

Abstract
Poly-ADP-ribosylation (PAR) of proteins by poly(ADP-ribose) polymerases (PARP) occurs after experimental traumatic brain injury (TBI) and modulates neurological outcome. Several promising pharmacological PARP inhibitors have been developed for use in humans, but there is currently no clinically relevant means of monitoring treatment effects. We therefore utilized an enzyme-linked immunosorbent assay (ELISA) to measure PAR-modified proteins in cerebrospinal fluid (CSF). CSF samples from 17 pediatric TBI and 15 control patients were plated overnight then incubated with polyclonal antibody against PAR. Histone-1, a PARP substrate, was incubated with active PARP, NAD, and nicked DNA, and served as the standard. Both peak and mean CSF PAR-modified protein were increased in TBI patients versus controls. Peak CSF PAR-modified protein levels occurred on day 1 and levels remained increased on day 2 after TBI. Increases in peak CSF PAR-modified protein concentrations were independently associated with age and male sex, but not initial Glasgow coma scale score, Glasgow outcome score, or mechanism of injury. The increase in PAR-modified proteins in CSF after TBI may be due to increased PARP activation, decreased PAR degradation, or both. Since PAR-modified protein concentration correlated with age and male sex, developmental and sex-dependent roles for PARP after TBI are implicated.
Keywords: ADP-ribosyltransferase, Biomarker, Enzyme-linked immunosorbent assay, Head injury, Poly(ADP-ribose) polymerase, Poly(ADP-ribose) synthetase
Poly-ADP-ribosylation (PAR) of proteins is a post-translational modification catalyzed by nuclear, cytosolic, and mitochondrial poly(ADP-ribose) polymerases (PARP) (Ueda and Hayaishi 1985; Virag and Szabo 2002; Du et al. 2003; Lai et al. 2008). During PARP activation, over 200 ADP-ribose moieties can be added to target proteins utilizing one molecule of NAD+ for each ADP-ribose moiety (Ueda and Hayaishi 1985). Polymerization of ADP-ribose moieties to target proteins confers a negative charge and alters protein function (Ueda and Hayaishi 1985; Ha et al. 2002; Virag and Szabo 2002; Lai et al. 2008), and has been used as a relatively quantitative biomarker of PARP activation (Yu et al. 2002; Du et al. 2003; Satchell et al. 2003). PARP serves important homeostatic roles in DNA repair, transcription, cell cycle regulation, and memory (de la Lastra et al. 2007). However, during periods of severe cellular stress and energy imbalance, such as after traumatic brain injury (TBI) or stroke, over-activation of PARP can result in depletion of NAD+, energy failure, and cell death and dysfunction (Eliasson et al. 1997; Endres et al. 1997; Whalen et al. 1999; Satchell et al. 2003; Clark et al. 2007). In addition to consumption of NAD+, PARP activation can also directly inhibit electron transport thereby reducing ATP production and energy repletion, compounding energy failure (Halmosi et al. 2001; Ha et al. 2002; Lai et al. 2007). PARP can also mediate apoptotic cell death via release of apoptosis-inducing factor (AIF) from mitochondria (Yu et al. 2002; Du et al. 2003), a process that occurs after TBI (Zhang et al. 2002). It follows that PARP inhibition has been targeted in the prevention of cellular injury and death after TBI and stroke, as well as other diseases where energy failure contributes to pathology.
Worldwide, TBI is a major cause of mortality and morbidity, and is the leading cause of death in children in the United States (Adekoya et al. 2002). Both pharmacologic and genetic PARP inhibition have been shown to be remarkably protective after experimental TBI (Cosi et al. 1996; Klaidman et al. 1996; Eliasson et al. 1997; Endres et al. 1997; Whalen et al. 1999; Satchell et al. 2003; Clark et al. 2007). Novel PARP inhibitors are being developed for clinical use both alone, and as chemosensitizers in combination with chemotherapy (de la Lastra et al. 2007). However, there is currently no efficient means for monitoring the pharmacological action of PARP inhibitors clinically. Here we report the use of an enzyme-linked immunosorbent assay (ELISA)-based method for the quantification of PAR-modified proteins—a surrogate biomarker of PARP activation—in cerebrospinal fluid (CSF) from pediatric patients after severe TBI.
