

Abstract
AIMS
To examine the allelic variation of three enzymes involved in 6-mercaptopurine/azathioprine (6-MP/AZA) metabolism and evaluate the influence of these polymorphisms on toxicity, haematological parameters and metabolite levels in patients with acute lymphoblastic leukaemia (ALL) or inflammatory bowel disease (IBD).
METHODS
Clinical data and blood samples were collected from 19 ALL paediatric patients and 35 IBD patients who were receiving 6-MP/AZA therapy. All patients were screened for seven genetic polymorphisms in three enzymes involved in mercaptopurine metabolism [xanthine oxidase, inosine triphosphatase (C94→A and IVS2+21A→C) and thiopurine methyltransferase]. Erythrocyte and plasma metabolite concentrations were also determined. The associations between the various genotypes and myelotoxicity, haematological parameters and metabolite concentrations were determined.
RESULTS
Thiopurine methyltransferase variant alleles were associated with a preferential metabolism away from 6-methylmercaptopurine nucleotides (P = 0.008 in ALL patients, P = 0.038 in IBD patients) favouring 6-thioguanine nucleotides (6-TGNs) (P = 0.021 in ALL patients). Interestingly, carriers of inosine triphosphatase IVS2+21A→C variants among ALL and IBD patients had significantly higher concentrations of the active cytotoxic metabolites, 6-TGNs (P = 0.008 in ALL patients, P = 0.047 in IBD patients). The study confirmed the association of thiopurine methyltransferaseheterozygosity with leucopenia and neutropenia in ALL patients and reported a significant association between inosine triphosphatase IVS2+21A→C variants with thrombocytopenia (P = 0.012).
CONCLUSIONS
Pharmacogenetic polymorphisms in the 6-MP pathway may help identify patients at risk for associated toxicities and may serve as a guide for dose individualization.
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
6-Mercaptopurine (6-MP) and azathioprine (AZA) are both inactive prodrugs that require intracellular activation into the active 6-thioguanine nucleotides (6-TGNs).
This metabolic process undergoes three different competitive pathways that are catalysed by three different enzymes; xanthine oxidase (XO), thiopurine methyltransferase (TPMT) and inosine triphosphatase (ITPA), all of which exhibit genetic polymorphisms.
Although the impact of genetic variation in the TPMT gene on treatment outcome and toxicity has been demonstrated, the role of other polymorphisms remains less well known.
WHAT THIS STUDY ADDS
New information on the allelic variation of these three enzymes (XO, TPMT and ITPA) and their influence on 6-MP/AZA metabolism and toxicity.
Confirmation of the association of TPMT polymorphism with haematological toxicity.
Identified potential genetic characteristics that may contribute to higher risk of adverse events (such as ITPA IVS2+21A→C mutation).
Keywords: 6-mercaptopurine, acute lymphoblastic leukaemia, azathioprine, inflammatory bowel disease, pharmacogenetics
Introduction
The thiopurine medication 6-mercaptopurine (6-MP) and its prodrug azathioprine (AZA) are cytotoxic and immunosuppressive drugs currently used in the treatment of several diseases, including childhood acute lymphoblastic leukaemia (ALL) [1, 2] and inflammatory bowel disease (IBD) [3–5]. Both are inactive prodrugs that require intracellular activation by hypoxanthine guanine phosphoribosyltransferase into thioinosine monophosphate (TIMP) and subsequently into thioguanine nucleotides (6-TGNs), the active cytotoxic metabolite [6] (Figure 1). Two other reactions compete with this process. Xanthine oxidase (XO) oxidizes 6-MP into 6-thioxanthine (6-TX) and subsequently into 6-thiouric acid (6-TU). In addition, thiopurine S-methyltransferase (TPMT) converts 6-MP into inactive 6-methyl-MP and also metabolizes TIMP into 6-methylmercaptopurine nucleotides (6-mMPNs), which are potent inhibitors of de novo purine synthesis [7].
Figure 1.
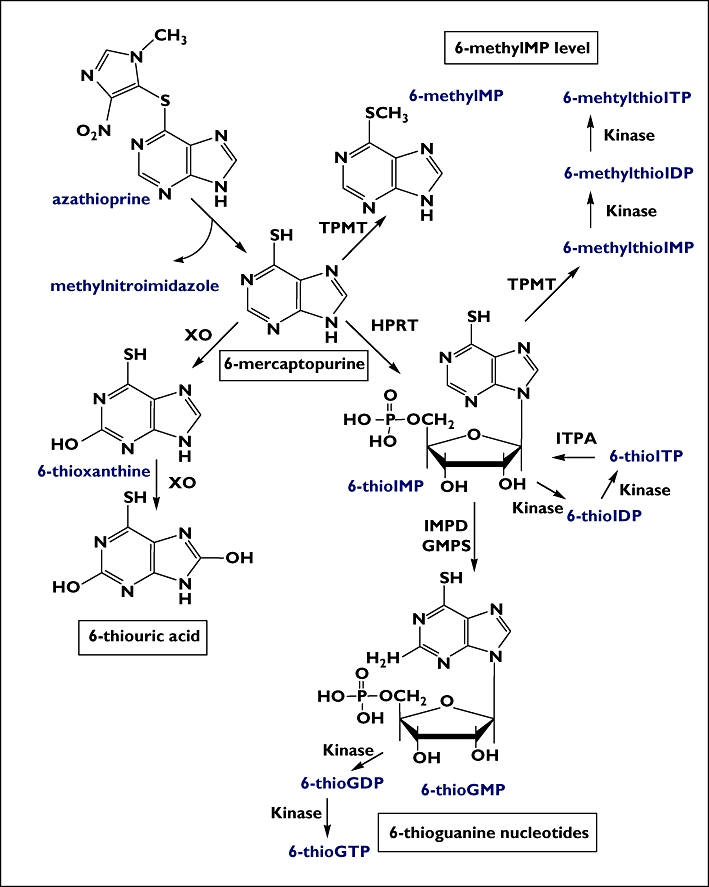
Thiopurine metabolism. 6-MP, mercaptopurine; IMP, inosine monophosphate; IDP, ioinosine diphosphate; ITP, ioinosine triphosphate; GMP, guanosine monophosphate; GDP, guanosine diphosphate; GTP, guanosine triphosphate; XO, xanthine oxidase; HPRT, hypoxanthine phosphoribosyltransferase; TPMT, thiopurine methyltransferase; IMPD, inosine monophosphate dehydrogenase; GMPS, guanosine monophosphate synthetase; ITPA, inosine triphosphate pyrophosphatase
The enzyme TPMT, which indirectly influences 6-TGN concentrations by shunting 6-MP metabolism away from 6-TGNs, is known to exhibit co-dominant genetic polymorphisms. To date, 20 mutant alleles responsible for TPMT deficiency have been described. TPMT*3A (3.2–5.7%) is the most common mutant allele in the White population, followed by TPMT*3C (0.2–0.8%), accounting for >95% of mutant alleles [8]. Genetic polymorphisms in such a key enzyme in 6-MP metabolism could lead to large interindividual variability in drug response and risk of toxicity. Hence, the prospective identification of the most prevalent TPMT mutant alleles (TPMT*3A, TPMT*3C) could identify individuals at higher risk of developing thiopurine-related adverse events. In addition to a high correlation between TPMT genotype and its phenotype [9], it has been reported that subjects with inherited TPMT deficiency treated with standard doses of thiopurines present higher levels of the active metabolites 6-TGNs and have increased risk of adverse events [10–12].
