

Abstract
Aims
Etravirine is a next-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) with activity against wild-type and NNRTI-resistant HIV. Proton pump inhibitors and H2-antagonists are frequently used in the HIV-negative-infected population, and drug–drug interactions have been described with other antiretrovirals. This study evaluated the effect of steady-state omeprazole and ranitidine on the pharmacokinetics of a single dose of etravirine.
Methods
In an open-label, randomized, one-way, three-period crossover trial, HIV-negative volunteers randomly received a single dose of 100 mg etravirine alone (treatment A); 11 days of 150 mg ranitidine b.i.d. (treatment B); and 11 days of 40 mg omeprazole q.d. (treatment C). A single dose of 100 mg etravirine was co-administered on day 8 of sessions 2 and 3. Each session was separated by a 14-day wash-out.
Results
Nineteen volunteers (seven female) participated. When a single dose of etravirine was administered in the presence of steady-state ranitidine, etravirine least squares means ratios (90% confidence interval) for AUClast and Cmax were 0.86 (0.76, 0.97) and 0.94 (0.75, 1.17), respectively, compared with administration of etravirine alone. When administered with steady-state omeprazole, these values were 1.41 (1.22, 1.62) and 1.17 (0.96, 1.43), respectively. Co-administration of a single dose of etravirine and ranitidine or omeprazole was generally safe and well tolerated.
Conclusions
Ranitidine slightly decreased etravirine exposure, whereas omeprazole increased it by approximately 41%. The increased exposure of etravirine when co-administered with omeprazole is attributed to CYP2C19 inhibition. Considering the favourable safety profile of etravirine, these changes are not clinically relevant. Etravirine can be co-administered with proton pump inhibitors and H2 antagonists without dose adjustments.
Keywords: Drug–drug interaction, etravirine, non-nucleoside reverse transcriptase inhibitor, omeprazole, ranitidine, TMC125
Introduction
HIV infection needs chronic, life-long treatment with a combination of several drugs, many of which display an interaction potential [1]. Due to gastrointestinal side-effects of antiretroviral therapy, acid-suppressing agents are frequently used by patients living with HIV [2]. In surveys of HIV-infected patients, 37–46% of patients treated with highly active antiretroviral therapy responded as having used acid-suppressing agents, of which a substantial part was obtained as over-the-counter medication [2, 3].
Acid-suppressing agents have the potential to affect the pharmacokinetics of co-administered drugs in several ways. Increased gastric pH generally facilitates absorption of weak acids and decreases the solubility and absorption of weak bases [4, 5]. Antiretroviral agents with a pH-dependent solubility and gastric absorption such as atazanavir showed significant decreases of plasma concentrations when co-administered with acid-reducing agents, despite boosting with low-dose ritonavir [6, 7]. No clinically significant interaction was observed when omeprazole or ranitidine was co-administered with boosted indinavir [8], fosamprenavir [6, 9], darunavir [10], tipranavir [11] or lopinavir [12–14]. Conversely, exposure to boosted saquinavir increased substantially when co-administered with omeprazole 40 mg q.d., regardless of whether the drugs were administered simultaneously or 2 h apart [15, 16]. The latter finding might be a consequence of increased solubility of the drug in a less acidic environment, but a positive effect on the absorption by delayed gastric emptying and/or an inhibitory effect on P-glycoprotein (P-gp) by omeprazole have also been suggested as possible mechanisms for this interaction.
In addition to these effects on absorption, systemic exposure to a co-administered drug can also be altered by changes in its metabolism. The H2 antagonist ranitidine has not been shown to have a clinically significant effect on drug metabolism by CYP1A2, CYP2D6 or CYP3A [17]. Omeprazole is known as an inhibitor of CYP2C19 [4, 5]. In vivo, competitive inhibition of CYP2C19 by omeprazole has been shown to reduce the clearance of a single dose of intravenous diazepam by 20–38% [4, 18]. This effect was not observed in poor metabolizers for CYP2C19, with a substantially lower baseline activity of this isoenzyme [19]. Evidence on the effect of omeprazole on CYP3A4 activity is conflicting. No major interaction of omeprazole with the CYP3A4 substrate ciclosporin was described previously [20]. In another study, a 40% reduction of the metabolite excretion of the CYP3A probe dapsone was shown, although a study in renal transplant patients has demonstrated a decrease of tacrolimus exposure when co-administered with omeprazole, attributed to the induction of intestinal CYP3A4 activity [21, 22].
Etravirine (ETR, TMC125) is a next-generation non-nucleoside reverse transcriptase inhibitor (NNRTI) with activity against both wild-type and NNRTI-resistant HIV-1 [23, 24]. Sustained efficacy of the drug has been demonstrated in a controlled Phase 2b dose-ranging trial in HIV-infected patients with documented genotypic resistance to the NNRTI and protease inhibitor classes after 24 and 48 weeks of treatment [25]. In two ongoing Phase 3 trials conducted in treatment-experienced patients with NNRTI resistance to other approved agents, treatment with etravirine achieved better virological suppression at weeks 24 and 48 than placebo, both given in combination with background antiretrovirals. Treatment with TMC125 was generally safe and well tolerated [26–29].
