

Abstract
BACKGROUND
Brugada syndrome (BS) and long QT syndrome (LQTS) are electrical disorders with a genetic background. They are revealed on surface electrocardiograms as either right bundle branch block and ST segment elevation in the right pericardial leads (V1, V2 and V3) in BS or as a long QTc interval on all 12 leads of the electrocardiogram in LQTS. Both BS and LQTS can lead to syncope and even sudden death. The R1193Q SCN5A variant was recently associated with LQTS, BS and cardiac conductance disease.
OBJECTIVE AND METHODS
The aim of the present study was to screen the SCN5A gene from two patients – one with BS and the other showing signs of a BS/LQTS phenotype – for mutations, and to characterize the effect of the mutations on channel function using the patch clamp technique.
RESULTS
A heterozygous mutation (R1193Q) was identified on the SCN5A gene in both patients. The R1193Q polymorphism was absent in 100 unrelated control alleles, suggesting that it has a low frequency in the French Canadian population. Mutant R1193Q expressed in tsA201 cells, which was studied using the patch clamp technique, had a persistent sodium current that could account for the QTc prolongation. A shift of steady-state inactivation toward more hyperpolarized voltages was observed that could explain the BS phenotype.
CONCLUSION
The R1193Q polymorphism is definitively associated with cardiac electrical abnormalities.
Keywords: Arrhythmia, Defibrillation, DNA, Ion channels, Sudden death, Ventricular fibrillation
Abstract
CONTEXTE
Le syndrome de Brugada (SB) et le syndrome du QT long (SQTL) sont des troubles électriques d’origine génétique. Ils se manifestent à l’électrocardiogramme de surface par un bloc de branche droit et un sus-décalage du segment ST dans les dérivations péricardiques droites (V1, V2 et V3) dans le cas du SB et par un allongement de l’intervalle QTc dans les 12 dérivations de l’électrocardiogramme dans le cas du SQTL. Les deux syndromes peuvent provoquer une syncope et parfois même entraîner la mort subite. La variante R1193Q du gène SCN5A a été associée dernièrement au SQTL, au SB et à des troubles de la conduction électrique dans le cœur.
BUTS ET MÉTHODE
La présente étude avait pour buts d’examiner le gène SCN5A chez deux patients, l’un atteint du SB et l’autre montrant des signes d’un phénotype du SB ou du SQTL, à la recherche de mutations et de caractériser l’effet de ces mutations sur le fonctionnement des canaux ioniques à l’aide de la technique du « patch clamp ».
RÉSULTATS
Une mutation hétérozygote (R1193Q) a été découverte sur le gène SCN5A chez les deux patients. Toutefois, l’absence du polymorphisme R1193Q dans 100 allèles témoins, non liés porte à croire que la fréquence de la mutation est peu élevée dans la population canadienne française. La mutation R1193Q qui s’exprimait dans les cellules tsA201, étudiée à l’aide de la technique du « patch clamp », produisait un courant sodique continuel, susceptible d’être mis en cause dans l’allongement de l’intervalle QTc. Par ailleurs, un déplacement de l’inactivation à l’état d’équilibre vers une plus grande hyperpolarisation des différences de potentiel pourrait expliquer le phénotype du SB.
CONCLUSION
Le polymorphisme R1193Q est, sans contredit, associé à des anomalies de la conduction électrique dans le cœur.
Brugada syndrome (BS) is characterized by a unique electrocardiographic (ECG) pattern of right bundle branch block with ST elevation in the right precordial leads and can result in malignant ventricular tachycardia and sudden cardiac death (1,2). Transient forms of the disease in which the ECG normalizes for a period of time have been described. Administration of sodium channel blockers unmasks abnormal ECGs in patients with transient normalized ECGs. Approximately 20% of BS patients have mutations in SCN5A, which encodes the alpha-subunit of the human cardiac voltage-dependent sodium channel (hNav1.5) (3). This implies that another or other genes are responsible for this syndrome, but their location and products are not yet known (4). However, a second locus has been recently identified that overlaps with the SCN5A gene. In this study (4), the involvement of SCN5A was excluded by direct sequencing.