Although the impact of genetic variation in the TPMT gene on treatment outcome and toxicity has been demonstrated, the role of other polymorphisms remains less well known [13]. For example, polymorphisms in the gene that encodes the XO enzyme, an important enzyme involved in the first-pass metabolism of 6-MP, have not been studied. This enzyme, predominantly expressed in the intestinal mucosa and liver, quickly metabolizes 84% of 6-MP into inactive thiouric acid, resulting in substantial reduction in 6-MP bioavailability. In a study using hepatectomy and liver biopsy specimens for the study of hepatocyte XO activity in adults, the average hepatic XO activity was 21% higher in samples from male patients than in those from female patients, and a fourfold interindividual variation was described for hepatic XO activity amongst subjects of each sex [14]. This variability in first-pass metabolism may contribute to the considerable variation in oral thiopurine pharmacokinetic parameters. Furthermore, the influence of interindividual variation in XO activity on the clinical outcome has yet to be clearly defined [15].
One theory has recently been proposed concerning the role of another polymorphic enzyme in the 6-MP pathway, inosine triphosphate (ITP) pyrophosphatase (ITPA). ITPA catalyses the pyrophosphohydrolysis of ITP into inosine monophosphate, thereby preventing the accumulation of ITP in normal cells. Deficiency of ITPA disrupts this cycle, leading to abnormal accumulation of the benign inosine nucleotide, ITP. However, in ITPA-deficient patients treated with AZA or 6-MP, accumulation of 6-thioITP is suggested, resulting in AZA or 6-MP toxicity (which is characterized by pancreatic toxicity, rash, neutropenia and gastrointestinal complaints) [16]. The most important mutations, identified to date, leading to ITPA deficiency are 94A→C and IVS2+21A→C [17]. The ITPA IVS2+21A→C allele frequency among Whites is about 13%, which is higher than that of 94A→C polymorphism (about 7%). Identification of such frequent mutations may have pharmacogenomic implications, since the abnormal metabolism of 6-MP/AZA in affected patients may lead to drug-related toxicity.
Numerous clinical studies have reported substantial interindividual differences in therapeutic response or toxicity of thiopurine therapy in ALL and IBD patients [18–25]. These differences are partly explained by the variable formation of active metabolites due to genetic polymorphisms of the key enzymes involved in thiopurine metabolism [8]. In this study, our aims were to examine the allelic variation of three enzymes involved in 6-MP metabolism (XO, TPMT and ITPA) and to evaluate the association between these variants and metabolite concentrations, the occurrence of adverse events or clinical outcome (as assessed by myelotoxicity and haematological parameters) in ALL and IBD patients receiving 6-MP/AZA therapy.
Methods
Study subjects
Acute lymphoblastic leukaemia
A total of 19 paediatric patients with ALL attending the Haematology and Oncology Outpatient Department at the Royal Belfast Hospital for Sick Children were prospectively enrolled in this study. Children were eligible for inclusion if they had started the continuous/maintenance phase of chemotherapy at least 1 month prior to enrolment and had received a constant daily dose of 6-MP for at least seven consecutive days. Patients who satisfied the above criteria but who had received intensification therapy or red blood cell (RBC) transfusion within the previous 2 months were excluded. The demographic details and clinical characteristics of ALL subjects are shown in Table 1.
Table 1.
The demographic and clinical characteristics of the study population
| Parameter* | ALL patients (n = 19) | IBD patients (n = 35) |
|---|---|---|
| Gender (F : M) | 6 : 13 (32% : 68%) | 19 : 16 (54% : 46%) |
| Age, median (range), years | 10 (3–17) | 39 (15–73) |
| Weight, median (range), kg | 33.4 (13.2–77.5) | 66.7 (33.6–104.3) |
| Surface area, median (range), m2 | 1.14 (0.59–2) | – |
| 6-MP daily dose, median (range), mg m−2 | 40 (5.88–76.47) | – |
| 6-MP daily dose, median (range), mg kg−1 | 1.42 (0.16–3.46) | – |
| AZA daily dose, median (range), mg kg–1† | – | 1.49 (0.47–3.17) |
| Haematological parameters | ||
| WBC, median (range), ×109 l−1 | 3.3 (1.2–9.1) | 6 (3.4–13.5) |
| PLT, median (range), ×109 l−1 | 282 (66–648) | 302 (148–601) |
| ANC, median (range), ×109 l−1 | 1.68 (0.3–8.3) | 4.24 (0.36–9.7) |
| Metabolite concentrations | ||
| 6-TGNs, median (range), pmol per 8 × 108 RBCs | 242.3 (56.1–1 114.15) | 241.7 (79.2–856.1) |
| 6-TU, median (range), pmol per 8 × 108 RBCs | 57.5 (14.3–251.7) | 33.2 (16.7–126.7) |
| 6-MP, median (range), pmol per 8 × 108 RBCs | 6.6 (2.6–43.8) | 5.2 (2.8–28.7) |
| 6-mMPNs, median (range) pmol per 8 × 108 RBCs | 5 466.1 (180.1–28 733.8) | 792.4 (100.8–13 837.8) |
F, female; M, male; WBC, white blood cell count; ANC, absolute neutrophil count; PLT, platelet count; RBCs, red blood cells; 6-MP, 6-mercaptopurine; AZA, azathioprine; 6-TGN, 6-thioguanine nucleotide; 6-TU, 6-thiouric acid; 6-mMPN, 6-methylmercaptopurine nucleotide.