Materials and methods
Cerebrospinal fluid samples
This study was approved by the Institutional Review Board at the University of Pittsburgh Medical Center. CSF was obtained from a series of seventeen infants and children after severe TBI obtained over a period of up to 4 days after injury, randomly selected from a TBI CSF tissue bank (Satchell et al. 2005). All patients were admitted to the Pediatric Intensive Care Unit at the Children’s Hospital of Pittsburgh and had an extra-ventricular drain placed for intracranial pressure monitoring and CSF drainage as standard-of-care for our institution. CSF was collected under sterile conditions from the graduated drip chamber of the closed extra-ventricular drain apparatus. CSF is measured in the drip chamber then drained hourly, thus the collected CSF is at room temperature for a maximum time of 1 h. Samples were centrifuged at 3,000 rpm for 10 minutes to remove cellular debris and supernatants were decanted and stored at -70°C until batch analysis. Control CSF represented excess CSF obtained via lumbar puncture from 15 infants and children who were found not to have meningitis based on cell count, culture, and Gram stain. Protein concentrations for each CSF sample were measured using the bicinchoninic acid method.
Clinical data collected included demographic information, mechanism of injury, Glasgow Coma Scale (GCS) score on admission, and Glasgow outcome score (GOS) determined at 6 months after injury. Inflicted TBI from child abuse was diagnosed by the Children’s Hospital of Pittsburgh Child Protection Team, and was independent of enrollment into the study and intensive care unit management.
Poly(ADP-ribose) modified protein standards
In order to quantify PAR-modified proteins a standard was produced using the known PARP substrate histone-1. A reaction mixture was prepared consisting of 16.6 μM histone-1 (Trevigen, Gaithersburg, MD), 2 μl active PARP enzyme (> 2 units/μl; Trevigen), 100 μM NAD+ (Sigma, St. Louis, MO), and 50 μg nicked DNA (Trevigen), and was incubated for 1 h at 37°. Serial dilutions of the PAR-modified histone mixture were used to generate standard curves, based on the known concentration of histone-1 and assuming close to 100% efficiency of the reaction. There was close agreement between duplicate samples, suggesting that the assay is reliable (r2 = 0.991; Fig. 1A). A typical standard curve with a range of 0 - 1,000 nM is shown in Figure 1B (r2 = 0.992). The lower limit of detection is 10 nM.
Figure 1.
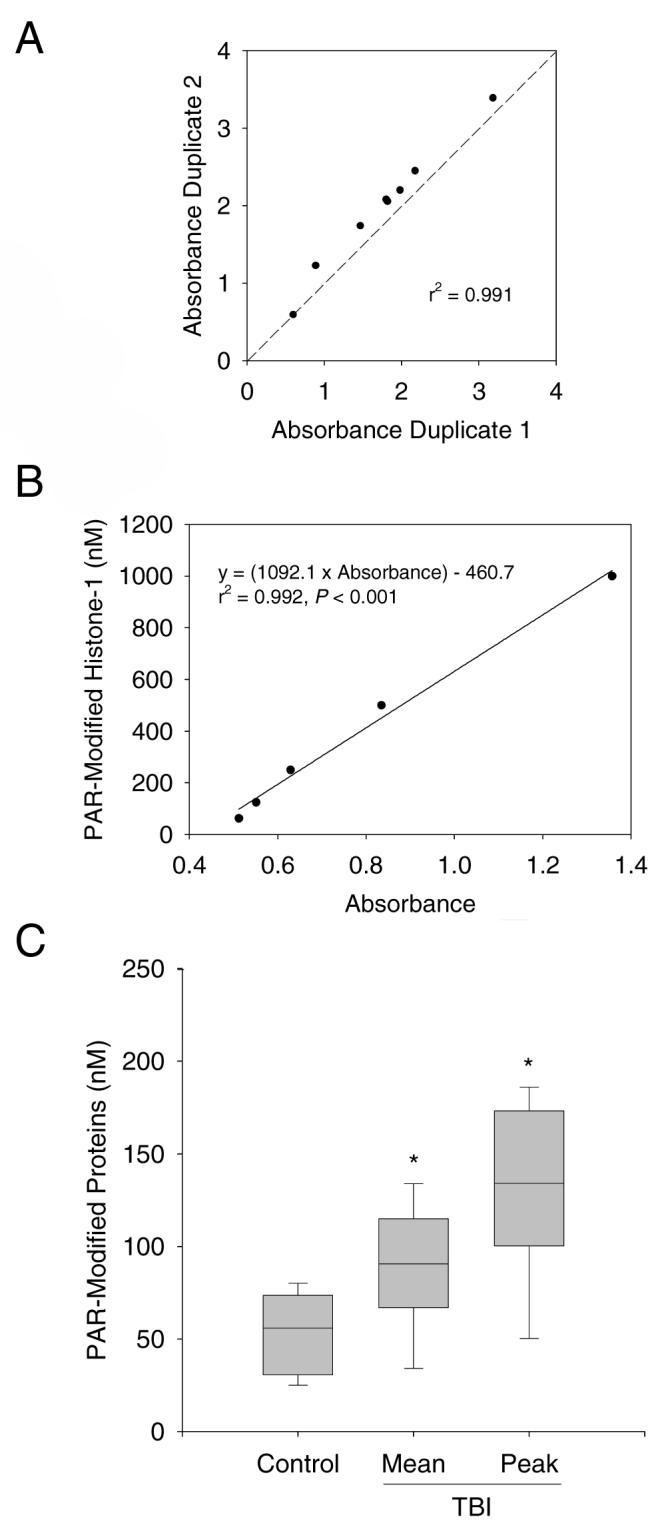