Patients with congenital long QT syndrome (LQTS) are also at risk of sudden death due to torsade de pointes ventricular arrhythmias (5,6). To date, over 200 mutations have been identified in six separate ion channel genes that encode sodium channels (SCN5A, which causes LQT3) and potassium channels (KCNQ1 and KCNH2, which cause LQT1 and LQT2, respectively) or their regulatory subunits KCNE1 and KCNE2 (which cause LQT5 and LQT6, respectively) (7). Recently, a mutation on ankyrin-B, a cytosolic protein, has also been reported to cause the fourth type of LQTS known as LQT4 (8,9).
Mixed LQTS and BS phenotypes have been reported in patients genotyped with mutations in the SCN5A gene (10), in whom genotyping showed the presence of a G-to-A mutation in exon 20 of SCN5A, which causes the substitution of an arginine (R) for a glutamine (Q) at position 1193 (R1193Q). Recently, the R1193Q mutation was reported to cause either LQTS (11,12) or BS (13,14) or to be associated with cardiac conductance abnormalities in a four-generation Chinese family (15). However, no direct links have been associated with the presence of the mutation and electrical abnormalities or sudden deaths in this family.
Voltage-gated sodium channels are transmembrane proteins that are responsible for the generation and propagation of action potentials and, therefore, the electrical activity of the heart. Mutations of this channel are responsible for several inherited cardiac diseases such as BS, LQTS and cardiac conduction disease.
We report cases of BS and BS/LQTS phenotype, respectively, in two patients genotyped with the R1193Q Nav1.5 mutation. This mutation was also characterized in vitro to determine its involvement in the physiopathology of BS and LQTS.
METHODS
Clinical evaluation
Neither patient had a history of ischemic heart disease, congestive heart failure or a family history of unexpected sudden cardiac death. Both had a normal physical examination and blood and exercise tests. Structural heart disease was excluded by echocardiography. They also received a sodium channel blocker (procainamide, 10 mg/kg over 10 min to 15 min, intravenously) to unmask a concealed form of BS (16). All tests were positive with a type 1 ECG (J wave amplitude greater than 2 mm with a coved ST-T configuration [17]). To increase the sensitivity, the V1 and V2 surface leads were positioned higher than usual (second intercostal space).
Molecular genetics
The present study was carried out in accordance with the guidelines of the Ethics Committee of Laval Hospital, Sainte-Foy, Quebec. Patients gave written informed consent to participate in the study.
Genomic DNA was extracted from peripheral lymphocytes. In both patients, exons 1 to 28 of SCN5A were amplified by polymerase chain reaction (PCR) using primers with intronic flanking sequences based on the gene sequences described by Wang et al (18). The PCR products were analyzed by cycle sequencing using an automated laser fluorescent DNA sequencer (ABI Prism 377, Applied Biosystems, USA).
Biophysical characterization
Wild-type and mutant channels were expressed in the tsA201 cell line, and sodium currents were recorded in the whole cell configuration as previously described (19).
RESULTS
Patient 1
The 40-year-old proband was a French Canadian man who was referred to our institution for possible ECG abnormalities. The patient was investigated for ataxia. During the examination, his ECG showed an ST segment elevation exhibiting a saddleback configuration suggesting a type 1 ECG pattern of BS. He had never had syncope and had no family history of syncope or sudden death. His 78-year-old father was in good health, while his 72-year-old mother had had a possible syncope three years previously with no obvious etiology. The physical examination, echocardiogram and Holter monitoring were normal. The intravenous procainamide (10 mg/kg over 10 min to 15 min) generated a type 1 pattern (Figure 1), suggesting BS. Because the patient was asymptomatic, he refused further investigation, including electrophysiological studies. After a 39-month follow-up, the patient remained asymptomatic.
Figure 1.