AZA or AZA equivalent dose if 6-MP was administered [6-MP dose was converted to AZA dose using a conversion factor of 2.07 (Sandborn, 1996)].
Maintenance chemotherapy for ALL patients consisted of daily oral 6-MP (given as a single dose) and weekly oral methotrexate. 6-MP dose was titrated to target protocol dose of 75 mg m−2 day−1, adjusted for each child according to leucocyte count and the presence of clinically relevant infections. Additionally, a monthly dose of intravenous vincristine was given to all children irrespective of blood counts. Continuation of maintenance chemotherapy is administered usually for 2 or 3 years. The children had their full blood counts assessed at each clinic visit (every 2 weeks) for evaluation of bone marrow toxicity.
Inflammatory bowel disease
A total of 35 IBD patients who attended the Gastroenterology Outpatient Department at the Antrim Area Hospital (n = 14) and the Royal Victoria Belfast Hospital (n = 21) agreed to participate. Patients were eligible for inclusion if they had been prescribed AZA or 6-MP therapy for at least 1 month and had received a stable daily dose of AZA or 6-MP for at least seven consecutive days. Patients who satisfied the above criteria but who had received RBC transfusion within the previous 2 months were excluded. The demographic and clinical characteristics of IBD subjects are shown in Table 1.
Immunosuppressive therapy for IBD patients consisted of oral AZA (2–3 mg kg−1 day−1) or 6-MP (1–2 mg kg−1 day−1) given twice or three times daily and adjusted for each patient according to their leucocyte count and the presence of clinically relevant infections. Patients had their full blood counts assessed at each clinic visit for indications of bone marrow toxicity and/or serum liver enzymes measured to test for hepatic toxicity.
Study design
Blood samples for the measurement of 6-MP metabolite concentrations were taken from patients during the course of their routine medication. In the case of children with ALL, blood samples were obtained at a phase of treatment when they had an indwelling cannula for vincristine therapy and at least 8 h after the preceding 6-MP dose (the blood sample was taken prior to the administration of vincristine to prevent its interference with 6-MP metabolite analysis). In the case of patients with IBD, blood samples were obtained during their routine clinic visit when blood is taken for full blood count and/or liver enzyme tests.
The blood samples (1.5 ml in ALL and 4 ml in IBD) were collected in ethylenediamine tetraaceticacid tubes and kept on ice until centrifugation. Plasma separated from RBC was frozen while RBCs were washed twice with balanced salt solution, then suspended at a density of 8 × 108 RBCs per 200 µl and kept frozen until required for further processing. These latter samples for the determination of metabolite content were taken on a maximum of five occasions, over a 4-month span, per patient during the study period (6 months in the case of ALL study and 1 year for IBD study).
In addition to accurate information on dosing and times of sampling, the following data were collected for each patient: age, weight, height, gender, ongoing pathology (e.g. renal and/or hepatic impairment), concomitant drug therapy, clinical laboratory test results and records of any side-effects experienced.
Ethical considerations
Separate ethical applications were submitted regarding each study (ALL; IBD) and both were approved by the National Health Services Office for Research Ethics Committees in Northern Ireland. Patients were included in the study only after they (or their parents in case of children) had been fully informed and had signed the study consent form. In addition, verbal assent was obtained from older children (>10 years) before enrolment.
Assay of 6-MP metabolites
RBC and plasma concentrations of 6-MP and its metabolites, 6-TGNs, 6-TU, 6-TX and 6-mMPNs, were measured by a reversed high-performance liquid chromatography methodology that was developed earlier [26]. The intraday and interday coefficient of variation (CV %) for the developed assay ranged between 1.6 and 3.5%, 3.2 and 10.9%, 2.2 and 8.5%, 0.8 and 5.3%, 3.0 and 4.3% for 6-TGNs, 6-TU, 6-MP, 6-TX and 6-mMPNs in erythrocytes, and between 0.2 and 1.8%, 1.4 and 3.5%, 3.0 and 8.7%, 0.7 and 2.7%, 0.8 and 4.2% for these compounds in plasma, respectively. The lower limits of quantification were 13, 14, 3, 2, 95 pmol per 8 × 108 in erythrocytes and 2, 5, 2, 3, 20 ng ml−1 in plasma for 6-TGNs, 6-TU, 6-MP, 6-TX and 6-mMPNs, respectively.
Genotyping of TMPT, ITPA and XO
All patients were screened for seven common polymorphisms in three enzymes involved in 6-MP metabolism (XO, TPMT and ITPA) that are potentially linked to the pharmacodynamics, toxicity and treatment outcome of 6MP/AZA; two polymorphisms in XO (A1936→G and A2107→G) and ITPA (C94→A and IVS2+21A→C); and three polymorphisms in TPMT (TPMT*3A, TPMT*3B and TPMT*3C).
Genomic DNA was extracted from peripheral blood (1-ml sample per patient) using QIAmp® DNA Blood Mini Kit (Qiagen, Hilden, Germany). The concentration of the extracted DNA was quantified using the Quanti-iT™ PicoGreen dsDNA Quantitation kit (Invitrogen, Eugene, OR, USA) according to the manufacturer's instructions. Detection of the various single nucleotide polymorphisms (SNPs) in the enzymes' genetic loci was carried out using a validated TaqMan® genotyping assay obtained from Applied Biosystems (Foster City, CA, USA). The TaqMan® primer/probe set designed for each SNP allele was included in the kits; their sequences and catalogue numbers are provided in Table 2.
