudi and P. berghei yoelii. Trans. R. Soc. Trop. Med. Hyg. 1966;60:633–649. doi: 10.1016/0035-9203(66)90010-1. [DOI] [PubMed] [Google Scholar]
- 36.Despommier DD. Parasitic diseases. Apple Tree Productions; New York: 2006. [Google Scholar]
- 37.Wysocka M, Robertson S, Riemann H, Caamano J, Hunter C, Mackiewicz A, Montaner LJ, Trinchieri G, Karp CL. IL-12 suppression during experimental endotoxin tolerance: dendritic cell loss and macrophage hyporesponsiveness. J. Immunol. 2001;166:7504–7513. doi: 10.4049/jimmunol.166.12.7504. [DOI] [PubMed] [Google Scholar]
- 38.Omer FM, Riley EM. Transforming growth factor beta production is inversely correlated with severity of murine malaria infection. J. Exp. Med. 1998;188:39–48. doi: 10.1084/jem.188.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peyron F, Burdin N, Ringwald P, Vuillez JP, Rousset F, Banchereau J. High levels of circulating IL-10 in human malaria. Clin. Exp. Immunol. 1994;95:300–303. doi: 10.1111/j.1365-2249.1994.tb06527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Souza JB, Williamson KH, Otani T, Playfair JH. Early gamma interferon responses in lethal and nonlethal murine blood-stage malaria. Infect. Immun. 1997;65:1593–1598. doi: 10.1128/iai.65.5.1593-1598.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stevenson MM, Tam MF, Wolf SF, Sher A. IL-12-induced protection against blood-stage Plasmodium chabaudi AS requires IFN-gamma and TNF-alpha and occurs via a nitric oxide-dependent mechanism. J. Immunol. 1995;155:2545–2556. [PubMed] [Google Scholar]
- 42.Su Z, Stevenson MM. Central role of endogenous gamma interferon in protective immunity against blood-stage Plasmodium chabaudi AS infection. Infect. Immun. 2000;68:4399–4406. doi: 10.1128/iai.68.8.4399-4406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackintosh CL, Beeson JG, Marsh K. Clinical features and pathogenesis of severe malaria. Trends Parasitol. 2004;20:597–603. doi: 10.1016/j.pt.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Gerard C, Bruyns C, Marchant A, Abramowicz D, Vandenabeele P, Delvaux A, Fiers W, Goldman M, Velu T. Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. J. Exp. Med. 1993;177:547–550. doi: 10.1084/jem.177.2.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Howard M, Muchamuel T, Andrade S, Menon S. Interleukin 10 protects mice from lethal endotoxemia. J. Exp. Med. 1993;177:1205–1208. doi: 10.1084/jem.177.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li C, Corraliza I, Langhorne J. A defect in interleukin-10 leads to enhanced malarial disease in Plasmodium chabaudi chabaudi infection in mice. Infect. Immun. 1999;67:4435–4442. doi: 10.1128/iai.67.9.4435-4442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boutlis CS, Yeo TW, Anstey NM. Malaria tolerance--for whom the cell tolls? Trends Parasitol. 2006;22:371–377. doi: 10.1016/j.pt.2006.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.deWalick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A, Kuns RD, MacDonald KP, Hill GR, Engwerda CR. Cutting Edge: conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J. Immunol. 2007;178:6033–6037. doi: 10.4049/jimmunol.178.10.6033. [DOI] [PubMed] [Google Scholar]
- 49.Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu. Rev. Immunol. 2004;22:307–328. doi: 10.1146/annurev.immunol.22.012703.104533. [DOI] [PubMed] [Google Scholar]
- 50.Grewal IS, Xu J, Flavell RA. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature. 1995;378:617–620. doi: 10.1038/378617a0. [DOI] [PubMed] [Google Scholar]
- 51.Martin E, O'Sullivan B, Low P, Thomas R. Antigen-specific suppression of a primed immune response by dendritic cells mediated by regulatory T cells secreting interleukin-10. Immunity. 2003;18:155–167. doi: 10.1016/s1074-7613(02)00503-4. [DOI] [PubMed] [Google Scholar]
- 52.MacLeod M, Kwakkenbos MJ, Crawford A, Brown S, Stockinger B, Schepers K, Schumacher T, Gray D. CD4 memory T cells survive and proliferate but fail to differentiate in the absence of CD40. J. Exp. Med. 2006;203:897–906. doi: 10.1084/jem.20050711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elliott SR, Kuns RD, Good MF. Heterologous immunity in the absence of variant-specific antibodies after exposure to subpatent infection with blood-stage malaria. Infect. Immun. 2005;73:2478–2485. doi: 10.1128/IAI.73.4.2478-2485.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]