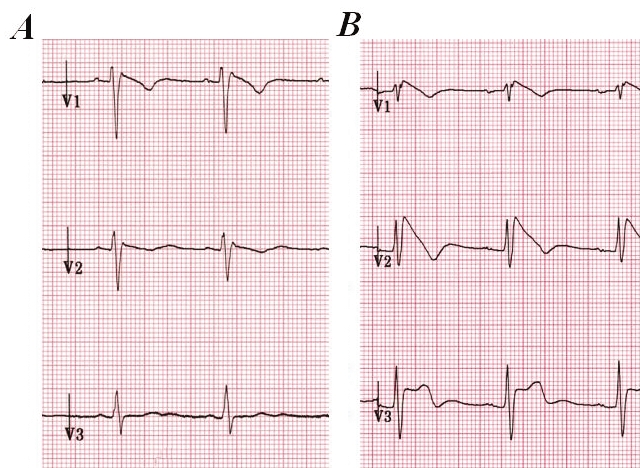
Precordial leads V1 to V3 of patient 1 with Brugada syndrome before (A) and after (B) the procainamide challenge. Note the dynamic electrocardiogram changes with a clear type 1 electrocardiogram pattern after procainamide superfusion
Patient 2
A 61-year-old man of Japanese origin was referred to our institution for a high probability of BS and LQTS. He had consulted for uncontrolled high blood pressure. During the examination, he had an abnormal ECG. The patient had been asymptomatic with no history of syncope. He was not under medication when these phenotypes occurred. The ECG revealed a borderline QTc prolongation of 480 ms (Figure 2). He received an intravenous administration of procainamide (10 mg/kg over 10 min to 15 min) to unmask BS. All tests were positive with a type 1 ECG characterized by a J wave amplitude greater than 2 mm with a coved ST-T configuration (Figure 3), which are signs of BS phenotypes.
Figure 2.
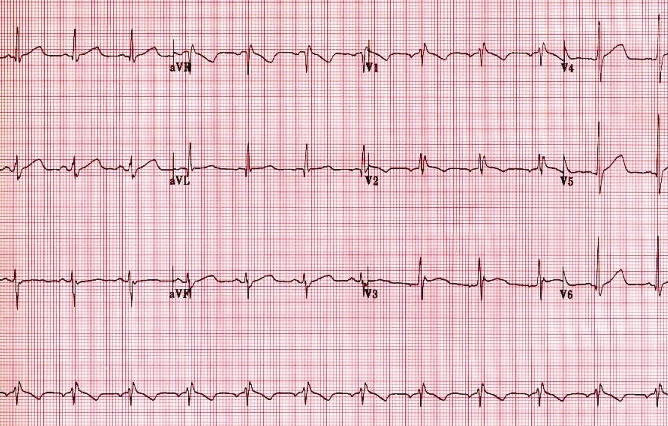
Twelve-lead electrocardiogram of patient 2 illustrating the 484 ms QTc prolongation
Figure 3.
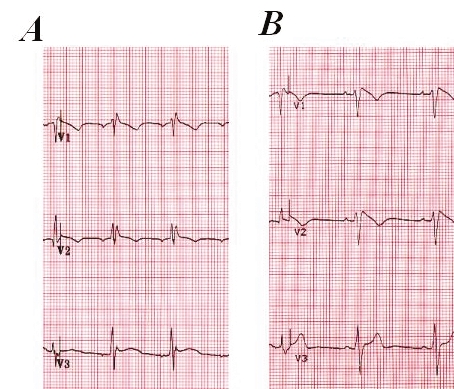
Precordial leads V1 to V3 of patient 2 with Brugada syndrome and long QT syndrome before (A) and after (B) the procainamide challenge. Note the dynamic electrocardiogram changes with a clear type 1 electrocardiogram pattern after procainamide superfusion
Because the patient was asymptomatic, he refused further investigation, including electrophysiological studies. After a follow-up more than 39 months later, the patient remained asymptomatic.
Genetic screening
Genetic testing for mutations on the SCN5A gene from the two patients revealed a G-to-A mutation at position 3578 in exon 20 of SCN5A, which causes the substitution of arginine (R), a positively charged amino acid, for a glutamine (Q), a neutral amino acid, at position 1193 (Figure 4). The R1193Q substitution occurred in the linker region between DII and DIII of the cardiac sodium channel Nav1.5. No tests were conducted on other family members because they declined genetic testing. In patient 1, mutations on KCNQ1, KCNH2, KCNE1 and KCNE2 were excluded.