Table 2.
Single nucleotide polymorphism (SNP) characteristics and sequences of TaqMan® probes designed for allele detection as supplied by the manufacturer*
| SNP | Location | NCBI reference | Probe sequence | Reporter1 Dye | Reporter 2 Dye |
|---|---|---|---|---|---|
| XO A(1936)→G | Chr2 | rs17323225 | GTCTCATCATTACAAATTCCAGTTA[C/T]GTTACTCCCAGGAACATCATCAGCG | VIC | FAM |
| XO A(2107)→G | Chr2 | rs17011368 | GGTCCATAAAAGGAGTTGTTCTTTA[C/T]AGCATCCTGAGGATCACAAAGAAGT | VIC | FAM |
| TPMT G(460)→A | Chr6 | rs1800460 | TCACCTGGATTGATGGCAACTAATG[T/C]TCCTCTATCCCAAATCATGTCAAAT | VIC | FAM |
| TPMT A(719)→G | Chr6 | rs1142345 | TCTCATTTACTTTTCTGTAAGTAGA[C/T]ATAACTTTTCAAAAAGACAGTCAAT | VIC | FAM |
| ITPA C(94)→A | Chr20 | rs1127354 | CGTTCAGATTCTAGGAGATAAGTTT[A/C]CATGCACTTTGGTGGCACAGAAAAT | VIC | FAM |
| ITPA IVS2+21A→C | Chr20 | rs7270101 | TGACCGTATGTCTCTGTTTTGTTTT[A/C]TTTTTAAAAGATGGTTGGATTTCTC | VIC | FAM |
XO, xanthine oxidase; TPMT, thiopurine S-methyl transferase; ITPA, inosine triphosphatase; Chr, chromosome; NCBI, National Centre for Biotechnology Information. VIC and FAM are two different fluorescent reporter dyes available commercially from Applied Biosystems® (P/N 4316033).
Polymerase chain reactions (PCR) were performed in a 96-well microplate format in a 10-µl reaction mixture containing TaqMan® universal PCR master mix and using an amplification protocol of 95 °C for 15 min, followed by 50 cycles of 95 °C for 15 s, then 60 °C for 1.5 min. The following allelic discrimination analysis was carried out using the DNA Engine Opticon® 2 System (MJ Research Inc., Waltham, MA, USA).
Statistical analysis
Statistical analysis was performed using SPSS® computer software (version 15; SPSS Inc., Chicago, IL, USA). Results were expressed as the median and range values or as frequencies (in the case of allele carriers in patient groups).
Standard statistical methods were used to evaluate the various univariate relationships; the differences in allele frequencies and genotype distribution of each polymorphism between patients who developed myelotoxic events vs. those who did not were analysed using χ2 test with one degree of freedom or two-sided Fisher's exact test, as appropriate. To determine the association between the various genotypes and 6-MP metabolite concentrations, the haematological parameters or the frequency of myelotoxicity per patient, the Kolmogorov–Smirnov Z test was used. Two-tailed P-values of <0.05 were considered statistically significant. P-values were corrected for multiple testing as per the Bonferroni justification.
Serial measurements of 6-MP metabolite concentrations in ALL patients [normalized per dose per patient surface area (SA)] were plotted against the time of sample collection (in months), and the area under the curve (AUC1–4 months) was calculated as a summary measure of metabolite concentrations for each subject [27]. Since most IBD patients had single measurements only, an alternative measure [the simple mean of the normalized concentrations as per dose per patient weight (Wt)] was used when more than one concentration was measured per patient. These summary measures (areas under the curve or simple means) were then compared between patients having the various genotypes using the Kolmogorov–Smirnov Z test.
Results
XO, TPMT and ITPA genotypes
Allelic variants of XO, TPMT and ITPA genes and their frequencies in ALL and IBD patients are shown in Table 3. Among the 19 ALL patients enrolled in this study, 16 (84.2%) did not carry any of the tested TPMT variant alleles and were considered to be wild type (TPMT*1/*1). Only TPMT*3A and TPMT*3C were identified among TPMT variant alleles studied, and no subjects were homozygous for a mutated TPMT gene. Seven patients were heterozygous (one with each of XO variants, two had TMPT*3C, one had TPMT*3A, three had ITPA C94→A and two had ITPA IVS2+21A→C variant allele). One patient was combined heterozygous for XO variants, TPMT*3A and ITPA C94→A variant alleles. Another patient was combined heterozygous for TPMT*3C and ITPA IVS2+21A→C. Only one patient was homozygous for the variant allele ITPA IVS2+21A→C.
Table 3.
Allelic variants of XO, TPMT and ITPA genes and their frequencies among (a) 19 paediatric ALL patients (n = 38 alleles) and (b) 35 IBD patients (n = 70 alleles)
| Allelic variants | Alleles (n) | Allelic frequency (%) | Heterozygotes/homozygotes (n) | Heterozygotes/homozygotes (%) |
|---|---|---|---|---|
| (a) ALL patients | ||||
| XO A1936→G | 1 | 2.6 | 1/0 | 5.3/0 |
| XO A2107→G | 1 | 2.6 | 1/0 | 5.3/0 |
| TPMT*3A | 1 | 2.6 | 1/0 | 5.3/0 |
| TPMT*3B | 0 | 0 | – | – |
| TPMT*3C | 2 | 5.3 | 2/0 | 10.5/0 |
| ITPA C94→A | 3 | 7.9 | 3/0 | 15.8/0 |
| ITPA IVS2+21A→C | 4 | 10.5 | 2/1 | 10.5/5.3 |
| (b)IBD patients | ||||
| XO A1936→G | 3 | 4.3 | 3/0 | 8.5/0 |
| XO A2107→G | 3 | 4.3 | 3/0 | 8.5/0 |
| TPMT*3A | 4 | 5.7 | 4/0 | 11.4/0 |
| TPMT*3B | 0 | 0 | – | – |
| TPMT*3C | 0 | 0 | – | – |
| ITPA C94→A | 4 | 5.7 | 2/1 | 5.7/1.4 |
| ITPA IVS2+21A→C | 9 | 12.9 | 5/2 | 14.3/2.9 |
XO, xanthine oxidase; TPMT, thiopurine S-methyl transferase; ITPA, inosine triphosphatase; ALL, acute lymphoblastic leukaemia; IBD, inflammatory bowel disease.