Etravirine is metabolized by cytochrome P450 (CYP) 3A4, CYP2C9 and CYP2C19 with subsequent glucuronidation of the metabolites. M8 and M12 have been shown to be the most abundant metabolites in plasma [30]. In vitro, etravirine is an inhibitor of CYP2C9 and P-gp. In vivo, etravirine is an inducer of CYP3A4 and an inhibitor of the CYP2C subfamily [31]. Etravirine has a terminal elimination half-life in plasma of approximately 30–40 h. The drug should be taken following a meal to enhance bioavailability [32]. Etravirine has low solubility and permeability and is categorized as a Class IV compound according to the Biopharmaceutics Classification System [33]. The compound is virtually insoluble in water and oils. The development of a solid dispersion tablet formulation with etravirine in an amorphous form, homogeneously dispersed in a polymer matrix, resulted in substantially improved oral bioavailability, as demonstrated in a single-dose comparative bioavailability study in healthy volunteers in fed state [34]. In vitro, no effect of pH on the solubility of the solid dispersion has been observed (Tibotec, data on file). This Phase 3 formulation was used for this study and is the formulation used in ongoing clinical trials with etravirine.
The primary objective of this study was to determine the steady-state effect of two acid-suppressing agents (the H2 antagonist ranitidine and the proton pump inhibitor [PPI] omeprazole) with different modes of action and different effects on drug metabolism on the pharmacokinetics of a single 100-mg tablet of etravirine in HIV-negative volunteers. Furthermore, the short-term safety and tolerability of the concomitant use of etravirine and ranitidine or omeprazole were also evaluated.
Methods
The final protocol was reviewed and approved by an Independent Ethics Committee according to specifications outlined in the applicable regulations (e.g. International Conference on Harmonisation/Good Clinical Practice, US Code of Federal Regulations). The trial was performed in accordance with the principles of Good Clinical Practice. All volunteers gave their written consent prior to any trial-related procedure.
Trial population
Eighteen healthy HIV-1-negative volunteers were to be enrolled in the trial, randomized into six panels of different treatment sequences as determined by the randomization schedule. A minimum of at least 16 evaluable subjects per treatment arm was considered sufficient to allow for relevant conclusions. With a minimum of 16 evaluable subjects, and expected within-subject variability on the logarithmic scale of 0.15 in AUClastfor etravirine, the range of the 90% confidence interval (CI) of the least squares means (LSmeans) ratio was estimated to be (−0.79; +1.27).
All volunteers had to be able to comply with the protocol requirements, aged 18–55 years and have a body mass index (BMI) between 18.0 and 30.0 kg m−2. Because of the significant effect of CYP2C19 on the pharmacokinetics of omeprazole, volunteers of a known poor metabolizer status to CYP2C19 or volunteers of Asian ethnicity (due to high incidence of poor metabolizers for CYP2C19 [35]) were excluded, as were smokers. Women of non-child-bearing potential could be included. All volunteers needed to be in good health, as determined by medical history, physical examination and clinical laboratory assessments. They needed to test HIV antibody negative, and not have participated in previous trials of investigational drugs within the past 90 days. Volunteers with a history or suspicion of current alcohol and/or drug abuse, or a suspected allergy to any of the trial medications were excluded. Volunteers with abnormalities that might have an effect on the absorption of drugs (such as chronic diarrhoea or gastric stasis, etc.), as well as those consuming solely plant-based diets were not allowed to participate. Volunteers were not allowed to use concomitant medication, including over-the-counter products and dietary supplements, from 7 days before the first administration of the trial medication. The use of grapefruit juice and beverages containing alcohol or quinine was prohibited 7 days and 24 h, respectively, prior to drug administration. Volunteers were advised to refrain from strenuous activities from day –1 until the last day of each session.
Trial design (Figure 1)
Figure 1.
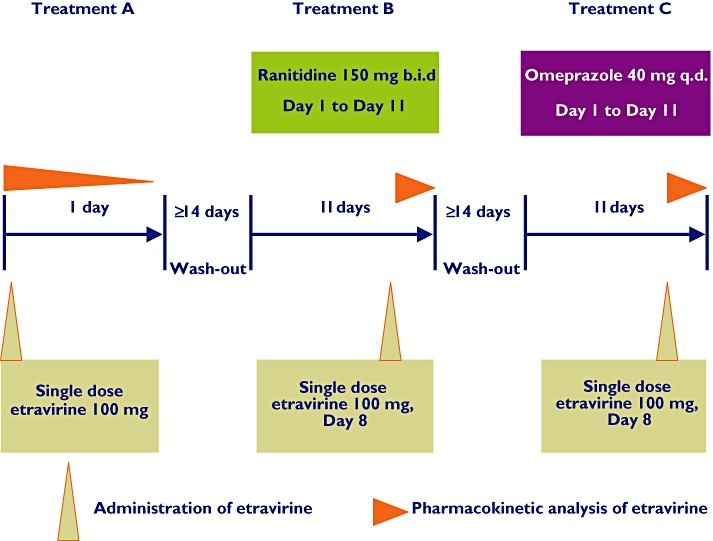
Trial design scheme
Screening of volunteers took place within 21 days before the first administration of trial medication.
For treatment A (etravirine alone) volunteers were admitted to the research unit on day –1 and remained confined until day 2. Both treatment B (ranitidine) and treatment C (omeprazole) started with a visit to the unit. Drug intake was continued at home and recorded, as well as the intake of relevant meals, in the subject's diary. Volunteers were readmitted to the research unit on day 7 and discharged on day 9. Etravirine was co-administered on day 8. Both ranitidine and omeprazole intakes were continued until day 11. Daily safety visits and pharmacokinetic sampling took place during each treatment up to 3 days after discharge. There was a wash-out period of minimally 14 days between each administration of etravirine and the start of the next period. Additional safety visits were planned on days 7, 9 and 11 after each administration of etravirine. The last follow-up visit was performed on day 31 after the last administration of etravirine.
