

Abstract
Increased lung CD8 cells and their expression of chemokine receptors CXCR3 and CCR5 have been previously reported in chronic obstructive pulmonary disease (COPD). Alterations of CD8-CCR3 and -CCR4 expression and their ligands in COPD patients have not been fully investigated. The objective of this study was to assess in COPD patients: (i) broncho-alveolar lavage (BAL) CD8 CCR3 and CCR4 expression in COPD patients; and (ii) airway levels of the CCR3 ligands, CCL11 and CCL5. Multi-parameter flow cytometric anlaysis was used to assess BAL CD3 and CD8-chemokine receptor expression in COPD patients, smokers and healthy non-smokers (HNS). CCL5 and CCL11 levels were measured in BAL, and from the supernatants of lung resection explant cultures. CD8-CCR3 and -CCR5 expression (means) were increased in COPD patients (22% and 46% respectively) and smokers (20% and 45%) compared with HNS (3% and 22%); P < 0·05 for all comparisons. CD3CXCR3 expression was raised in smokers and COPD while CD8CXCR3 and CD3 and CD8 CCR4 expression was similar between groups. CD8CCR5 expression correlated to smoking pack years (r = 0·42, P = 0·01). COPD explants released more CCL5 compared with smokers (P = 0·02), while there was low level CCL11 production. CD8CCR3 and CCR5 expression appear to be regulated by cigarette smoke exposure. We show that COPD lung tissue released more CCL5, suggesting a role for CCL5–CCR3 signalling in pulmonary CD8 recruitment in COPD.
Keywords: CD8 cells, chemokine receptors, COPD
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by progressive airway obstruction and pulmonary inflammation. Increased numbers of neutrophils, macrophages and lymphocytes are found in lungs of COPD patients 1]. A hallmark feature of COPD is the increased numbers of CD8 cells in the airways, and it has been proposed that auto-immunity contributes to the pathogenesis of COPD [2,3]. The antigenic stimulation driving lymphocyte activation may either be from pathogens such as viruses or bacteria, or altered presentation of self antigens caused by cigarette smoke [2–4].
Chemokines are proteins involved in the recruitment of inflammatory cells from the circulation, and their positioning within tissues. Chemokines signal through G-protein coupled serpentine receptors (reviewed [5]). Chemokine receptors are categorized broadly into the CXC and CC families. Chemokine receptor expression may change due to specific inflammatory signals within tissue micro-environments [6].
Pulmonary lymphocytes express high levels of CXCR3 and CCR5 [6]. Studies of lymphocyte chemokine receptors in COPD have focused mainly on the role of CXCR3 and CCR5 in the pulmonary recruitment of CD8 cells. It has been shown by immunohistochemistry that expression of CXCR3 is increased on CD8 cells in the small airway walls of COPD patients compared with non-smoking controls [7]. Using flow cytometric analysis of dispersed cells from lung resections, increased CD3 lymphocyte CXCR3 and CCR5 expression in patients with emphysema compared with ex-smoking controls has been reported [8].
CCR3 and CCR4 are both known to play a role in lymphocyte chemotaxis [9]. Exacerbations of chronic bronchitis are associated with raised CCR3 levels on pulmonary eosinophils and lymphocytes [10], while CCR4 is thought to play a role in the pulmonary recruitment of lymphocytes in asthmatics [11]. CCR3 and CCR4 have been understudied in COPD. Our hypothesis was that CCR3 and CCR4 are involved in the pulmonary recruitment of CD8 cells in COPD.
This paper reports the expression of CCR3 and CCR4 on CD8 cells in COPD patients compared with controls. The expression of the more commonly studied receptors CCR5 and CXCR3 was also investigated. Our results showed an increase in CCR3 expression in COPD patients and smokers with normal lung function compared with healthy non-smokers (HNS). We therefore also investigated levels of the CCR3 ligands, CCL11 chemokine (Eotaxin) and CCL5 chemokine (Rantes), to determine if CCR3 ligands as well as the receptor itself are up-regulated in COPD.
Materials and methods
Subjects
Patients undergoing clinical investigational bronchoscopies or lung surgery for tumour resection were recruited. Subjects with any other pulmonary conditions, including infections 6 weeks or less prior to bronchoscopy and/or a history of asthma, were excluded. COPD was diagnosed based on a history of smoking (> 10 pack years), typical symptoms and airflow obstruction (forced expired volume in 1 s (FEV1) < 80% predicted, and FEV1/forced vital capacity (FVC) ratio < 0·7) [12]. Subjects with a smoking history but normal pulmonary function were categorized as ‘smokers’. All subjects gave written informed consent and the study was approved by the local research ethics committee.