Among the 35 IBD patients studied, 12 were heterozygous [three (8.5%) with each of the two XO variants, four (11.4%) had the allele TPMT*3A, two (5.7%) had ITPA C94→A, and five (14.3%) had the allele ITPA IVS2+21A→C]. Homozygosity was found in three patients (one for ITPA C94→A and two for ITPA IVS2+21A, but none for the TPMT variant alleles). Two patients were combined heterozygotes for ITPA IVS2+21A and XO variant alleles.
6-MP metabolite concentrations
On average, four blood samples per patient were assayed for 6-MP and its metabolite concentrations in children with ALL. The concentrations of 6-MP and its metabolites (6-TGNs, 6-TU, 6-TX and 6-mMPNs) were measured in both RBCs and plasma. Concentrations of 6-MP and 6-TX in plasma and of 6-TX in RBCs were very low (at or below the lower limit of quantification of the method of analysis) in comparison with other metabolites. Other 6-MP metabolites in plasma where slightly above the lower limit of quantification. The median concentrations of 6-MP metabolites in RBCs for ALL patients are shown in Table 1.
In the IBD population, 1.3 samples per patient were assayed on average. Plasma concentrations of 6-MP, 6-TX and 6-TU in IBD patients were 4–15-fold higher than in ALL patients due to the more frequent dosing and, therefore, the shorter sampling time in the IBD population (samples were collected at a time closer to dosage administration). However, 6-TX concentrations in RBCs were very low in comparison with the other metabolites. Medians and ranges of RBC concentrations of 6-MP metabolites for IBD patients are also shown in Table 1.
Correlation of XO, TPMT and ITPA genotypes with 6-MP metabolite concentrations
Both ALL and IBD populations were divided into different groups according to the various polymorphisms present in each patient. Summary measures (AUC1–4 months or means) of 6-MP and its metabolite concentrations (adjusted per dose/SA or dose/Wt) were compared among these groups to evaluate the association of 6-MP and its metabolite concentrations with XO, TPMT and ITPA genotypes among the two populations (Table 4). Figure 2 details potentially relevant associations between 6-MP metabolites and the various polymorphisms discussed below.
Table 4.
Significance of the association of 6-MP metabolite concentrations summary measures with XO, TPMT or ITPA variant genotypes in (a) ALL patients and (b) IBD patients treated with 6-MP/AZA*
| (a) ALL patients genotype | AUC of RBC metabolite concentrations (AUC1–4 months) | |||
|---|---|---|---|---|
| 6-TGNs | 6-mMPNs | 6-TU | 6-MP | |
| TPMT*3 heterozygote | 0.021 | 0.008 | 0.021 | 0.041 |
| ITPA C94→A heterozygote | 0.439 | 0.972 | 0.950 | 0.359 |
| ITPA IVS2+21A→C mutant† | 0.008 | 0.660 | 0.660 | 0.660 |
| (b)IBD patients Genotype | Mean RBC metabolite concentrations | |||
|---|---|---|---|---|
| 6-TGNs | 6-mMPNs | 6-TU | 6-MP | |
| XO heterozygote | 0.337 | 0.670 | 0.497 | 0.060 |
| TPMT*3 heterozygote | 0.866 | 0.038 | 0.187 | 0.670 |
| ITPA C94→A mutant | 0.670 | 0.154 | 0.822 | 0.497 |
| ITPA IVS2+21A→C mutant | 0.047 | 0.960 | 0.305 | 0.427 |
Numbers represent P-values for the corresponding association adjusted as per the Bonferroni justification for multiple testing.
Mutant refers to any mutant (homozygote or heterozygote). 6-MP, 6-mercaptopurine; XO, xanthine oxidase; TPMT, thiopurine S-methyl transferase; ITPA, inosine triphosphatase; ALL, acute lymphoblastic leukaemia; IBD, inflammatory bowel disease; AZA, azathioprine; 6-TGN, 6-thioguanine nucleotide; 6-mMPN, 6-methylmercaptopurine nucleotide; 6-TU, 6-thiouric acid.
Figure 2.
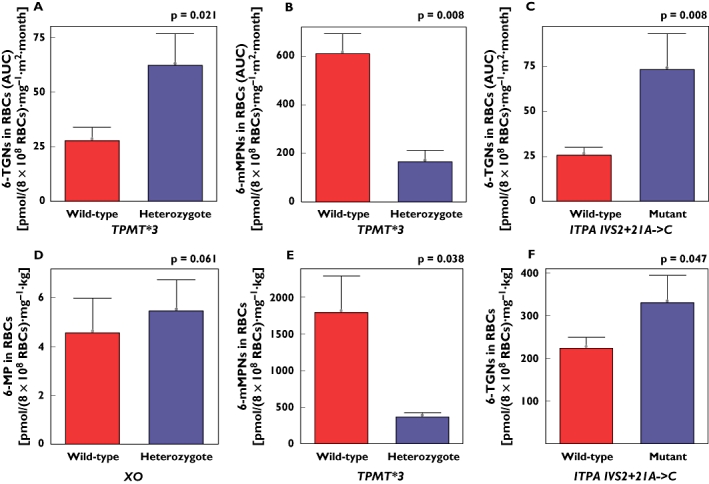
Differences in mercaptopurine (6-MP) metabolite concentrations (mean ± SE) among the various polymorphisms studied in acute lymphoblastic leukaemia (ALL) (A,B,C) and inflammatory bowel disease (IBD) (D,E,F) patients. Mutant refers to any mutant (homozygote or heterozygote). P-values were adjusted for multiple testing as per the Bonferroni justification
In the ALL population, AUC values of 6-TU RBC concentrations was lower in the patient who had XO variants when compared with the rest of the group (AUC = 1.41 vs. 4.95 pmol per 8 × 108 RBCs mg−1 m−2 month−1). Concentrations of 6-TGNs in RBCs, however, were higher in that patient (AUC = 37.25 vs. 32.98 pmol per 8 × 108 RBCs mg−1 m−2 month−1). In the IBD population, there was a trend toward higher RBC concentrations of 6-MP in patients having XO variants (P = 0.061).