Etravirine (treatment A) was administered as a single 100-mg tablet of the Phase 3 formulation and was to be ingested within 10 min after the completion of a standardized breakfast. In treatment B, ranitidine (Zantac®) was administered at the current recommended oral dose regimen of 150 mg twice daily, 1 h before breakfast or dinner. In treatment C, omeprazole was administered as one 40-mg tablet of the controlled release formulation Losec MUPS 40® 1 h before breakfast. All drug intakes took place with approximately 200 ml of water.
Safety assessments
Adverse events (AEs) were evaluated throughout the trial. During drug administration at home, volunteers were requested to record all AEs in their diary. Severity and drug relationship of AEs to etravirine, ranitidine and/or omeprazole were assessed. Laboratory assessments, ECG, vital signs assessments and physical examinations were performed at specific time points. AEs were coded according to the Medical Dictionary for Regulatory Activities dictionary (version 8.0); laboratory data were evaluated using the National Institute of Allergy and Infections Diseases Division of AIDS toxicity scale.
Pharmacokinetic and statistical analyses
Etravirine
Plasma samples for the bioanalysis of etravirine were collected on days 1–5 of treatment A, and on days 8–12 of treatment B and C before dosing, at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 32, 48, 72 and 96 h after dose administration. Plasma samples were stored at ≤−18°C until assayed. Plasma concentrations of etravirine were determined using validated liquid chromatography mass spectrometry/mass spectrometry (LC-MS/MS) methods. The internal standard (IS) used was a stable isotope labelled etravirine-d4. Plasma was extracted once using tertiary butyl methyl ether under alkaline conditions. Isocratic chromatographic separation was achieved at 40 °C on a 3.5-µm Symmetry Shield RP18 (Waters, Milford, MA, USA; 2.1 i.d. × 50 mm) at 0.3 ml min−1. The elution mixture consisted of a formic acid solution (0.1%) and acetonitrile (20:80 v/v ratio). Detection was done by tandem MS (API3000 instrument; Applied Biosystems, Foster City, CA, USA) in the electrospray positive multiple reaction monitoring (MRM) mode. MRM transitions were m/z 435 to 165 and m/z 440 to 164 for etravirine and the IS, respectively. The effective linear range was 2.00–5000 ng ml−1 with a lower limit of quantification of 2.00 ng ml−1. Intrabatch precision varied between 1.5 and 7.7% (CV%) and intrabatch accuracy varied between 93.3 and 108.3%.
Metabolites of etravirine (M8 and M12)
Pharmacokinetic samples taken during treatment with etravirine alone (treatment A) and during co-administration with omeprazole (treatment C) at time points predose, 2, 4, 6, 8, 12 and 24 h were analysed post hoc using LC-MS/MS methodology. Fifty-microlitre aliquots of plasma were precipitated using methanol, followed by acetonitrile. After thorough vortex mixing and centrifugation, 2 µl of the clear supernatant was injected onto an high-performance liquid chromatography-MS/MS system (API4000; Applied Biosystems). Chromatographic separation was done on a 3.5-µm X-bridge C18 column (Waters; 4.6 mm i.d. × 50 mm) at 1.2 ml min−1, applying gradient elution. The elution mixture consisted of formic acid in water (0.1%, v/v) and acetonitrile. Quantification was based on MRM mass spectrometric detection: for M8 m/z 451.1 to 353, and for M12 m/z 469.1 to 369. The linear range was set at 2.00–2000 ng ml−1 for each metabolite. Study samples were preceded by the calibration curve in every analytical batch and bracketed by sets of quality control (QC) samples, which were independently prepared, at four different concentration levels. An analytical batch was accepted when at least 2/3 (66.7%) of all the QC results and at least 50% at each concentration were within 15.0% of their respective nominal value. Intrabatch accuracy and precision results at the level of the QC samples were 92.7–112.6% and 0.4–10.1% (CV%) for M8 and 84.7–115.3% and 2.1–16.9% (CV%) for M12, respectively.
Pharmacokinetic and statistical analyses of plasma concentrations of etravirine (planned) and its metabolites (post hoc), and the statistical analysis of the pharmacokinetic parameters were performed using WinNonlin Professional (version 4.1; Pharsight Corp., Mountain View, CA, USA), Microsoft Excel® (Microsoft, Redmond, WA, USA) and SAS (SAS Institute Inc., Cary, NC, USA). Noncompartmental analysis model 200 (extravascular input, plasma data) was applied for the pharmacokinetic analysis. The maximum plasma concentration (Cmax) and time to reach Cmax (tmax) were obtained by inspection of the plasma concentration–time profiles. The value of the area under the plasma concentration–time curve from the time of administration up to the last time point with measurable plasma concentrations (AUClast) was determined using the linear trapezoidal rule.
Descriptive statistics were calculated for the plasma concentrations and pharmacokinetic parameters of etravirine and metabolites. Statistical analyses were performed for etravirine using co-administration with ranitidine or omeprazole as test (treatment B or C) and treatment with etravirine alone (treatment A) as reference. The primary pharmacokinetic parameters used in the statistical analysis were Cmax, and AUClast on the logarithmic scale. All available observations were included in the statistical analysis. The least squares means (LSmeans) of the primary parameters for both treatments were estimated with a linear mixed effects model, controlling for treatment, sequence and period as fixed effect, and subject as a random effect. A 90% CI was constructed around the difference between the LSmeans of test and reference. Both the difference between the LSmeans and the 90% confidence limits were retransformed to the original scale. Safety and pharmacodynamic parameters were evaluated by means of descriptive statistics and frequency tabulations.
Results
The trial was conducted in one centre in the Netherlands. Seven female and 12 male volunteers with a median age of 49 years (range 27–54) were enrolled. The median BMI was 26 kg m−2 (range 21–30); 16 (84.2%) volunteers were White, one was Black, one of mixed (Black/White) and one of Hispanic origin. At screening, all volunteers tested negative for hepatitis, HIV, drug screen tests and alcohol breath test. No clinically relevant findings in the medical history or currently active disorders were reported.