Study design
Chemokine receptor expression
Bronchoalveolar lavage (BAL) samples from 34 subjects were assayed by flow cytometry for CD3 and CD8 cell chemokine receptor expression. These 34 subjects were part of a cohort of 53 subjects who had CD3, CD4, CD8 and T regulatory cell data analysed, the results of which have been published elsewhere [13]. Fifteen COPD patients and 11 smokers with normal lung function who were undergoing clinical investigational bronchoscopies (demographics shown in Table 1A) were recruited. The reasons for bronchoscopy were haemoptysis (COPD 4/15, smokers 2/11), unexplained shortness of breath or weight loss (COPD 4/15, smokers 1/11), and investigation of abnormal chest X-ray findings suggestive of lung cancer (COPD, 7/15; smokers, 8/11). Five HNS were recruited specifically to undergo a bronchoscopy for research purposes, and three HNS undergoing a clinical bronchoscopy for haemoptysis were also recruited.
Table 1A.
Subject demography: chemokine receptor expression.
| COPD n = 15 | Smokers n = 11 | HNS n = 8 | |
|---|---|---|---|
| Sex (F/M) | 4/11 | 4/7 | 4/4 |
| Age (years) | 64·6 (45–76) | 60 (39–82) | 64·2 (43–75) |
| FEV1 | 1·7 (1·1–2·4) | 2·5 (1·1–4) | 2·7 (2·4–4·2) |
| FEV1 predicted | 64 (36–85) | 94·6 (77–158) | 118·5 (100–150) |
| FEV1/FVC ratio | 67 (40–78) | 78·8 (72–86) | 76·2 (75–87) |
| Pack year history | 43·7 (25–78) | 33·1 (3·6–84) | 0 |
| Current/ex smokers | 9/6 | 5/6 | N/A |
| ICS users | 5 | N/A | N/A |
| BAL yield (ml) | 29·2 (11·7) | 37·6 (8·1) | 157·8 (112) |
Results expressed as mean (range). ICS, inhaled corticosteroid.
Chemokine levels
The levels of CCL11 and CCL5 were studied in BAL fluid from a separate cohort of COPD patients (n = 10), smokers (n = 12) and HNS (n = 6), (demographics shown in Table 1B). The reasons for bronchoscopy were haemoptysis (COPD, 1/10; smokers, 2/12), unexplained shortness of breath or weight loss (COPD 3/10, smokers 5/12), and investigation of abnormal chest X-ray findings suggestive of lung cancer (COPD, 6/10; smokers, 5/12). Four of the HNS were recruited specifically to undergo a bronchoscopy for research purposes. Two were being clinically investigated for haemoptysis.
Table 1B.
Subject demography: chemokines.
| COPD n = 10 | Smokers n = 12 | HNS n = 6 | ||
|---|---|---|---|---|
| BAL levels | Sex (F/M) | 5/5 | 5/7 | 3/3 |
| Age (years) | 66·2 (55–83) | 56·5 (20–81) | 45·6 (24–65) | |
| FEV1 | 1·6 (1·1–2·5) | 2·1 (1·9–4·2) | 2·8 (1·6–4·1) | |
| FEV1 predicted | 64·6 (60–75) | 90 (75–113) | 92·6 (86–107) | |
| FEV1/FVC ratio | 64·7 (48–70) | 80 (71–87) | 75·8 (71–82) | |
| Pack year history | 41·8 (16–80) | 22·2 (3·7–50) | 0 | |
| Current/Ex smokers | 7/3 | 6/6 | N/A | |
| ICS users | 5 | 0 | 0 |
| n = 6 | n = 6 | Not done | ||
|---|---|---|---|---|
| Lung explant release | Sex (F/M) | 3/3 | 2/4 | N/A |
| Age (years) | 64·5 (50–75) | 62·8 (53–73) | ||
| FEV1 | 1·7 (0·9–2·8) | 2·2 (1·7–3·4) | ||
| FEV1 predicted | 61·2 (42–81) | 90 (73–128) | ||
| FEV1/FVC ratio | 57·3 (45–68) | 81 (75–85) | ||
| Pack year history | 46·8 (35–58) | 31 (10–53) | ||
| Current/Ex smokers | 4/2 | 2/4 | ||
| ICS users | 4 | 0 |
Results expressed as mean ICS, inhaled corticosteroid; P, lung explant study; B, BAL study.