ALL patients with TPMT polymorphisms were found to have lower RBC concentrations of 6-mMPNs (AUC mean = 165.49 vs. 609.71 pmol per 8 × 108 RBCs mg−1 m−2 month−1; P = 0.008), but higher concentrations of 6-TGNs in RBCs (AUC mean = 62.18 vs. 27.78 pmol per 8 × 108 RBCs mg−1 m−2 month−1; P = 0.021). This was also evident in IBD patients having TPMT polymorphisms who showed significant association with lower concentrations of 6-mMPNs in RBCs (mean = 363.18 vs. 1788.95 pmol per 8 × 108 RBCs mg−1 kg−1; P = 0.038).
Carriers of ITPA IVS2+21A→C variant allele among the ALL population had significantly higher concentrations of 6-TGNs in RBCs (AUC mean = 73.47 vs. 25.66 pmol per 8 × 108 RBCs mg−1 m−2 month−1; P = 0.008) and plasma (AUC mean = 0.41 vs. 0.13 ng ml−1 mg−1 m−2 month−1; P = 0.072). The latter reached borderline significance only.
The cohort of IBD subjects with ITPA IVS2+21A→C allelic variants had also higher concentrations of 6-TGNs in RBCs (mean = 330.21 vs. 223.25 pmol per 8 × 108 RBCs mg−1 kg−1; P = 0.047). On the other hand, carriers of ITPA C94→A variant allele had lower RBC concentrations of 6-TGNs in ALL and IBD patients, but this did not reach statistical significance.
6-MP-related myelotoxicity
Myelotoxicity in ALL and IBD patients has been defined according to the following criteria: leucopenia (white blood count <3.0 × 109 l−1), neutropenia (absolute neutrophil count <1.5 × 109 l−1) and thrombocytopenia (platelet count <150 × 109 l−1). Among the 75 samples collected from the ALL population over 6 months, 29 samples (38.7%) indicated the presence of leucopenia, 22 (29.3%) indicated the presence of neutropenia (19 of those indicated the presence of both leucopenia and neutropenia) and nine (12%) showed the development of thrombocytopenia at some stage during the study. Details regarding ALL patients who developed myelotoxicity at least twice during the study period (6 months), their genotyping details and the lowest values of their haematological parameters are shown in Table 5.
Table 5.
Details for ALL patients who developed myelotoxicity at least twice during the study (n = 13)*
| Patient no. | WBC (×109 l−1) | ANC (×109 l−1) | PLT (×109 l−1) | XO A1936G | A2107G | TPMT genotype | ITPA C94A | IVS2+21AC |
|---|---|---|---|---|---|---|---|---|
| 1 | 1.7 | 0.86 | 356 | A/A | A/A | *1/*1 | C/C | A/A |
| 2 | 1.2 | 0.54 | 253 | A/A | A/A | *1/*1 | C/C | A/A |
| 3 | 2 | 1.24 | 196 | A/A | A/A | *1/*3C | C/C | A/C |
| 4 | 1.5 | 1.08 | 93 | A/A | A/A | *1/*1 | C/C | A/C |
| 5 | 2.5 | 1.84 | 203 | A/A | A/A | *1/*1 | C/C | A/A |
| 6 | 2.2 | 1.48 | 174 | A/A | A/A | *1/*1 | C/A | A/A |
| 7 | 1.7 | 0.3 | 253 | A/A | A/A | *1/*3C | C/C | A/A |
| 8 | 2.2 | 0.68 | 310 | A/A | A/A | *1/*1 | C/C | A/A |
| 9 | 2.3 | 0.98 | 213 | A/A | A/A | *1/*1 | C/C | A/A |
| 10 | 2.7 | 1.81 | 133 | A/A | A/A | *1/*1 | C/C | C/C |
| 11 | 1.3 | 0.83 | 232 | A/A | A/A | *1/*1 | C/C | A/A |
| 12 | 3 | 1.26 | 104 | A/A | A/A | *1/*1 | C/C | A/A |
| 13 | 1.4 | 0.94 | 144 | A/A | A/A | *1/*1 | C/C | A/A |
Values for WBC, ANC, PLT, and Hb are the lowest recorded during the study. WBC, white blood cell count; ANC, absolute neutrophil count; PLT, platelet count; ALL, acute lymphoblastic leukaemia; XO, xanthine oxidase; TPMT, thiopurine S-methyl transferase; ITPA, inosine triphosphatase. Mutations are highlighted in bold.
Only three IBD patients (8.8%), however, were found to have myelotoxicity during the study (one had leucopenia, another neutropenia and a third thrombocytopenia).
In the following two sections, the prevalence of the various forms of drug-related myelotoxicity along with the median values of the haematological parameters tested are compared between patients having XO, TPMT or ITPA variant alleles and those who did not. In addition, the frequency of myelotoxicity reoccurrence in each patient is compared between these two groups.
Correlation of XO, TPMT and ITPA genotypes with myelotoxicity
The investigated associations between myelotoxicity and the different combinations of TPMT, ITPA and XO mutations in ALL patients are summarized in Table 6. The total TPMT deficiency-associated alleles tested were significantly associated with both neutropenia (P = 0.006) and leucopenia (P = 0.045) and predicted both. In addition, when the frequency of myelotoxic events per patient was investigated, higher numbers of neutropenic and leucopenic events were found among ALL patients having these variant alleles (P = 0.024 and P = 0.053, respectively), the latter being at borderline significance only. The total TPMT variant alleles, however, did not predict myelotoxicity overall or individual forms of it.
Table 6.