Two volunteers were prematurely discontinued from the trial on day 8 of treatment B and C, respectively, due to a dosing error, and one volunteer withdrew consent after treatment A (replaced). One volunteer was noncompliant with the trial medication intake during treatment B. Full pharmacokinetic profiles of etravirine were available for 18 volunteers for treatment A, 16 volunteers for treatment B and 17 for treatment C.
Pharmacokinetics of etravirine
The mean plasma concentrations of etravirine were slightly lower when co-administered with ranitidine and higher when etravirine was given with omeprazole (Figure 2). On average, the first measurable etravirine plasma concentrations were observed 1 h post dose and maximum concentrations were reached from 2 to 6 h after the administration of etravirine in all treatments. Plasma concentration–time curves were similar after the administration of etravirine alone and when co-administered with ranitidine. After co-administration with omeprazole, the plasma concentrations of etravirine were in general higher compared with administration of etravirine alone.
Figure 2.
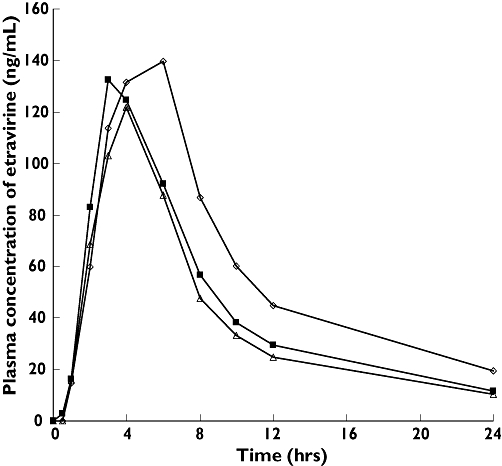
Mean plasma concentration–time curves of a single dose of 100 mg etravirine with or without co-administration of ranitidine 150 mg b.i.d. or omeprazole 40 mg q.d. etravirine 100 mg alone (n = 18), (▪); ranitidine 150 mg b.i.d + etravirine 100 mg (n = 16), (▵); omeprazole 400 mg q.d. + etravirine 100 mg (n = 17), (◊)
The mean value of AUClast was slightly lower when etravirine was co-administered with ranitidine and higher after co-administration with omeprazole (Table 1). Based on the ratio of LSmeans, AUClast was decreased by 14% when etravirine was given with ranitidine and increased by 41% after co-administration with omeprazole. No significant changes in Cmax were observed in any of the treatments. Interindividual variability of the primary pharmacokinetic parameters was comparable for treatments A and B (46–55%), but lower for treatment C (31–34%).
Table 1.
Pharmacokinetic parameters of etravirine without (treatment A) and with ranitidine (treatment B) or omeprazole (treatment C) after administration of a single 100-mg tablet of etravirine
| Pharmacokinetic parameter* | Etravirine 100 mg alone Treatment A (reference) | Ranitidine + etravirine 100 mg Treatment B (test) | Omeprazole + etravirine 100 mg Treatment C (test) | LSmeans ratio test/reference (90% CI) | |
|---|---|---|---|---|---|
| Ranitidine B vs. A< | Omeprazole C vs. A | ||||
| n | 18 | 16 | 17 | – | – |
| tmax, h | 3.0 (2.0–6.0) | 4.0 (2.0–6.0) | 4.0 (3.0–6.0) | – | – |
| Cmax, ng ml−1 | 146 ± 69 | 141 ± 78 | 165 ± 51 | 0.94 (0.75, 1.17) | 1.17 (0.96, 1.43) |
| AUClast, ng h ml−1 | 1501 ± 686 | 1257 ± 653 | 2113 ± 670 | 0.86 (0.76, 0.97) | 1.41 (1.22, 1.62) |
Values are shown as mean ± SD except tmax, which is shown as median (range).
Pharmacokinetics of the metabolites of etravirine (M8 and M12)
Metabolite M12 showed slightly higher mean plasma concentrations when etravirine was given alone compared with when co-administered with omeprazole. In contrast, plasma concentrations of metabolite M8 were substantially lower when etravirine was given concomitantly with omeprazole than when given alone (data not shown). The AUC24h of M12 was approximately 35% higher and that of M8 67% lower after administration with omeprazole. The parent/metabolite ratio for the AUC24h of M12 showed no relevant change: 6.14 ± 1.68 (mean ± SD) when etravirine was administered alone vs. 6.30±1.55 when etravirine was given with omeprazole. The parent/metabolite ratio for metabolite M8 increased from 3.22 (SD ± 1.66) to 14.12 (SD ± 7.95) (Figure 3) when etravirine was administered alone or with omeprazole, respectively.
Figure 3.