Six smokers and six COPD patients who had undergone tumour resection surgery as part of their normal clinical care (demographics shown on Table 1B) were recruited for bronchial explant cultures.
Methods
Cell collection
Bronchoalveolar lavage was collected from a lobe not affected by radiographic or endobronchial abnormalities: The bronchoscope was wedged in the bronchus and a maximum of 4 × 60 ml aliquots of pre-warmed sterile 0·9% NaCl solution were instilled. The aspirated fluid was stored on ice before filtration (100 µm filter, Becton Dickenson). The filtrate was centrifuged (400 g/10 min at 4°C) and the cell pellet washed in RPMI 1640 medium supplemented with 2 mM L-glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. Five millilitres of supernatant were stored at −20oC for later chemokine ELISA. Viable cell counts were determined by trypan blue exclusion (Neubauer hemocytometer). The cell count was adjusted to 1 × 106/ml in supplemented RPMI 1640 medium with additional 10% (vol/vol) fetal calf serum. This cell suspension was then used for flow cytometry.
Flow cytometry
Four hundred µl aliquots of cell suspension were dispensed into FACS tubes for antibody labelling. Cells were labelled with three directly conjugated antibodies (BD Biosciences, Oxford) for 45 min at 4oC in the dark. The following antibodies and IgG isotype controls were used: (i) PE-Cy5: CD4, CD8, IgG; (ii) FITC: CD3, CCR5, IgG; (iii) PE: CCR3, CCR4, CCR5, CXCR3, IgG. Cells were washed [phosphate-buffered saline (PBS) containing 1% bovine serum albumin] and fixed (PBS containing 0·5% paraformaldehyde). Using EPICS XL-MCL flow cytometer (Beckman-Coulter, Buckinghamshire), lymphocytes were gated using forward scatter and side scatter profiles while ensuring FL1, FL2 and FL3 isotype control co-plots remained in the lower left quadrants. Ten thousand BAL lymphocyte events were collected. The percentage of CD3, CD4 and CD8 cells expressing chemokine receptors was determined.
Cell culture
Following surgery, lung parenchyma explants were isolated from regions of the lung that were distal (> 10 cm) from the tumour lesion. Samples were processed within 3 h of collection, having been placed immediately in RPMI at 4°C. Samples were washed × 2 in RPMI, weighed and adjusted to 30 mg then cultured; 1 per triplicate well in 24-well plates for 24 h in 800 µl RPMI 1640 media containing 10% FCS, penicillin/streptomycin and L-glutamine at 37°C, 5% CO2. Parallel triplicate wells were stimulated with the Toll receptor 2 and 4 ligand LPS (extracted from E. coli, Sigma; 1 µg/ml) for the 24-h culture period. Supernatant was subsequently removed and stored at −20°C for later ELISA analysis.
ELISA
Bronchoalveolar lavage fluid and lung explant supernatants were analysed for CCL5 and CCL11 levels using ELISA duosets purchased from R&D systems according to the manufacturer's instructions.
Data analysis
The percentage of leukocytes (in 10 000 events) expressing CD3, CD4 and CD8 were determined. This figure was adjusted by the total number of cells retrieved/ml of BAL to calculate the total number of CD3, CD4, CD8/ml of BAL. Cell number data were not normally distributed, so three-way comparisons between groups were performed using Dunn non-parametric anova, and Mann–Whitney U-tests for two-way comparisons. Chemokine receptor expression data were normally distributed, so comparisons between groups (HNS versus smokers versus COPD and inhaled corticosteroid users versus non-users) were performed using Tukey parametric anova. Subsequently, two-way between-group comparisons (non-paired t-tests) were performed. Spearmans coefficient was used to analyse the relationship between lymphocytes and %FEV1 and %FEV1 with BAL yield. CCL5 and CCL11 data from BAL and lung explants were normally distributed, so non-paired-T tests wereperformed to compare COPD patients and smokers. Paired t-tests were performed to assess LPS effects. P < 0·05 was considered statistically significant in all analysis.
Results
The CD3, CD4 and CD8 data from the 34 subjects undergoing flow cytometry analysis has already been reported elsewhere [13] as part of a cohort of 53 subjects. As previously reported [14], retrieved BAL yield (volume) was lower in COPD than smokers, which was lower than HNS (Table 1A), and the BAL yield was related to FEV1 (P = 0·008, r = 0·55). Similar to the previous data, COPD patients had significantly more CD3 cells/ml BAL compared with smokers and HNS (P < 0·05); median CD3 cell numbers × 102/ml BAL were COPD 92·3, smokers 15·8, HNS 28. There were a numerically greater number of CD8 cells/ml BAL (× 102) in COPD patients (30·9) compared with both smokers (6·9) and HNS (6), with statistical significance being reached in comparison with HNS (P = 0·02) but not smokers (P = 0·1). Current smoking had no significant effect on lymphocyte subset numbers (P > 0·05 for CD3, CD4 and CD8) and similarly inhaled corticosteroid treatment in COPD patients also had no effect (P > 0·05 for CD3, CD4 and CD8).