A. Agreement between duplicate samples (r2 = 0.991, P < 0.001). B. A typical standard curve for PAR-modified histone-1. Serial dilutions based on the concentration of histone-1 were plotted against absorbance at 450 nm. The range for the assay is 10 - 1,000 nM. For this curve r2 = 0.992, P < 0.001. C. Box plot depicting CSF PAR-modified proteins in pediatric control (n = 15) and TBI (n = 17) patients. *P < 0.05 vs. control, median (line), 25-75th (box), 5-95th (whisker) percentiles.
Poly(ADP-ribose) modified protein enzyme-linked immunosorbent assay
PAR-modified histone-1 standards and samples of CSF from TBI and control patients (100 μl) were diluted 1:2 with phosphate buffered saline (PBS) then placed into 96 well polystyrene microtiter plates with high protein binding properties (catalogue #3590, Corning Inc., Corning, NY) in duplicate. Samples were incubated overnight at 4°C to allow for protein binding to the microtiter plate. A commercially made ELISA blocking solution (catalogue #80160, Alpha Diagnostic, San Antonio, TX) was then added to each well for 2 h at room temperature. To identify PAR-modified proteins captured on the microtiter plate, a 1:1000 dilution of rabbit polyclonal antibody against PAR (catalogue # 4336-BPC-100, Trevigen) was added to each well and incubated at room temperature for 30 min, followed by incubation in a 1:2000 dilution of secondary anti-rabbit antibody conjugated to horseradish peroxidase for 30 min at room temperature. A colorimetric detection system was used and absorbance was determined at 450 nm. CSF PAR-modified protein concentrations were calculated against the PAR-modified histone-1 standards.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM) or median [range] where appropriate. The mean and peak CSF PAR-modified protein levels were determined for each patient. Comparisons between TBI and control groups were made using a Mann-Whitney rank sum test since the data were not normally distributed. Comparisons between CSF PAR-modified protein levels for each day after injury were made using ANOVA on ranks. Comparisons between peak CSF PAR-modified protein levels and the clinical variables age, sex, admission GCS, 6 mo GOS, and accidental vs. inflicted TBI were examined using linear regression or Spearman rank tests for parametric and nonparametric data, respectively. A multivariate linear regression model was used that included age, sex, admission GCS, 6 mo GOS, and accidental vs. inflicted TBI mechanism, to determine independent predictors of CSF PAR-modified protein levels. Accidental vs. inflicted TBI were analyzed because inflicted TBI as a mechanism of injury is known to have a unique biomarker profile and unfavorable outcome vs. accidental injury (Clark et al. 2000; Berger et al. 2002; Satchell et al. 2005). Multivariate analysis was also performed using only independent variables with a P-value < 0.2 on univariate analysis.