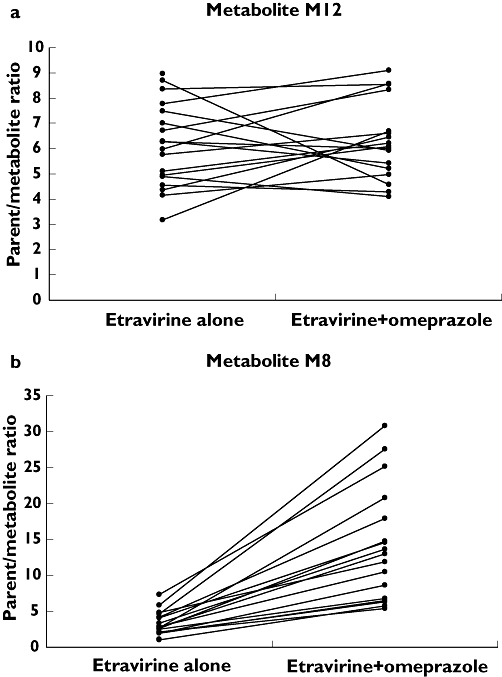
Ratio of AUC24h of the parent drug vs. the etravirine metabolites M12 (a) and M8 (b) after the administration of a single dose of 100 mg etravirine alone and when co-administered with omeprazole 40 mg q.d. on day 8
Safety
No volunteers discontinued the trial due to an AE. Most AEs were mild or moderate in severity. The two most frequently reported AEs during the trial were headache (12 volunteers, 63%) and somnolence (eight volunteers, 42%), most of which occurred during omeprazole treatment. Two volunteers reported a grade 3 (severe) AE: one case of diarrhoea in the wash-out period 8 days after treatment with etravirine alone, doubtfully related to etravirine, and one case of increased lipase (grade 3) during the co-administration of etravirine and omeprazole, possibly related to both agents. Both events resolved without intervention. No grade 4 or serious AEs were reported. There were no consistent or clinically relevant changes in physical examinations, laboratory assessments, vital signs or ECG parameters.
Discussion
In our study, co-administration of a single tablet of 100 mg etravirine in HIV-negative volunteers treated with ranitidine 150 mg b.i.d. or omeprazole 40 mg once daily resulted in 14% lower and 41% higher exposure to etravirine, respectively, compared with administration alone, with no change or only a slight increase in Cmax, respectively.
The pharmacodynamic effects of the H2 receptor antagonist ranitidine on gastric pH are typically less pronounced than those of omeprazole. Ranitidine suppresses gastric acid production by competitive histamine receptor blockade, whereas omeprazole directly and irreversibly inhibits gastric acid secretion by suppressing the activity of the H+/K+ ATPase, or proton pump [36, 37]. Moreover, a rapid decline of acid inhibitory effect (tachyphylaxis) occurs during treatment with H2 receptor antagonists. Tolerance has been described already after 5–10 days of oral treatment with ranitidine 150–300 mg q.d. [36, 38]. Komazawa et al. have shown that after 14 days’ treatment with ranitidine 150 mg b.i.d. especially the nocturnal acid-inhibiting activity of ranitidine declined [39]. In our study, in order to allow enough time for enzyme induction or inhibition, a 7-day treatment regimen was chosen, which was potentially sufficient to develop tachyphylaxis to ranitidine. As no intragastric pH measurements were performed, the development of tolerance after 7 days of ranitidine treatment could not be assessed.
PPIs have been shown to be substrates and inhibitors of P-gp in vitro[40]. As no appreciable influence of P-gp on the uptake of etravirine has been demonstrated in vitro (data on file, Tibotec), the role of P-gp as a causative factor in the increased exposure to etravirine when co-administered with omeprazole is unlikely.
A 15–40% delay in t1/2 for gastric emptying during omeprazole treatment has been reported in the literature [41]. The rate of gastric emptying may play a role in drug absorption, enabling longer dissolution and mixing of the administered drug [42]. Since in this study etravirine was administered after the ingestion of a standardized breakfast, it is unlikely that the effect of omeprazole on gastric emptying as a single factor would lead to an additional 41% increase of exposure, although a minor effect cannot be excluded.
In order to gain a better understanding of the effect of omeprazole on etravirine pharmacokinetics, we performed a post hoc exploratory analysis of the two major metabolites of etravirine, M8 and M12. The formation of these metabolites has been shown to be catalysed by CYP2C19 and CYP3A4 [30]. An increase of the exposure to etravirine when co-administered with omeprazole was accompanied by an increase of exposure to metabolite M12 of approximately the same magnitude. Consequently, no change of the parent/metabolite ratio of AUC24h for M12 was demonstrated after the administration of etravirine with omeprazole. On the other hand, a substantial decrease of the AUC24h of M8 was observed, whereas exposure to the parent compound increased when etravirine was given in steady-state for omeprazole. This resulted in a more than four-fold increase of the parent/metabolite ratio for M8 (Figure 3). The effect of omeprazole on CYP3A4 has been shown to be either conflicting or demonstrating an induction [4, 22], which would ultimately result in a decrease of plasma concentrations of the parent compound and an increase of the metabolites, with a decrease of the metabolic ratio. Our findings therefore suggest a role of CYP2C19 in the metabolism of etravirine due to inhibition by omeprazole, affecting metabolite M8, rather than an effect on CYP3A4. Of note, an effect of reduced gastric acidity resulting in a slight reduction in the absorption of etravirine, as was shown with ranitidine but masked by metabolic changes when omeprazole is co-administered, can not be excluded.
Metabolic interactions with other PPIs will depend on the interaction potential of the co-administered drug. Evidence suggests that other drugs of this class have a weaker potential for interactions than omeprazole [4], therefore the magnitude of the interaction is not expected to be higher when etravirine is co-administered with these agents.
The 41% increase of exposure to etravirine is not considered to be clinically relevant. In both ongoing Phase 3 trials (DUET-1 and DUET-2) in 577 treatment-experienced patients, the safety and tolerability profile of etravirine, administered as 200 mg b.i.d. for 24 and 48 weeks, was generally comparable to placebo, except for rash [26–29]. Median exposure to etravirine assessed by population pharmacokinetic methods was 4451 ng h ml−1 (range 458–56 279 ng h ml−1). The use of omeprazole was allowed in both trials. No relationship was observed between the incidence and severity of AEs and pharmacokinetic parameters of etravirine [43, 44].