Significance of the association of myelotoxicity with TPMT or ITPA variant genotypes in ALL patients treated with 6-MP (n = 75 samples in 19 ALL subjects)*
| Genotype | Leucopenia (n = 29) | Neutropenia (n = 22) | Thrombocytopenia (n = 11) |
|---|---|---|---|
| TPMT*3 heterozygote | 0.045 | 0.006 | 0.336 |
| ITPA C94→A heterozygote | 0.342 | 1.000 | 0.582 |
| ITPA IVS2+21A→C mutant† | 1.000 | 1.000 | 0.012 |
Numbers represent P-values for the corresponding association adjusted as per the Bonferroni justification for multiple testing.
Mutant refers to any mutant (homozygote or heterozygote). TPMT, thiopurine S-methyl transferase; ITPA, inosine triphosphatase; ALL, acute lymphoblastic leukaemia.
ITPA IVS2+21A→C deficiency-associated allelic variants were significantly associated with thrombocytopenia (P = 0.012). In contrast, ITPA C94→A and XO mutant alleles did not predict any type of myelotoxicity in ALL patients.
In IBD patients, however, myelotoxicity was found in only three patients, with XO mutant alleles present in one of them. Due to the small number of IBD patients having myelotoxicity, investigation of a possible association between myelotoxicity and mutations in this group was not possible.
Correlation of XO, TPMT and ITPA genotypes with haematological parameters
In order to assess further the relationship between myelotoxicity and the studied genotypes, the median values of the haematological parameters studied (white cell count, neutrophil count and platelet count) among the various polymorphisms were compared.
In ALL patients, TPMT*3C was significantly associated with lower counts of neutrophils (P = 0.003) and white blood cells (P = 0.012) (Table 7). In addition, there was a significant association of the total TPMT deficiency-associated alleles with lower neutrophil counts (P = 0.039).
Table 7.
Association of haematological parameters with the various polymorphisms studied in ALL patients (n = 75 samples from 19 subjects)*
| Genotype | WBC (×109 l−1) | ANC (×109 l−1) | PLT (×109 l−1) |
|---|---|---|---|
| TPMT*3C heterozygote | 0.012 | 0.003 | 0.864 |
| ITPA C94→A heterozygote | 0.981 | 1.000 | 0.534 |
| ITPA IVS2+21A→C mutant† | 0.618 | 0.702 | 0.003 |
Numbers represent P-values for the corresponding association adjusted as per the Bonferroni justification for multiple testing.
Mutant refers to any mutant (homozygote or heterozygote). ALL, acute lymphoblastic leukaemia; WBC, white blood cell count; ANC, absolute neutrophil count; PLT, platelet count; TPMT, thiopurine S-methyl transferase; ITPA, inosine triphosphatase.
No significant association was observed between ITPA C94→A variant allele and haematological parameters. In contrast, carriers of ITPA IVS2+21A→C variant allele had significantly lower counts of platelets (P = 0.003).
XO variant alleles, however, were not significantly associated with any haematological parameters. In the studied IBD population, there was no correlation between XO, TPMT or ITPA genotypes and haematological parameters.
Discussion
In this study, the allelic frequencies of seven potentially important polymorphisms of three different enzymes involved in 6-MP/AZA metabolism (TPMT, ITPA and XO) have been evaluated. Minor allele frequencies of the different mutations studied were similar to those reported in the National Centre for Biotechnology Information genetic database. The association of these polymorphisms with toxicity, haematological parameters and 6MP/AZA metabolite concentrations were examined.
The cytotoxic effect of 6-MP is related to the formation and intracellular accumulation of 6-TGNs [28, 29]. One well-defined source of variability in 6-TGN concentrations includes inherited differences in the TPMT gene [30, 31]. Low TPMT enzyme activity results in greater production of 6-TGNs at the expense of 6-mMPNs, leading to increased toxicity. A number of SNPs that cause reduced or absent TPMT enzyme activity have been identified. The frequency of most of these variant alleles is very low except for the TPMT*3A and TPMT*3C alleles. In the present study, TPMT*3A and TPMT*3C were the only TPMT variants identified in the ALL subjects and, as anticipated, both were strongly associated with lower RBC concentrations of 6-mMPNs and higher concentrations of 6-TGNs in RBCs. The TPMT genotype could therefore be an important factor in determining the delicate balance between the potentially hepatotoxic 6-mMPNs and the principle cytotoxic metabolites (6-TGNs).
Lower 6-TU RBC concentrations in patients with TPMT mutations, on the other hand, could be explained by the fact that some patients having TPMT variant alleles had also XO variants, which would in turn lead to lower concentrations of 6-TU due to lower XO enzymatic activity. This was supported by the fact that in the IBD population, where TPMT mutations were not combined with XO variant alleles, such a correlation between TPMT mutations and lower 6-TU concentration in RBCs was not present.
ITPA mutant alleles were associated with higher RBC concentrations of 6-TGNs in ALL and IBD patients. This is an interesting finding, since 6-TGNs are considered the active cytotoxic metabolites of 6-MP and hence directly contribute to its toxicity and efficacy. In ALL patients, ITPA mutant alleles were also associated with higher concentrations of 6-TG in plasma. This could be explained by the fact that thiopurine metabolites in plasma circulate in the form of free bases, whereas intracellular metabolites predominantly exist as nucleotides. This is due to the presence of a charged phosphate moiety that entraps nucleotides inside cells. Upon loss of the phosphate moiety, however, the corresponding bases are free to enter the plasma and, hence, plasma concentrations could be in equilibrium with the intracellular concentrations of the corresponding nucleotides. Higher plasma concentrations of 6-TG in these patients, therefore, could be due to the higher concentrations of 6-TGNs within RBCs.
XO is an important enzyme involved in the first-pass metabolism of 6-MP [32]. This enzyme, predominantly expressed in the intestinal mucosa and liver, converts 6-MP to 6-TX and consequently to the inactive metabolite 6-TU [33, 34], a principle pathway of 6-MP catabolism. The presence of lower concentrations of 6-TU and higher concentrations of 6-TGNs in ALL patients who had XO variant alleles is consistent with the role of this enzyme in catabolism of 6-MP, but it may be related to concordant variability of XO in other tissues (e.g. the liver). However, the relationship between XO mutants and 6-TU should be interpreted with caution, for two reasons: first, XO mutants were combined with TPMT polymorphisms; second, ALL subjects were taking methotrexate in association with 6-MP, which has inhibitory effects on XO activity [35]. In IBD patients, there was a trend toward lower RBC concentrations of 6-MP in patients with XO variants, which coincides with the above explanation.