Figure 4.

Sequencing panels of exon 20 of SCN5A. Automated DNA sequencing panels from a control allele (top panel) and from patient 1 (bottom left panel) and patient 2 (bottom right panel) illustrating the presence of the heterozygote guanine-to-adenine polymorphism (arrow) resulting in R1193Q
The presence of the R1193Q mutation was evaluated in 100 control patients. No mutations were found in these control alleles (data not shown). This suggests a potential involvement of the R1193Q mutation in the observed clinical phenotypes.
Biophysical characterization
Wild-type and mutant Nav1.5 channels were expressed in tsA201 cells, and sodium currents were recorded in the whole cell configuration using the patch clamp method (Figure 5). A persistent sodium current was detected, which could account for the QTc prolongation (Figure 5D). Biophysical analyses of the Nav1.5/R1193Q mutant channels expressed in tsA201 cells showed that the voltage dependence of activation was comparable to the wild-type (V1/2=–59.9±0.9 mV and kv=6.9±0.2, n=8, for the wild-type channels and V1/2=57.9±1.2 mV and kv=6.7±0.3, n=6, for Nav1.5/R1193Q mutant channels) (Figure 6A). However, the steady-state inactivation exhibited a shift of 5 mV to more hyperpolarized voltages (V1/2=107.8±1.8 mV and kv=5.4±0.4, n=8, for the wild-type channels and V1/2=113±1.3 mV and kv=5.4±0.1, n=6, for Nav1.5/R1193Q mutant channels) (Figure 6B). The intermediate slow inactivation was not affected by this mutation (Figure 6C). The time constants of slow inactivation were as flow, τ=214±12 for wild-type and τ=240±13 for Nav1.5/R1193Q mutant channels; they were not statistically different from each other.
Figure 5.

Sodium current traces from the cardiac voltage-dependent sodium channel (Nav1.5)/wild-type (A) and the Nav1.5/R1193Q (B) mutation. Panel C represents currents at –30 mV from Nav1.5/wild-type channels without a residual current, and panel D illustrates the presence of a residual current in Nav1.5/R1193Q mutant channels
Figure 6.
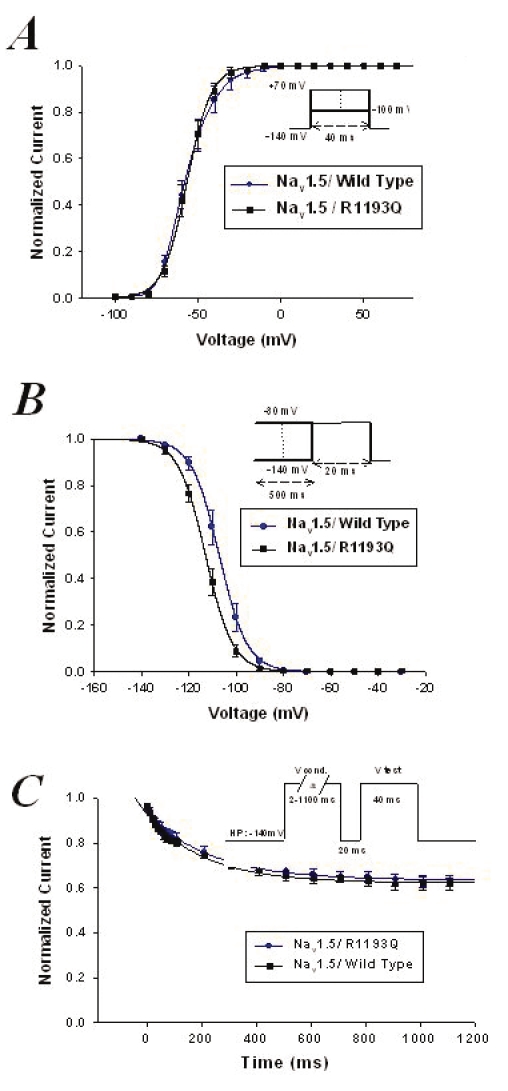
Activation (A) and inactivation (B) curves of cardiac voltage-dependent sodium channel (Nav1.5)/wild-type and Nav1.5/R1193Q mutant channels (see text for the V1/2 and kv values). C Slow inactivation (see text for time constants values)
DISCUSSION
While LQTS is related to QTc prolongation, BS is characterized by a typical ECG pattern of right bundle branch block with an ST-segment elevation in leads V1 to V3. In BS, ECG manifestations are often transient in many patients and class 1 antiarrhythmic drugs such as procainamide can unmask a typical ECG pattern in patients with a normal baseline ECG. In the present study, we used this highly specific test to reveal BS in two patients. The genotyping showed the presence of a G-to-A mutation in exon 20 of SCN5A, which causes the substitution of an arginine (R) for a glutamine (Q) at position 1193 (R1193Q).