In conclusion, co-administration with drugs that increase gastric pH has no clinically relevant effect on the pharmacokinetics of etravirine. The administration of etravirine after pretreatment of 8 days with ranitidine resulted in a slight, but not clinically relevant 14% decrease of exposure to etravirine. Exposure to etravirine was 41% higher when co-administered with omeprazole. This increase is attributed to a metabolic interaction due to CYP2C19 inhibition by omeprazole and is clinically not relevant, considering the favourable safety profile of etravirine and the lack of any relationship of etravirine pharmacokinetics with adverse effects. Etravirine can therefore be co-administered with PPIs and H2 antagonists without dose adjustments.
Competing interests
All authors are employees of Tibotec, except for M-P.B., who is an employee of Johnson & Johnson.
Funding for this study was provided by Tibotec. The results of this study have been partly presented at the XVIth International AIDS Conference, Toronto, Canada, 13–18 August 2006, poster TUPE0082.
REFERENCES
- 1.Béïque L, Giguère P, la Porte C, Angel J. Interactions between protease inhibitors and acid-reducing agents: a systematic review. HIV Med. 2007;8:335–45. doi: 10.1111/j.1468-1293.2007.00482.x. [DOI] [PubMed] [Google Scholar]
- 2.Van Lunzen J, Liess H, Arasteh K, Walli R, Daut B, Schürmann D. Concomitant use of gastric acid-reducing agents is frequent among HIV-1-infected patients receiving protease inhibitor-based highly active antiretroviral therapy. HIV Med. 2007;8:220–5. doi: 10.1111/j.1468-1293.2007.00456.x. [DOI] [PubMed] [Google Scholar]
- 3.Luber A, Garg V, Gharakhanian S, Vertex HIV Program Team Survey of medication used by HIV-infected patients that affect gastrointestinal (GI) acidity and potential for negative drug interactions with HAART. 7th International Congress on Drug Therapy in HIV Infection. Glasgow, UK, November 2004 [Abstract 206.
- 4.Blume H, Donath F, Warnke A, Schug BS. Pharmacokinetic drug interaction profiles of proton pump inhibitors. Drug Saf. 2006;29:769–84. doi: 10.2165/00002018-200629090-00002. [DOI] [PubMed] [Google Scholar]
- 5.Gerson LB, Triadafilopoulos G. Proton pump inhibitors and their drug interactions: an evidence-based approach. Eur J Gastroenterol Hepatol. 2001;13:611–6. doi: 10.1097/00042737-200105000-00025. [DOI] [PubMed] [Google Scholar]
- 6.Luber AD, Brower R, Kim D, Silverman R, Peloquin CA, Frank I. Steady-state pharmacokinetics of once-daily fosamprenavir/ritonavir and atazanavir/ritonavir alone and in combination with 20 mg omeprazole in healthy volunteers. HIV Med. 2007;8:457–64. doi: 10.1111/j.1468-1293.2007.00496.x. [DOI] [PubMed] [Google Scholar]
- 7.Agarwala S, Gray K, Wang Y, Grasela D. Pharmacokinetic effect of omeprazole on atazanavir with ritonavir in healthy subjects. 12th Conference on Retroviruses and Opportunistic Infections; February; Boston, MA. Abstract. [Google Scholar]
- 8.Tappouni HL, Rublein JC, Donovan BJ, Hollowell SB, Tien HC, Min SS, Theodore D, Rezk NL, Smith PC, Tallman MN, Raasch RH, Kashuba AD. Effect of omeprazole on the plasma concentrations of indinavir when administered alone and in combination with ritonavir. Am J Health Syst Pharm. 2008;65:422–8. doi: 10.2146/ajhp070226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ford SL, Wire MB, Lou Y, Baker KL, Stein DS. Effect of antacids and ranitidine on the single-dose pharmacokinetics of fosamprenavir. Antimicrob Agents Chemother. 2005;49:467–9. doi: 10.1128/AAC.49.1.467-469.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sekar VJ, Lefebvre E, De Paepe E, De Marez T, De Pauw M, Parys W, Hoetelmans RMW. Pharmacokinetic interaction between darunavir boosted with ritonavir and omeprazole or ranitidine in human immunodeficiency virus-negative healthy volunteers. Antimicrob Agents Chemother. 2007;51:958–61. doi: 10.1128/AAC.01203-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.La Porte CJL, Cameron DW, Sabo JP, Murray GE, Fagan N, Bosisio M, Jones P, De Perri G. The effect of omeprazole, food and formulation on the pharmacokinetics of tipranavir coadministered with ritonavir (TPV/r). 8th International Workshop on Clinical Pharmacology of HIV Therapy; Budapest, Hungary, April 2007 [Abstract 59. [Google Scholar]
- 12.Klein C, Chiu YL, Cai Y, Beck K, King K, Causemaker S, Doan T, Esslinger HU, Podsadecki T, Hanna G. Lack of effect of acid reducing agents on the pharmacokinetics of lopinavir/ritonavir tablet. 13th CROI; Denver, CO, February 2006 [Abstract 578. [Google Scholar]
- 13.Overton ET, Tschampa J, Klebert M, Rodriguez M, Spitz T, Kim G, Mondy K, Acosta E. Acid reduction with a proton pump inhibitor does not effect pharmacokinetics of lopinavir or ritonavir in HIV-infected subjects on lopinavir/ritonavir-based therapy. 8th International Workshop on Clinical Pharmacology of HIV Therapy; Budapest, Hungary, April 2007 [Abstract 60. [Google Scholar]