Results
Patient demographic data are shown in Table 1. The age range for TBI patients was 2 mo to 11 y. Ten of the seventeen TBI patients were male. The median GCS assigned on admission to the emergency department was 7. The majority of patients had TBI as a consequence of motor vehicle-related accidents(MVA; collisions, pedestrian or bicycle struck by motor vehicle), followed by inflicted injury due to child abuse. The survival in this cohort of TBI patients was 94%. Control patients were younger (range 1 mo to 7 y) than TBI patients (P = 0.002), and reflect the predominance of lumbar punctures to rule out infection in infants compared with older children.
Table 1.
Patient demographics
| Controls | Traumatic Brain Injury | |
|---|---|---|
| n | 15 | 17 |
| Age (years) | 1.4 ± 0.5 | 4.3 ± 0.8* |
| Male:Female | 6:9 | 10:7 |
| Admission Glasgow coma scale score | - | 7 [3 - 15] |
| Mechanism of injury (%) | ||
| Motor vehicle-related accident | - | 7 (41) |
| Inflicted injury (child abuse) | - | 5 (29) |
| Fall | - | 3 (18) |
| Struck by object | - | 1 (6) |
| Bicycle accident | - | 1 (6) |
| 6 month Glasgow outcome score1 | - | 5 [1-5] |
| Survived:died (%) | - | 16:1 (94) |
P = 0.002 vs. controls
1 = dead, 2 = vegetative, 3 = severe disability, 4 = moderate disability, 5 = normal
Figure 1C shows the mean and peak CSF PAR modified protein levels for TBI patients vs. the CSF PAR modified protein levels for controls, presented as median, 25-75th, and 5-95th percentiles. The mean and peak CSF PAR-modified protein levels were increased in TBI patients compared with control patients (88.1 ± 8.1, 132.5 ± 11.5, and 53.6 ± 5.2 nM, respectively; mean ± SEM, P < 0.05). CSF PAR-modified protein levels peaked on day 1 and remained increased on day 2 vs. controls (Fig. 2A, P < 0.05). Table 2 and Figure 2 show relationships between peak CSF PAR-modified protein levels and clinical variables. Peak CSF PAR-modified protein levels increased with patient age and were higher in male vs. female patients (Fig. 2B and C, P < 0.05 by univariate analysis). In the control group, CSF PAR-modified protein levels also correlated with age (Fig. 2B, P < 0.05), but there was no difference between males and females (Fig. 2C, P > 0.05). Both age and male sex were independently associated with peak CSF PAR-modified protein level in TBI patients by multivariate analysis, using either a model that included all independent variables or a model that only included independent variables with a P-value < 0.2 (age, sex, GCS). The following derived multivariate model predicts peak CSF PAR-modified protein levels with an r2 of 0.667: CSF PAR-modified protein = 30.578 + (10.319 × age in years) + (57.748 × [1 if male, 0 if female]) + (3.769 × GCS) + (7.532 × [0 if inflicted injury, 1 if accidental injury]) + (1.783 × GOS). There was no association between peak CSF PAR-modified protein levels and admission GCS, accidental vs. inflicted TBI, or 6 mo GOS.