The R1193Q mutation has recently been identified in patients with acquired LQTS, BS and cardiac conductance disease (11–13,15).
Because the cytosines in the CpG sequence are frequently methylated in mammals, it has been suggested that methylated cytosine residues are hotspots for mutations in mammalian DNA (20). Because the R1193Q mutation is in the arginine codon sequence of the CpG dimer, it is likely that the CpG dimer is also a hotspot for mutations.
The frequency of R1193Q polymorphisms is 12% in the Chinese population, which is relatively high (15). To determine the prevalence of the R1193Q polymorphism in our population, the genomic DNAs from 100 unrelated subjects with no sign of ECG abnormalities were amplified by PCR and directly sequenced using the automatic sequencer. No R1193Q mutations were found in the control subjects. This suggests that this polymorphism is rare in the French Canadian population, unlike in the Chinese population, which points to a pathological origin.
Our study connects this mutation to BS and BS/LQTS, which is the first report of such a correlation in the French Canadian population.
Neither patient was taking drugs, which is an important issue given that the LQTS observed in patient 2 can be acquired by the use of certain drugs. While the biophysical characterization explained the involvement of R1193Q in LQTS, its involvement in BS remains to be fully established. The biophysical characterization of R1193Q revealed a defect in inactivation with the presence of a residual current typical of LQT3 mutations (21). The shift of 5 mV observed in the presence of R1193Q mutation of steady-state inactivation to more hyperpolarized voltages could explain the loss of function of sodium current and therefore gives a background to explain the BS clinical phenotypes.
It is noteworthy that this mutation was found in two unrelated patients and caused BS/LQTS in patient 2 and solely BS in patient 1.
Study limitations
Although environmental factors have not been established in our two patients, earlier reports on the R1193Q SCN5A polymorphism and the biophysical data showed that this polymorphism could be implicated in both BS and LQTS. Because these diseases are associated with high risk of syncope and sudden death, all gene carriers should be educated about the syndrome; this includes avoiding sodium channel blockers and antidepressants. Patients with this polymorphism may have increased risk of LQTS, and drugs that are suspected to increase the QT interval should be avoided. This is the least we can do for these patients while waiting for more details on genetic studies and the environmental factor that could trigger the disease in patients carrying the R1193Q SCN5A polymorphism.
CONCLUSIONS
The R1193Q variant is definitively associated with ECG abnormalities, and patients with this variant should be kept under close observation. Particular care must be taken when prescribing drugs to these patients.
ACKNOWLEDGEMENTS
This study was supported by grants from the Heart and Stroke Foundation of Quebec (HSFQ) and the Canadian Institutes of Health Research (CIHR). M Chahine is an Edwards Senior Investigator (Joseph C Edwards Foundation).