Chemokine receptor expression
CD8 chemokine receptor expression (Fig. 1a)
Fig. 1.

Chemokine receptor expression on: (a) % BAL CD8 cells, (b) % BAL CD3 cells. Blocks show mean group values, error bars represent SEM. *(P < 0·05), **(P < 0·01), ***P < 0·001. HNS (n = 8), smokers without airway obstruction (n = 11), COPD patients (n = 15).
The proportion of CD8 cells expressing CCR3 was significantly greater in COPD patients (P = 0·02) and smokers (P = 0·008) compared with HNS. The proportion of CD8 cells expressing CCR5 was significantly greater in COPD patients (P = 0·009) and smokers (P = 0·04) compared with HNS. For CCR3 and CCR5, there was no difference in CD8 chemokine receptor expression between COPD patients and smokers. CCR4 and CXCR3 expression levels were similar in all three subject groups (P > 0·05).
CD3 lymphocyte chemokine receptor expression (Fig. 1b)
The proportion of CD3 cells expressing CCR3 was significantly greater in COPD patients (P = 0·02) and smokers (P = 0·01) compared with HNS. The proportion of CD3 cells expressing CCR5 was significantly greater in COPD patients (P ≤ 0·001) and smokers (P = 0·01) compared with HNS. In contrast to CD8 cells CXCR3 expression on CD3 lymphocytes was greater in COPD patients (P = 0·03) and smokers (P = 0·01) compared with HNS. For CCR3, CCR5 and CXCR3, there was no difference in CD3 chemokine receptor expression between COPD patients and smokers. CD3CCR4 expression levels were similar in all three subject groups (P > 0·05).
Current smoking and inhaled corticosteroid effects
CCR5 expression on COPD and smokers BAL CD8 and CD3 lymphocytes was significantly higher in current smokers compared with ex-smokers (Table 2). Additionally, there was a positive correlation between BAL CD8CCR5 expression with pack year history (Fig. 2). Current smoking and pack year history did not influence the BAL expression levels of any other CD8 chemokine receptors.
Table 2.
Interaction of smoking and BAL CD8 and CD3 chemokine receptor expression.
| Current smoking effects Current versus ex-smokers: Data from COPD patients and smokers are combined | ||||||
|---|---|---|---|---|---|---|
| BAL CD8 cells | BAL CD3 cells | |||||
| Current n = 14 | Ex n = 12 | Non-paired t-test P value | Current n = 14 | Ex n = 12 | Non-paired t-test P value | |
| CCR3 | 19·9 (5·8) | 22·3 (6·9) | 0·39 | 11·7 (4) | 9·5 (2·3) | 0·32 |
| CCR4 | 33·4 (6·7) | 20·1 (4·4) | 0·06 | 20·6 (4·2) | 10·6 (3·1) | 0·07 |
| CCR5 | 55·4 (7·5) | 36·3 (7·9) | 0·04 | 33·3 (4·1) | 21·3 (5·3) | 0·04 |
| CXCR3 | 45·4 (6·7) | 37·2 (8·4) | 0·22 | 29·3 (4·8) | 21·9 (6) | 0·17 |
| Analysis of ex- and non-smokers: COPD ex-smokers versus ex-smokers (Hex) versus HNS | ||||||||
|---|---|---|---|---|---|---|---|---|
| BAL CD8 cells | BAL CD3 cells | |||||||
| COPD ex n = 6 | Hex n = 6 | HNS n = 8 | Three-way anovaP value | COPD ex n = 6 | Hex n = 6 | HNS n = 8 | Three-way anovaP value | |
| CCR3 | 30·1 (12·3) | 14·5 (5·6) | 3·4 (1·4) | 0·03 | 12·2 (3·8) | 6·9 (2·3) | 1·1 (0·3) | 0·019 |
| CCR4 | 27·9 (6·9) | 12·4 (4) | 21·5 (5·6) | 0·23 | 14·4 (5·5) | 6·8 (2·8) | 7 (1·5) | 0·26 |
| CCR5 | 41·8 (12·9) | 30·9 (9·8) | 21·6 (6·4) | 0·32 | 22·8 (7·8) | 19·9 (8) | 6·5 (2·8) | 0·18 |
| CXCR3 | 31·7 (9·1) | 42·7 (14·7) | 36 (10·5) | 0·82 | 17·3 (5·9) | 26·4 (10·7) | 9·5 (2·7) | 0·24 |
Mean per cent cell chemokine receptor expression (standard error of the mean). Hex, ex-smoker with normal lung function.