Figure 2.
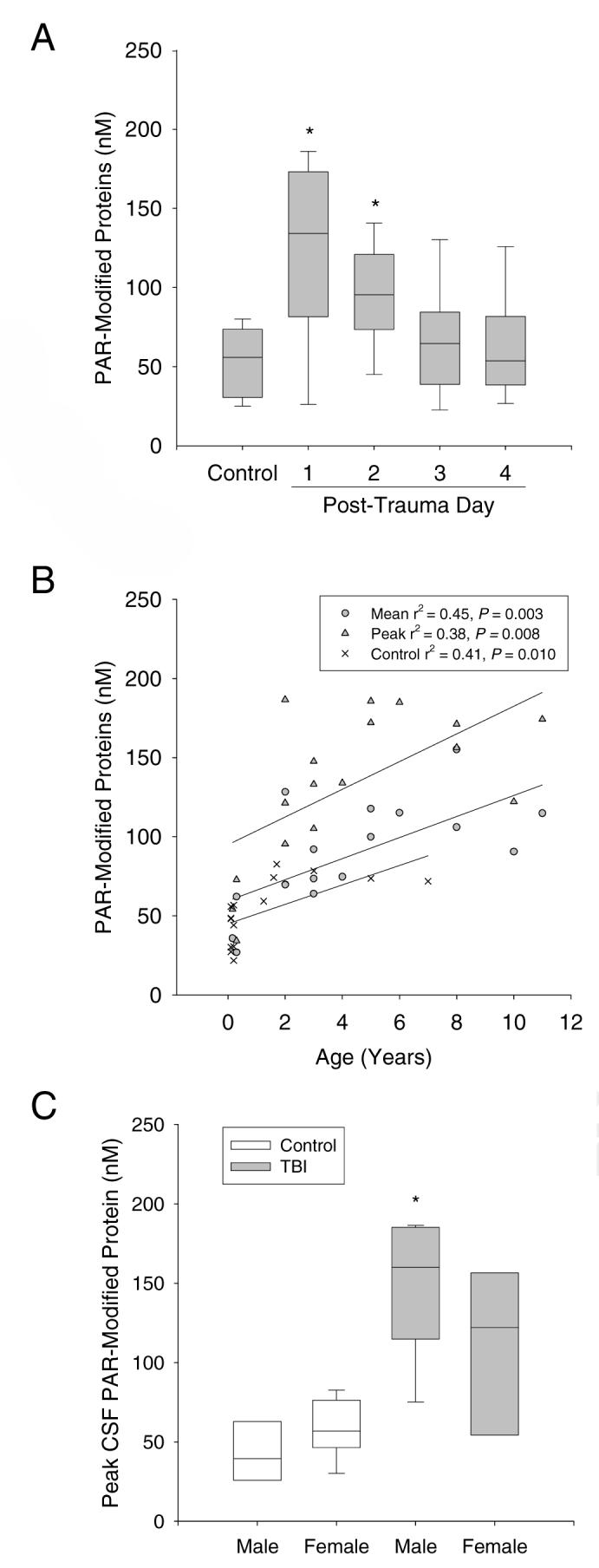
CSF PAR-modified protein levels and clinical variables. A. Temporal distribution of CSF PAR-modified protein levels after TBI (*P < 0.05 vs. control). B. Relationship between mean and peak CSF PAR-modified protein levels and age in control and TBI patients. The lower line represents the linear regression for the control values (×, r2 = 0.41, P < 0.05), the middle line represents the linear regression for the mean TBI values (circles, r2 = 0.45, P < 0.05), and the upper line represents the linear regression for the peak TBI values (triangles, r2 = 0.38, P < 0.05). C. Relationship between peak CSF PAR-modified protein levels and patient sex in control and TBI patients (*P < 0.05 vs. female). Median (line), 25-75th (box), 5-95th (whisker) percentiles.
Table 2.
Univariate and multivariate analysis of peak cerebrospinal fluid poly(ADP-ribose) modified protein levels an clinical variables
|
Univariate analysis |
Multivariate analysis1 |
Multivariate analysis2 |
|
|---|---|---|---|
| Age | r2 = 0.38, P = 0.008 | P = 0.004 | P = 0.001 |
| Male vs. female sex | rs = 0.39, P = 0.119 | P = 0.017 | P = 0.006 |
| Admission Glasgow coma scale | rs = 0.09, P = 0.737 | P = 0.185 | P = 0.181 |
| (GCS) score | |||
| Accidental vs. inflicted injury | rs = -0.29, P = 0.254 | P = 0.795 | - |
| 6 month Glasgow outcome score | rs = 0.21, P = 0.409 | P = 0.875 | - |
| (GOS)2 |
including all independent variables, r2 = 0.67, power = 0.990 with alpha = 0.05
including independent variables where P < 0.2 on univariate analysis, r2 = 0.66, power = 0.988 with alpha = 0.05
1 = dead, 2 = vegetative, 3 = severe disability, 4 = moderate disability, 5 = normal
Discussion
We report a reliable method for detection and quantification of PAR-modified proteins in CSF. In so much as PAR-modified proteins reflect PARP activity (Yu et al. 2002; Duet al. 2003; Satchell et al. 2003), this assay may prove useful for the evaluation of the role of PARP in diseases such as TBI, as well as for therapeutic drug monitoring of pharmacological PARP inhibitors. Using this assay, we were able to show an increase in CSF PAR-modified proteins and associations between male sex and increasing patient age.