- 14.Chiu YL, Klein CE, Woodward WC, King KR, Naylor C, Awni W, Brun S. Lack of effect gastric acid-reducing agents on the pharmacokinetics of lopinavir/ritonavir in HIV-infected patients. AIDS Patient Care STDS. 2007;21:247–51. doi: 10.1089/apc.2006.0120. [DOI] [PubMed] [Google Scholar]
- 15.Winston A, Back D, Fletcher C, Robinson L, Unsworth J, Tolowinska I, Schutz M, Pozniak AL, Gazzard B, Boffito M. Effect of omeprazole on the pharmacokinetics of saquinavir-500 mg formulation with ritonavir in healthy male and female volunteers. AIDS. 2006;20:1401–6. doi: 10.1097/01.aids.0000233573.41597.8a. [DOI] [PubMed] [Google Scholar]
- 16.Singh K, Dickinson L, Chaikan A, Back D, Fletcher C, Pozniak A, Moyle G, Nelson M, Gazzard B, Herath D, Boffito M. Pharmacokinetics and safety of saquinavir/ritonavir and omeprazole in HIV-infected subjects. Clin Pharmacol Ther. 2007 doi: 10.1038/sj.clpt.6100375. Sep 26 [E pub ahead of print], PMID: 17898705. [DOI] [PubMed] [Google Scholar]
- 17.Martinez C, Albet C, Agundez JAG, Herrero E, Carrillo JA, Marquez M, Benitez J, Ortiz JA. Comparative in vitro and in vivo inhibition of cytochrome P450 CYP1A2, CYP2D6 and CYP3A by H2-receptor antagonists. Clin Pharmacol Ther. 1999;65:369–76. doi: 10.1016/S0009-9236(99)70129-3. [DOI] [PubMed] [Google Scholar]
- 18.Andersson T, Cederberg C, Edvardsson G, Heggelund A, Lundborg P. Effect of omeprazole treatment on diazepam plasma levels in slow versus normal rapid metabolisers of omeprazole. Clin Pharmacol Ther. 1990;47:79–85. doi: 10.1038/clpt.1990.12. [DOI] [PubMed] [Google Scholar]
- 19.Ishizaki T, Chiba K, Manabe K, Koyama E, Hayashi M, Yasuda S, Horai Y, Tomono Y, Yamato C, Toyoki T. Comparison of the interaction potential of a new proton pump inhibitor, E3810, versus omeprazole with diazepam in extensive and poor metabolizers of S-mephenytoin 4′hydroxylation. Clin Pharmacol Ther. 1995;58:155–64. doi: 10.1016/0009-9236(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 20.Blohme I, Idström JP, Andersson T. A study of the interaction between omeprazole and cyclosporine in renal transplant patients. Br J Clin Pharmacol. 1993;35:156–60. [PMC free article] [PubMed] [Google Scholar]
- 21.Caraco Y, Wilkinson GR, Wood AJJ. Differences between white subjects and Chinese subjects in the in vivo inhibition of cytochrome P450s 2C19, 2D6, and 3A by omeprazole. Clin Pharmacol Ther. 1996;60:396–404. doi: 10.1016/S0009-9236(96)90196-4. [DOI] [PubMed] [Google Scholar]
- 22.Lemahieu PD, Maes BD, Verbeke K, Vanrenterghem Y. Impact of gastric acid suppressants on cytochrome P450 3A4 and P-glycoprotein: consequences for FK506 assimilation. Kidney Int. 2005;67:1152–60. doi: 10.1111/j.1523-1755.2005.00182.x. [DOI] [PubMed] [Google Scholar]
- 23.Andries K, Azijn H, Thielemans T, Ludovici D, Kukla M, Heeres J, Janssen P, De Corte B, Vingerhoets J, Pauwels R, de Bethune MP. TMC125, a novel next-generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2004;48:4680–6. doi: 10.1128/AAC.48.12.4680-4686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vingerhoets J, Azijn H, Fransen E, De Baere I, Smeulders L, Jochmans D, Andries K, Pauwels R, de Bethune MP. TMC125 displays a high genetic barrier to the development of resistance: evidence from in vitro selection experiments. J Virol. 2005;79:12773–82. doi: 10.1128/JVI.79.20.12773-12782.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen C, Steinhart CR, Ward DJ, Ruane P, Vingerhoets J, Peeters M, White A, Baeten B, de Bethune M, Woodfall B. Efficacy and safety results at 48 weeks with the novel NNRTI, TMC125, and impact of baseline resistance on the virologic response in study TMC125-C223. 16th International AIDS Conference; 2006; Toronto, Canada. [Abstract TUPE0061. [Google Scholar]
- 26.Madruga JV, Cahn P, Grinsztejn B, Haubrich R, Lalezari J, Mills A, Pialoux G, Wilkin T, Peeters M, Vingerhoets J, De Smedt G, Leopold L, Trefiglio R, Woodfall B. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-1: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet. 2007;370:29–38. doi: 10.1016/S0140-6736(07)61047-2. [DOI] [PubMed] [Google Scholar]
- 27.Lazzarin A, Campbell T, Clotet B, Johnson M, Katlama C, Moll A, Towner W, Trottier B, Peeters M, Vingerhoets J, De Smedt G, Baeten B, Beets G, Sinha R, Woodfall B. Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-2: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet. 2007;370:39–48. doi: 10.1016/S0140-6736(07)61048-4. [DOI] [PubMed] [Google Scholar]