Fig. 2.
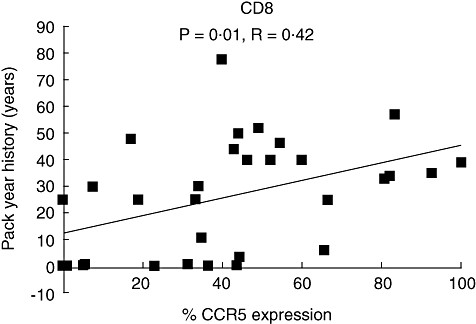
Pack year history correlation with BAL CD8-CCR5 expression. HNS (n = 8), smokers (n = 11) and COPD patients (n = 15).
In a subgroup analysis, we excluded all current smokers to remove any confounding effect of this variable (see Table 2). Significantly raised CCR3 levels were observed on CD8 and CD3 cells in the COPD group compared with HNS (P = 0·01, and 0·005 respectively). The levels in ex-smokers with normal pulmonary function were numerically lower than COPD patients, although this was not statistically significant.
CD8 chemokine receptor expression in COPD patients who were inhaled corticosteroid (ICS) users was not significantly different from the non-users. The mean % expression levels for ICS versus non-users, respectively, were: CCR3 14·1 versus 25·2, P = 0·21; CCR5 49·9 versus 45·5, P = 0·38; CCR4 20 versus 31·5, P = 0·18; and CXCR3 31 versus 37, P = 0·32.
Bronchoalveolar lavage CCL11 and CCL5 Levels
Having found increased CCR3 levels in BAL CD8 cells from COPD patients and smokers, levels of the CCR3 ligands CCL11 and CCL5 in BAL fluid were assessed in a separate cohort of COPD patients, smokers and HNS (see Fig. 3a). CCL5 levels in smokers and COPD patients were numerically higher than HNS (mean pg/ml: 30 and23 and 5 respectively), but these differences did not reach statistical significance (P = 0·1 and 0·09 respectively). CCL11 levels were significantly higher in smokers compared with HNS (mean pg/ml: 4·1 and 3·1 respectively, P = 0·03), but the numerical difference between COPD patients and HNS did not reach statistical significance (mean pg/ml: 5·7 and 3·1 respectively, P = 0·08). For CCL5 and CCL11, there was no difference between COPD patients and smokers. CCL5 and CCL11 levels in COPD BAL were similar in ICS users compared with non-users (P = 0·42 and 0·15 respectively).
Fig. 3.
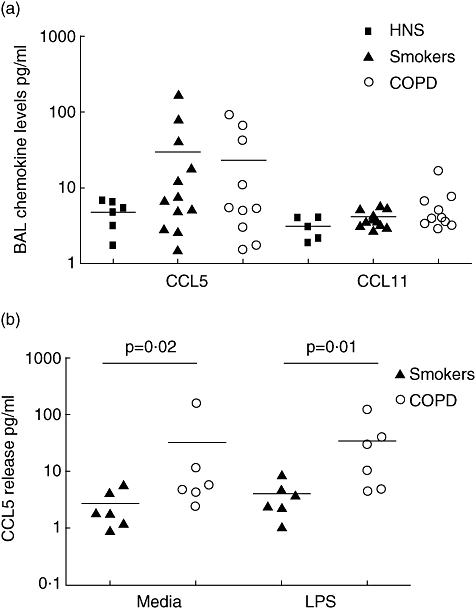
CCL11 and CCL5 levels (pg/ml) determined by ELISA. (a) Chemokine levels detected in retrieved BAL fluid of HNS (n = 6), smokers (n = 12) and COPD subjects (n = 10). (b) CCL5 levels detected in lung parenchymal 24-h culture supernatants of HNS (n = 6) and COPD subjects (n = 6). Horizontal lines indicate mean group values.
Lung explant cultures
After 24-h culture, CCL5 levels (mean pg/ml) were significantly raised in COPD subjects compared with smokers (30·4 and 2·6 respectively, P = 0·02). LPS was used to stimulate culture to maximize chemokine release, but levels were not increased by LPS stimulation (34·3 and 3·8 respectively, see Fig. 3b).