Increased PARP activation has been shown in multiple experimental models of TBI, including controlled cortical impact in mice and rats, and fluid percussion injury in rats (LaPlaca et al. 1998; Satchell et al. 2003; Lai et al. 2007). Importantly, a detrimental role for PARP over activation after TBI has been shown using both PARP-1 knockout mice (Whalen et al. 1999) and pharmacological PARP-1 inhibitors (LaPlaca et al. 2001; Satchell et al. 2003; Clark et al. 2007). While to our knowledge there have been no clinical studies implicating a role for PARP activation after TBI, there is a report showing increased PAR in human brain after stroke (Love et al. 1999). Based on pre-clinical studies, testing the effect of PARP inhibitors in patients with severe TBI appears justified. It logically follows that use of this (or a similar) assay to verify therapeutic effectiveness of PARP inhibition and perhaps tailor therapy would improve the scientific rigor of such a study.
The association between PAR-modified protein levels and male sex in TBI but not control patients is in agreement with experimental in vitro and in vivo studies. Our laboratory showed that while baseline PARP levels in primary neurons from male and female rats were similar, male neurons were more sensitive to the potent PARP activator peroxynitrite than female neurons (Du et al. 2004). In addition, the inflammatory response to endotoxin shows a preferential modulation by PARP in male animals (Mabley et al. 2005). Perhaps most directly relevant to the present study, Hagberg et al. used a neonatal model of hypoxia-ischemia in PARP-1 knockout mice, and showed that male, but not female, mice were protected (Hagberg et al. 2004). Taken together, it would be important to take into account patient sex when designing clinical trials using PARP inhibitors, and the sex of the animal in experimental studies evaluating the role of PARP.
The basis for the association between PAR-modified protein levels and increasing age is less clear. Possible explanations for this finding include increased or more mature PARP enzymes, or decreased PAR hydrolysis by PARG, with age. As one popular theory for aging is that DNA damage increases and accumulates with time, one could speculate that PARP enzymes are also increasing in amount or activity with age (Messripour et al. 1994). It has been reported that the amount of PARP-1 activity correlates with species-specific life span (Grube and Burkle 1992). However, there does appear to be an upper limit, as elderly humans have less constitutive PARP-1 and PARP-2 compared with young humans; although in healthy centenarians amounts of PARP-1 and PARP-2 appear to be preserved (Chevanne et al. 2007). In whole brain homogenates of late fetal rats, PARP activity was found to be 2.5 times that of post-natal day 4 rats, implicating a role for PARP in CNS cell differentiation, synaptogenesis, and development (Shambaugh et al. 1988). While, the effect of PARP activation on outcome after brain injuries sustained during development is less clear, the degree of PARP activation in neonatal rats after cerebral hypoxia-ischemia correlates with the degree of cerebral injury (Martin et al. 2005).
The finding that CSF PAR-modified protein levels in both control and TBI patients positively correlated with age raises a limitation of this study, since age differed between groups, with the control patients being much younger. As such, the difference in PAR-modified protein levels between TBI and control groups could potentially be explained by age alone and not TBI. However, peak CSF PAR-modified protein levels in 14 of the 17 TBI patients were above the value of the highest control patient (82.6 nM, 1.7 y.o.; Fig. 2b). Certainly, increasing the number of control CSF samples from older patients would be ideal, but these were not available. We also recognize that with a sample size of 17 patients our multivariate analysis is exploratory. The sample size of 17 reported here is above the minimum number generally accepted for multivariate analysis that includes 3 independent variables (at least 5 per variable); however, studies in a larger sample are needed to confirm these findings and determine whether or not GCS, GOS, and mechanism of injury are also predictive variables. There are also caveats related to the assay, for example, we did not control for hydrolysis of PAR. In tissues with active poly(ADP-ribose) glycohydrolase (PARG), the primary enzyme responsible for metabolizing PAR, or other hydrolytic enzymes, this assay would not reflect the degree of PARP activity. Nonetheless, this ELISA appears capable of quantifying PAR-modified proteins in CSF, and testing in other tissues and diseases appears warranted.
In conclusion, age- and sex-dependent increases in CSF PAR-modified proteins are seen during the first 2 days after TBI in infants and children. The increase in PAR-modified proteins may be due to increased PARP activation, decreased PAR degradation, or both. This assay may represent a useful method for evaluating the role for PARP enzymes, and the potency and bioavailability of PARP inhibitors, in TBI and other disorders.