- 28.Haubrich R, Cahn P, Grinsztejn B, Lalezari J, Madruga J, Mills A, Peeters M, Vingerhoets J, Iveson K, De Smedt G. DUET-1: week 48 results of a phase III randomized double-blind trial to evaluate the efficacy and safety of TMC125 versus placebo in 612 treatment-experienced HIV-1 infected patients. 15th CROI; Boston, MA, February 2008 [Poster 790. [Google Scholar]
- 29.Johnson M, Campbell T, Clotet B, Katlama C, Lazzarin A, Towner W, Peeters M, Vingerhoets J, Bollen S, De Smedt G. DUET-1: week 48 results of a phase III randomized double-blind trial to evaluate the efficacy and safety of TMC125 versus placebo in 591 treatment-experienced HIV-1 infected patients. 15th CROI; February 2008; Boston, MA. [Poster 791. [Google Scholar]
- 30.Schöller-Gyüre M, Raoof A, Mannens G, Lachau-Durand S, Woodfall B, Peeters M, Vyncke V, Vandermeulen K, Hoetelmans RM. Mass-balance of 14C-labelled TMC125 in healthy volunteers. 8th International Workshop of Clinical Pharmacology; April 2006; Budapest, Hungary. [Abstract 78. [Google Scholar]
- 31.Kakuda TN, Schöller-Gyüre M, Woodfall BJ, De Smedt G, Peeters M, Vandermeulen K, Hoetelmans R. TMC125 in combination with other medications: summary of drug–drug interactions. 8th International Congress on Drug Therapy in HIV Infection; November 2006; Glasgow, UK. [Abstract PL5.2. [Google Scholar]
- 32.Schöller-Gyüre M, Leemans R, Vyncke V, Vandermeulen K, Peeters M, Baeten B, Debroye C, Woodfall B, Hoetelmans R. Effect of food on the oral bioavailability of the phase III formulation of TMC125. 7th International Workshop of Clinical Pharmacology; April 2006; Lisbon, Portugal. [Abstract 80. [Google Scholar]
- 33.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–20. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 34.Schöller M, Hoetelmans R, Beets G, Vandermeulen K, Peeters M, Bastiaanse L, Leemans R, Debroye C, Woodfall B. Substantial improvement of oral bioavailability of TMC125 using new tablet formulations in healthy volunteers. 6th International Workshop of Clinical Pharmacology; April 2005; Quebec, Canada. [Abstract 6.8. [Google Scholar]
- 35.Caraco Y, Lagerstrom PO, Wood AJJ. Ethnic and genetic determinants of omeprazole disposition and effect. Clin Pharmacol Ther. 1996;60:157–67. doi: 10.1016/S0009-9236(96)90131-9. [DOI] [PubMed] [Google Scholar]
- 36.Ley LM, Becker A, Lühmann R, Sander P, Lücker PW. Pharmacodynamic effects of 3-day intravenous treatment with pantoprazole or ranitidine after 10 days of oral ranitidine. Methods Find Exp Clin Pharmacol. 2005;27:25–9. doi: 10.1358/mf.2005.27.1.875433. [DOI] [PubMed] [Google Scholar]
- 37.Blum RA, Shi H, Karol MD, Greski-Rose PA, Hunt RH. The comparative effects of lansoprazole, omeprazole, and ranitidine in suppressing gastric acid secretion. Clin Ther. 1997;19:1013–23. doi: 10.1016/s0149-2918(97)80053-7. [DOI] [PubMed] [Google Scholar]
- 38.Lachmann L, Howden CW. Twenty-four-hour intragastric pH: tolerance within 5 days of continuous ranitidine administration. Am J Gastroenterol. 2000;95:57–61. doi: 10.1111/j.1572-0241.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 39.Komazawa Y, Adachi K, Mihara T, Ono M, Kawamura A, Fujishiro H, Kinoshita Y. Tolerance to famotidine and ranitidine treatment after 14 days of administration in healthy subjects without Helicobacter pylori infection. J Gastroenterol Hepatol. 2003;18:678–82. doi: 10.1046/j.1440-1746.2003.03041.x. [DOI] [PubMed] [Google Scholar]
- 40.Pauli-Magnus C, Rekersbrink S, Klotz U, Fromm MF. Interaction of omeprazole, lanzoprazole and pantoprazole with P-glycoprotein. Naunyn Schmiedebergs Arch Pharmacol. 2001;346:551–7. doi: 10.1007/s00210-001-0489-7. [DOI] [PubMed] [Google Scholar]
- 41.Tougas G, Earnest DL, Chen Y, Vanderkoy C, Rojavin M. Omeprazole delays gastric emptying in healthy volunteers: an effect prevented by tegaserod. Aliment Pharmacol Ther. 2005;22:59–65. doi: 10.1111/j.1365-2036.2005.02528.x. [DOI] [PubMed] [Google Scholar]
- 42.Fleisher D, Li C, Zhou Y, Pao L, Karim A. Drug, meal and formulation interactions influencing drug absorption after oral administration. Clin Pharmacokinet. 1999;36:233–54. doi: 10.2165/00003088-199936030-00004. [DOI] [PubMed] [Google Scholar]
- 43.Kakuda TN, Schöller-Gyüre M, Peeters M, Vingerhoets J, Corbett C, Woodfall BJ, Hoetelmans RM. Pharmacokinetics and pharmacodynamics of TMC125 in HIV-infected patients with non-nucleoside reverse transcriptase inhibitor and protease inhibitor resistance: TMC125-C223. 14th CROI; February 2007; Los Angeles, CA. [Abstract L-131. [Google Scholar]
- 44.Kakuda TN, Wade JR, Snoek E, Peeters M, Corbett C, De Smedt G, Leopold L, Vingerhoets J, Woodfall B, Hoetelmans RM. Pharmacokinetics (PK) and pharmacodynamics (PD) of the non-nucleoside reverse transcriptase inhibitor (NNRTI) TMC125 in treatment-experienced HIV-1 infected patients: pooled 24-week results of DUET-1 and DUET-2. 15th CROI; 2008; Boston, MA. [Abstract L-160. February. [DOI] [PubMed] [Google Scholar]