CCL11 production in smokers and COPD was low and similar (mean pg/ml: 1·8 and 1·0 respectively) and was not altered by LPS stimulation (mean pg/ml: 2 and 1·1 respectively).
Discussion
Smokers, with and without COPD, had increased CCR3 and CCR5 expression on BAL CD3 lymphocytes and specifically on CD8 cells compared with healthy non-smoking controls. The expression of CCR3 and CCR5 on BAL CD8 cells was not different between COPD patients and smokers, suggesting that smoking history, and not the development of airflow obstruction, alters the expression of these chemokine receptors. This is the first study to suggest a role for CCR3 in the recruitment of CD8 cells into the lungs of smokers and COPD patients.
To further investigate the role of CCR3 signalling in CD8 recruitment, we assessed the levels of the CCR3 ligands CCL5 and CCL11 in two distinct compartments of the lung; BAL to assess chemokine levels in the airway lumen produced by the epithelia and luminal inflammatory cells, and explant cultures to assess chemokine production from parenchymal cells. CCL5 and CCL11 levels were numerically raised in BAL from smokers and COPD subjects compared with HNS, but only CCL11 levels in smokers were significantly higher compared with HNS. We suggest that our sample size was too small to prove statistically significant differences in all cases. However, our data support previous observations of increased BAL and induced sputum CCL5 and CCL11 levels in COPD patients compared with controls [15,16]. It would certainly be of interest to investigate airway levels of other CCR3 ligands such as MCP 2, 3 and 4.
Explant cultures used to investigate the spontaneous and LPS-induced release of CCL5 and CCL11 from lung parenchyma showed CCL5 levels were increased in COPD patients compared with smokers, while CCL11 levels were low and not different between groups. This suggests that CCL5 production is up-regulated specifically in COPD patients. Altered CCL5–CCR3 signalling may be a key mechanism for CD8 recruitment in COPD. A disadvantage of the explant model is that we could not recruit HNS, as there were no patients of this phenotype undergoing lung surgery. The low levels of CCL11 production from lung parenchyma, in conjunction with our BAL findings and other published findings showing increased levels of this chemokine in the airway lumen [15,16], suggest that CCL11 is predominantly produced by the airway epithelia [17] and/or luminal inflammatory cells in COPD patients.
CCL5 may play a role in lymphocyte recruitment via CCR3, and can also signal through CCR5 [18]. CCL5 may also play a role in eosinophil recruitment through CCR3, and it is known that a subset of COPD patients have significant airway eosinophilia [19]. Other possible roles for CCL5 include the promotion of TH1 IFNγ producing lymphocytes [20]. Although the current study was focused on CD8 recruitment, our data regarding CCL5 may have wider implications in the pathophysiology of COPD.
CCR3 is a member of the inducible CC family [5], with expression levels regulated by inflammation in the surrounding microenvironment [10]. CCR3 has been implicated in CD4 recruitment in atopy, with TH2 cytokines such as IL-4 capable of increasing CD4CCR3 expression levels [21]. CCR3 expressing lymphocytes are thought to be TH2 cells [9]. It is known that COPD lymphocytes have a TH1 profile [22,23]. However, there is also evidence that airway lymphocytes from COPD patients can secrete more TH2 cytokines, notably IL-4 and IL-13, compared with lymphocytes from controls [24,25], and that some COPD patients have significant airway eosinophilia [19]. These observations support our findings that CCR3 expressing TH2 CD8 cells may be involved in the pathophysiology of COPD. We speculate that CCR3 up-regulation on BAL CD8 cells in smokers and COPD patients is caused by inflammation and/or oxidative stress.
It has previously been reported in patients with chronic bronchitis that CCR3 expression is increased on lung tissue lymphocytes during exacerbations [10]. In contrast, Grumelli et al.[8] found low-level expression of CCR3 and CCR4 in COPD pulmonary lymphocytes dispersed from lung tissue and analysed by flow cytometry.
In agreement we also observed very low CCR3 levels in our control group of HNS. The differences between our findings in smokers and COPD patients and those of Grumelli et al. may be due to the following. (i) It is possible that the lymphocyte dispersion methodology altered the ability to detect CCR3 and CCR4. (ii) Alternatively, it is entirely possible that the CD8CCR3 population is specifically recruited into the airway lumen/alveolar spaces, leading to lower expression levels in the tissue. It has been reported that CCR3 expression was not observed in sputum lymphocytes [26]. Again, this may be suggestive of different recruitment mechanisms into separate lung compartments.