REFERENCES
- 1.Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3: A marker for sudden death in patients without demonstrable structural heart disease. Circulation. 1998;97:457–60. doi: 10.1161/01.cir.97.5.457. [DOI] [PubMed] [Google Scholar]
- 2.Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. J Am Coll Cardiol. 1992;20:1391–6. doi: 10.1016/0735-1097(92)90253-j. [DOI] [PubMed] [Google Scholar]
- 3.Chen Q, Kirsch GE, Zhang D, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 4.Weiss R, Barmada MM, Nguyen T, et al. Clinical and molecular heterogeneity in the Brugada syndrome: A novel gene locus on chromosome 3. Circulation. 2002;105:707–13. doi: 10.1161/hc0602.103618. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz PJ. The long QT syndrome. Curr Probl Cardiol. 1997;22:297–351. doi: 10.1016/s0146-2806(97)80009-6. [DOI] [PubMed] [Google Scholar]
- 6.Vincent GM. The molecular genetics of the long QT syndrome: Genes causing fainting and sudden death. Annu Rev Med. 1998;49:263–74. doi: 10.1146/annurev.med.49.1.263. [DOI] [PubMed] [Google Scholar]
- 7.Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. N Engl J Med. 1997;336:1562–7. doi: 10.1056/NEJM199705293362204. [DOI] [PubMed] [Google Scholar]
- 8.Mohler PJ, Schott JJ, Gramolini AO, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–9. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 9.Schott JJ, Charpentier F, Peltier S, et al. Mapping of a gene for long QT syndrome to chromosome 4q25–27. Am J Hum Genet. 1995;57:1114–22. [PMC free article] [PubMed] [Google Scholar]
- 10.Bezzina C, Veldkamp MW, van Den Berg MP, et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–13. doi: 10.1161/01.res.85.12.1206. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Chen S, Chen Q, et al. The common SCN5A mutation R1193Q causes LQTS-type electrophysiological alterations of the cardiac sodium channel. J Med Genet. 2004;41:e66. doi: 10.1136/jmg.2003.013300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Q. Author’s reply: link of SCN5A SNP R1193Q to long QT syndrome. J Med Genet. 2005;42:e8. doi: 10.1136/jmg.2004.027995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vatta M, Dumaine R, Varghese G, et al. Genetic and biophysical basis of sudden unexplained nocturnal death syndrome (SUNDS), a disease allelic to Brugada syndrome. Hum Mol Genet. 2002;11:337–45. doi: 10.1093/hmg/11.3.337. [DOI] [PubMed] [Google Scholar]
- 14.Skinner JR, Chung SK, Montgomery D, et al. Near-miss SIDS due to Brugada syndrome. Arch Dis Child. 2005;90:528–9. doi: 10.1136/adc.2004.058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hwang HW, Chen JJ, Lin YJ, et al. R1193Q of SCN5A, a Brugada and long QT mutation, is a common polymorphism in Han Chinese. J Med Genet. 2005;42:e7. doi: 10.1136/jmg.2004.027995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brugada R, Brugada J, Antzelevitch C, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. 2000;101:510–5. doi: 10.1161/01.cir.101.5.510. [DOI] [PubMed] [Google Scholar]
- 17.Wilde AAM, Antzelevitch C, Borggrefe M, et al. Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation. 2002;106:2514–9. doi: 10.1161/01.cir.0000034169.45752.4a. [DOI] [PubMed] [Google Scholar]
- 18.Wang Q, Li Z, Shen J, Keating MT. Genomic organization of the human SCN5A gene encoding the cardiac sodium channel. Genomics. 1996;34:9–16. doi: 10.1006/geno.1996.0236. [DOI] [PubMed] [Google Scholar]
- 19.Deschênes I, Baroudi G, Berthet M, et al. Electrophysiological characterization of SCN5A mutations causing long QT (E1784K) and Brugada (R1512W and R1432G) syndromes. Cardiovasc Res. 2000;46:55–65. doi: 10.1016/s0008-6363(00)00006-7. [DOI] [PubMed] [Google Scholar]
- 20.Barker D, Schafer M, White R. Restriction sites containing CpG show a higher frequency of polymorphism in human DNA. Cell. 1984;36:131–8. doi: 10.1016/0092-8674(84)90081-3. [DOI] [PubMed] [Google Scholar]
- 21.Keller DI, Acharfi S, Delacrétaz E, et al. A novel mutation in SCN5A, delQKP 1507–1509, causing long QT syndrome: Role of Q1507 residue in sodium channel inactivation. J Mol Cell Cardiol. 2003;35:1513–21. doi: 10.1016/j.yjmcc.2003.08.007. [DOI] [PubMed] [Google Scholar]