Acknowledgments
Supported by NINDS RO1 NS38620 and P50 NS30318, NICHD T32 HD40686 and RO1 HD045968, RO1 GM060915, and the Children’s Hospital of Pittsburgh.
REFERENCES
- Adekoya N, Thurman DJ, White DD, Webb KW. Surveillance for traumatic brain injury deaths--United States, 1989-1998. Morbidity & Mortality Weekly Report. Surveillance Summaries. 2002;51:1–14. [PubMed] [Google Scholar]
- Berger RP, Pierce MC, Wisniewski SR, Adelson PD, Clark RS, Ruppel RA, Kochanek PM. Neuron-specific enolase and S100B in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatrics. 2002;109:E31. doi: 10.1542/peds.109.2.e31. [DOI] [PubMed] [Google Scholar]
- Chevanne M, Calia C, Zampieri M, Cecchinelli B, Caldini R, Monti D, Bucci L, Franceschi C, Caiafa P. Oxidative DNA damage repair and parp 1 and parp 2 expression in Epstein-Barr virus-immortalized B lymphocyte cells from young subjects, old subjects, and centenarians. Rejuvenation Res. 2007;10:191–204. doi: 10.1089/rej.2006.0514. [DOI] [PubMed] [Google Scholar]
- Clark RS, Vagni VA, Nathaniel PD, Jenkins LW, Dixon CE, Szabo C. Local administration of the poly(ADP-ribose) polymerase inhibitor INO-1001 prevents NAD+ depletion and improves water maze performance after traumatic brain injury in mice. Neurotrauma. 2007;24:1399–1405. doi: 10.1089/neu.2007.0305. [DOI] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Adelson PD, Bell MJ, Carcillo JA, Chen M, Wisniewski SR, Janesko K, Whalen MJ, Graham SH. Increases in bcl-2 protein in cerebrospinal fluid and evidence for programmed cell death in infants and children after severe traumatic brain injury. J Pediatr. 2000;137:197–204. doi: 10.1067/mpd.2000.106903. [DOI] [PubMed] [Google Scholar]
- Cosi C, Colpaert F, Koek W, Degryse A, Marien M. Poly(ADP-ribose) polymerase inhibitors protect against MPTP-induced depletions of striatal dopamine and cortical noradrenaline in C57B1/6 mice. Brain Res. 1996;729:264–269. [PubMed] [Google Scholar]
- de la Lastra CA, Villegas I, Sanchez-Fidalgo S. Poly(ADP-ribose) polymerase inhibitors: new pharmacological functions and potential clinical implications. Curr Pharm Des. 2007;13:933–962. doi: 10.2174/138161207780414241. [DOI] [PubMed] [Google Scholar]
- Du L, Bayir H, Lai Y, Zhang X, Kochanek PM, Watkins SC, Graham SH, Clark RSB. Innate gender-based proclivity in response to cytotoxicity and programmed cell death pathway. J Biol Chem. 2004;279:38563–38570. doi: 10.1074/jbc.M405461200. [DOI] [PubMed] [Google Scholar]
- Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabo C, Clark RSB. Intra-mitochondrial poly-ADP-ribosylation contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP- ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Grube K, Burkle A. Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc Natl Acad Sci USA. 1992;89:11759–11763. doi: 10.1073/pnas.89.24.11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha HC, Hester LD, Snyder SH. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc Natl Acad Sci U S A. 2002;99:3270–3275. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagberg H, Wilson MA, Matsushita H, Zhu C, Lange M, Gustavsson M, Poitras MF, Dawson TM, Dawson VL, Northington F, Johnston MV. PARP-1 gene disruption in mice preferentially protects males from perinatal brain injury. J Neurochem. 2004;90:1068–1075. doi: 10.1111/j.1471-4159.2004.02547.x. [DOI] [PubMed] [Google Scholar]
- Halmosi R, Berente Z, Osz E, Toth K, Literati-Nagy P, Sumegi B. Effect of poly(ADP-ribose) polymerase inhibitors on the ischemia-reperfusion-induced oxidative cell damage and mitochondrial metabolism in Langendorff heart perfusion system. Mol Pharmacol. 2001;59:1497–1505. doi: 10.1124/mol.59.6.1497. [DOI] [PubMed] [Google Scholar]