We performed a subgroup analysis to compare chemokine receptor expression in subjects who were not current smokers. We observed that CCR3 expression was increased in the COPD patients compared with HNS, providing some evidence that the increase in the expression levels of this chemokine receptor in COPD patients is not only dependant on smoking history. The numbers in this subgroup analysis were small, but nevertheless the difference between COPD patients and controls was large.
Previous studies of CCR5 and CXCR3 expression on pulmonary CD8 cells in COPD patients have evaluated lung parenchymal cells. Chemokine receptor expression may change as CD8 cells traverse across the epithelium into the airway lumen, so the results from these studies may differ compared with the current study. Previous work has shown that CD8CCR5 expression is increased in COPD patients compared with ex-smokers [8], which is supported by our data; CD8CCR5 expression was associated with acute cigarette smoke exposure and the overall burden of cigarette smoke pack years. We also found similar CD8CCR5 levels in COPD patients and smokers that were increased when compared with HNS, suggesting that smoking was the key determinant of CCR5 expression.
We did not find any difference between COPD patients and smokers in pulmonary CD8CXCR3, in agreement with the findings of Saetta et al.[7]. Similarly to our study, the study by Grumelli et al.[8] reported increased CD3CXCR3 in COPD patients compared with ex-smokers, but the specific data for CD8CXCR3 were not shown.
A possible limitation of the lung explant sub-study was the small sample size. However, the differences for CCL5 were striking, and we feel that this is unlikely to be a chance observation. Another issue is that we cannot totally rule out any effects lung cancer may have had on our results, but we ensured that sampling occurred at a different site to any endobronchial or radiological abnormality. Additionally, it is possible that some of our smokers had subclinical emphysema, and we were not able to perform high resolution CT scanning to rule this out. However, even if we had done this, the patients would still have been categorized as not having COPD according to GOLD criteria [12].
In conclusion, BAL CD8 chemokine receptor expression appears to be regulated by cigarette smoke exposure, rather than the development of airflow obstruction in smokers. CCR3 and CCR5 were the most highly regulated BAL CD8 receptors. We also present data to support a role for CCR3 and CCR5 ligands in the recruitment of CD8 cells into the airways, and in particular we show that production of CCL5 from airway tissue is increased in COPD patients compared with smokers. Therapeutic approaches designed to interfere with these recruitment mechanisms may reduce the burden of pulmonary inflammation in COPD patients.
References
- 1.Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in COPD. N Engl J Med. 2004;24:2645–53. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 2.Agusti A, MacNee W, Donaldson K, Cosio M. Hypothesis: does COPD have an autoimmune component? Thorax. 2003;58:832–4. doi: 10.1136/thorax.58.10.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee SH, Goswami S, Grudo A, et al. Antielastin autoimmunity in tobacco smoking-induced emphysema. Nat Med. 2007;5:567–9. doi: 10.1038/nm1583. [DOI] [PubMed] [Google Scholar]
- 4.Saetta M, Turato G, Maestrelli P, Mapp CE, Fabbri LM. Cellular and structural bases of COPD. Am J Respir Crit Care Med. 2001;163:1304–9. doi: 10.1164/ajrccm.163.6.2009116. [DOI] [PubMed] [Google Scholar]
- 5.Proudfoot AEI. Chemokine receptors: multifaceted therapeutic targets. Nat Rev Immunol. 2002;2:106–15. doi: 10.1038/nri722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunkel EJ, Boisvert J, Murphy K, et al. Expression of the chemokine receptors CCR4, CCR5, and CXCR3 by human tissue-infiltrating lymphocytes. Am J Pathol. 2002;160:347–55. doi: 10.1016/S0002-9440(10)64378-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saetta M, Mariani M, Panina-Bordignon P, et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with COPD. Am J Respir Crit Care Med. 2002;15:1404–9. doi: 10.1164/rccm.2107139. [DOI] [PubMed] [Google Scholar]
- 8.Grumelli S, Corry DB, Song LZ, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004;1:e8, 75–83. doi: 10.1371/journal.pmed.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D'ambrosio D, Mariani M, Panina-Bordignon P. Chemokines and their receptors guiding T lymphocyte recruitment in lung inflammation. Am J Respir Crit Care Med. 2001;164:1266–75. doi: 10.1164/ajrccm.164.7.2103011. [DOI] [PubMed] [Google Scholar]
- 10.Bocchino V, Bertorelli G, Bertrand CP, et al. CCL11 and CCR3 are up-regulated in exacerbations of chronic bronchitis. Allergy. 2002;57:17–22. [PubMed] [Google Scholar]
- 11.Panina-Bordignon P, Papi A, Mariani M, et al. The C-C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen-challenged atopic asthmatics. J Clin Invest. 2001;107:1357–64. doi: 10.1172/JCI12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Executive Summary: Global Strategy for the diagnosis, management and prevention of COPD. 11. Available at: http://www.goldcopd.com.
- 13.Smyth LJ, Starkey C, Vestbo J, Singh D. CD4-regulatory cells in COPD patients. Chest. 2007;132:156–63. doi: 10.1378/chest.07-0083. [DOI] [PubMed] [Google Scholar]
- 14.Löfdahl JM, Cederlund K, Nathell L, Eklund A, Sköld CM. Bronchoalveolar lavage in COPD: fluid recovery correlates with the degree of emphysema. Eur Respir J. 2005;25:275–81. doi: 10.1183/09031936.05.00033504. [DOI] [PubMed] [Google Scholar]
- 15.Miller M, Ramsdell J, Friedman PJ, Cho JY, Renvall M, Broide DH. Computed tomographic scan–diagnosed COPD – emphysema: CCL11-1 is associated with bronchodilator response and extent of emphysema. J Allergy Clin Immunol. 2007;120:1118–25. doi: 10.1016/j.jaci.2007.08.045. [DOI] [PubMed] [Google Scholar]
- 16.Costa C, Rufino R, Traves SL, Lapa E Silva JR, Barnes PJ, Donnelly LE. CXCR3 and CCR5 chemokines in induced sputum from patients with COPD. Chest. 2008;133:26–33. doi: 10.1378/chest.07-0393. [DOI] [PubMed] [Google Scholar]
- 17.Witherden IR, Vanden Bon EJ, Goldstraw P, Ratcliffe C, Pastorino U, Tetley TD. Primary human alveolar type II epithelial cell chemokine release. Effects of cigarette smoke and neutrophil elastase. Am J Resp Cell Mol Biol. 2004;30:500–9. doi: 10.1165/rcmb.4890. [DOI] [PubMed] [Google Scholar]
- 18.Blanpain C, Buser R, Power CA, et al. A chimeric MIP-1α/CCL5 protein demonstrates the use of different regions of the CCL5 protein to bind and activate its receptors. J Leukocyte Biol. 2001;69:977–85. [PubMed] [Google Scholar]
- 19.Saha S, Brightling CE. Eosinophilic airway inflammation in COPD. Int J Chron Obstruct Pulmon Dis. 2006;1:39–47. doi: 10.2147/copd.2006.1.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grayson MH, Holtzman MJ. Chemokine complexity: the case for CCL5. Am J Respir Cell Mol Biol. 2006;35:143–6. doi: 10.1165/rcmb.f318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Francis JN, Lloyd CM, Sabroe I, Durham SR, Till SJ. T lymphocytes expressing CCR3 are increased in allergic rhinitis compared with non-allergic controls and following allergen immunotherapy. Allergy. 2007;62:59–65. doi: 10.1111/j.1398-9995.2006.01253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hodge G, Nairn J, Holmes M, Reynolds PN, Hodge S. Increased intracellular T helper 1 proinflammatory cytokine production in peripheral blood, bronchoalveolar lavage and intraepithelial T cells of COPD subjects. Clin and Exp Immunology. 2007;150:22–9. doi: 10.1111/j.1365-2249.2007.03451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Majori M, Corradi M, Caminati A, Cacciani G, Bertacco S, Pesci A. Predominant TH1 cytokine pattern in peripheral blood from subjects with COPD. J Allergy Clin Immunol. 1999;103:458–62. doi: 10.1016/s0091-6749(99)70471-9. [DOI] [PubMed] [Google Scholar]
- 24.Barczyk A, Pierzchala W, Kon OM, Cosio B, Adcock IM, Barnes PJ. Cytokine production by bronchoalveolar lavage T lymphocytes in chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2006;117:1484–92. doi: 10.1016/j.jaci.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Barceló B, Pons J, Fuster A, et al. Intracellular cytokine profile of T lymphocytes in patients with chronic obstructive pulmonary disease. Clin Exp Immunol. 2006;145:474–9. doi: 10.1111/j.1365-2249.2006.03167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leckie MJ, Jenkins GR, Khan J, et al. Sputum T lymphocytes in asthma, COPD and healthy subjects have the phenotype of activated intraepithelial T cells (CD69+ CD103+) Thorax. 2003;58:23–9. doi: 10.1136/thorax.58.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]