- Klaidman LK, Mukherjee SK, Hutchin TP, Adams JD. Nicotinamide as a precursor for NAD+ prevents apoptosis in the mouse brain induced by tertiary-butylhydroperoxide. Neurosci Lett. 1996;206:5–8. doi: 10.1016/0304-3940(96)12446-0. [DOI] [PubMed] [Google Scholar]
- Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM, Jenkins LW, Szabo C, Clark RS. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem. 2008;104:1700–1711. doi: 10.1111/j.1471-4159.2007.05114.x. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Raghupathi R, Saatman KE, McIntosh TK. Poly (ADP-ribose) polymerase activity following brain injury in the rat: an indictor of post-traumatic DNA damage. J Neurotrauma. 1998;15:878. [Google Scholar]
- LaPlaca MC, Zhang J, Raghupathi R, Li JH, Smith F, Bareyre FM, Snyder SH, Graham DI, McIntosh TK. Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma. 2001;18:369–376. doi: 10.1089/089771501750170912. [DOI] [PubMed] [Google Scholar]
- Love S, Barber R, Wilcock GK. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer’s disease. Brain. 1999;122(Pt 2):247–253. doi: 10.1093/brain/122.2.247. [DOI] [PubMed] [Google Scholar]
- Mabley JG, Horvath EM, Murthy KG, Zsengeller Z, Vaslin A, Benko R, Kollai M, Szabo C. Gender differences in the endotoxin-induced inflammatory and vascular responses: potential role of poly(ADP-ribose) polymerase activation. J Pharmacol Exp Ther. 2005;315:812–820. doi: 10.1124/jpet.105.090480. [DOI] [PubMed] [Google Scholar]
- Martin SS, Perez-Polo JR, Noppens KM, Grafe MR. Biphasic changes in the levels of poly(ADP-ribose) polymerase-1 and caspase 3 in the immature brain following hypoxia-ischemia. Int J Dev Neurosci. 2005;23:673–686. doi: 10.1016/j.ijdevneu.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Messripour M, Weltin D, Rastegar A, Ciesielski L, Kopp P, Chabert M, Mandel P. Age-associated changes of rat brain neuronal and astroglial poly(ADP-ribose) polymerase activity. J Neurochem. 1994;62:502–506. doi: 10.1046/j.1471-4159.1994.62020502.x. [DOI] [PubMed] [Google Scholar]
- Satchell MA, Zhang X, Kochanek PM, Dixon CE, Jenkins LW, Melick JA, Szabo C, Clark RS. A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14-3-3gamma. J Neurochem. 2003;85:697–708. doi: 10.1046/j.1471-4159.2003.01707.x. [DOI] [PubMed] [Google Scholar]
- Satchell MA, Lai Y, Kochanek PM, Wisniewski SR, Fink EL, Siedberg NA, Berger RP, DeKosky ST, Adelson PD, Clark RS. Cytochrome c, a biomarker of apoptosis, is increased in cerebrospinal fluid from infants with inflicted brain injury from child abuse. J Cereb Blood Flow Metab. 2005;25:919–927. doi: 10.1038/sj.jcbfm.9600088. [DOI] [PubMed] [Google Scholar]
- Shambaugh GE, 3rd, Koehler RR, Radosevich JA. Developmental pattern of poly (ADP-ribose) synthetase and NAD glycohydrolase in the brain of the fetal and neonatal rat. Neurochem Res. 1988;13:973–981. doi: 10.1007/BF00970771. [DOI] [PubMed] [Google Scholar]
- Ueda K, Hayaishi O. ADP-ribosylation. Annu Rev Biochem. 1985;54:73–100. doi: 10.1146/annurev.bi.54.070185.000445. [DOI] [PubMed] [Google Scholar]
- Virag L, Szabo C. The therapeutic potential of poly(ADP-Ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Clark RS, Dixon CE, Robichaud P, Marion DW, Vagni V, Graham SH, Virag L, Hasko G, Stachlewitz R, Szabo C, Kochanek PM. Reduction of cognitive and motor deficits after traumatic brain injury in mice deficient in poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab. 1999;19:835–842. doi: 10.1097/00004647-199908000-00002. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabo C, Watkins SC, Clark RS. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82:181–191. doi: 10.1046/j.1471-4159.2002.00975.x. [DOI] [PubMed] [Google Scholar]