To our knowledge, this is the first report in the literature correlating XO genetic polymorphisms with XO metabolic activity and metabolite concentrations. However, more studies of larger populations are needed to evaluate further the potential importance of these polymorphisms.
Since the genetic polymorphisms examined were associated with different 6-MP metabolite concentrations, they were likely to identify patients at risk of toxicity or differences in treatment outcome. The associations of these polymorphisms with side-effects and haematological parameters were, therefore, evaluated in this study.
The association between low TPMT activity and excessive haematological toxicity is now well recognized [36, 37]. However, the clinical importance of intermediate TPMT activity (or heterozygous genotype) has not been fully elucidated. A study of adult subjects receiving AZA therapy for rheumatological disorders has found that patients with a heterozygous TPMT genotype were forced to discontinue therapy within 1 month of initiation due to haematological toxicity [38]. However, this was not observed in another study on children with ALL [1]. Children with a wild-type or heterozygous TPMT variant genotype spent a similar number of weeks during the maintenance period unable to receive therapy due to haematological toxicity. The inverse was also true (the number of weeks when full-dose 6-MP could be administered was the same). This may reflect, in part, the differences in treatment philosophy for leukaemia vs. rheumatological disease, in that the accepted level of bone marrow suppression is greater for ALL [1]. In our study, myelotoxicity for ALL and IBD patients was defined according to the criteria usually applied in clinical practice for IBD patients, which represented a good cut-off point (defining myelotoxic/nonmyelotoxic events) and reflected the effect of various polymorphisms on different forms of haematological toxicity.
The total TPMT deficiency-associated alleles tested in the present study were significantly associated with both leucopenia and neutropenia in ALL patients. However, TPMT genotype alone failed to explain all cases of haematological impairment. Methotrexate or cotrimoxazole may have contributed to the observed toxicity. It is also possible that the children may have had impaired TPMT activity due to one or more of the recently identified, less common variant TPMT alleles [39]. Some authors have criticised TPMT genotyping because it is not able to explain all impairment of bone marrow function [40]. However, the fact that TPMT pharmacogenetic testing would allow us to understand, anticipate and avoid some adverse effects in a subset of patients clearly represents a significant clinical advance [39].
On the other hand, it has been unquestionably demonstrated that TPMT genotyping is important in the case of homozygous TPMT mutants, who will experience early and severe myelosuppression when treated with standard 6-MP or AZA dosages. TPMT geno- or phenotyping prior to initiation of therapy is the only way of identifying these patients at high risk [41, 42]. However, no subject homozygous for TPMT mutated alleles was found among our sample.
ITPA deficiency has been shown to be another risk factor for 6-MP or AZA toxicity [43]. ITPA deficiency is a clinically benign condition characterized by abnormal accumulation of ITP in erythrocytes. However, in ITPA-deficient patients receiving 6-MP or AZA, possible accumulation of 6-TITP in could occur. One study of 62 IBD patients observed an increase in adverse events in patients carrying the C94A mutation; however, a risk for leucopenia was not detected [16]. Furthermore, a recent study investigating the ITPA C94→A frequency in 41 patients found no increased risk for the development of leucopenia [44]. Even though we were unable to investigate the association of ITPA C94→A or other polymorphisms with myelotoxicity in the present study due to the small number of IBD patients who had myelotoxic events (three patients only), we have investigated for the first time the association of ITPA variant alleles with myelotoxic adverse events in ALL children receiving 6-MP therapy. In line with the recent study in IBD patients, ITPA C94→A mutation was not associated with leucopenia or any other form of myelotoxicity in the ALL population.
Our study has also evaluated a second genetic change in ITPA. A significant association, however, was present between ITPA IVS2+21A→C variant alleles and thrombocytopenia. These results showed for the first time an association between ITPA IVS2_21A→C mutation and myelotoxicity. This may be of interest to clinicians, as it has the potential to generally identify patients at increased risk for drug intolerance.
Neither of the XO mutant alleles in ALL patients was associated with any form of myelotoxicity. However, the small cohort size may have contributed to the lack of association between XO mutations and toxicity in this study.
In order to assess further the relationship between the examined genotypes and myelotoxicity, the median values of the haematological parameters tested were compared among the various polymorphisms. Reports evaluating the potential difference in haematological parameters (white cell counts, neutrophil counts and platelet counts) amongst different genotypes have not been published before.
A significant association was found between TPMT deficiency-associated alleles and lower neutrophil counts. In addition, TPMT*3C variant alleles were significantly associated with lower white blood cell counts and neutrophil counts in ALL patients. This is in agreement with the association of TPMT mutations with both neutropenia and leucopenia, as stated above. This may be due to the association of TPMT variant alleles with higher concentrations of RBC 6-TGNs, which have been shown to correlate positively with decreased white cell counts and neutrophil counts (relative to the initial value before onset of treatment) [45].
ITPA IVS2+21A→C variant alleles were significantly associated with lower platelet counts in ALL subjects. Again, this is in line with the association of these variant alleles with thrombocytopenia. In the studied IBD population, however, there was no correlation between XO, TPMT or ITPA genotypes and haematological parameters, which could be explained by the lack of multiple tests for haematological parameters within this group. Only one to two blood tests per patient were performed during the study period, which might not be enough to detect all myelotoxic events or trends in haematological parameters within the IBD population. More frequent blood tests are therefore warranted if the association between haematological parameters and mutations is to be investigated.
Conclusions
The present study has contributed to a clearer understanding of 6-MP metabolism and has shown important pharmacogenetic associations. Various significant relationships were demonstrated between the different polymorphisms examined and 6-MP metabolite concentrations. The association of TPMT polymorphism with toxicity has been confirmed in this study. Furthermore, the study has identified potential genetic characteristics that may contribute to higher risk of adverse events (such as ITPA IVS2+21A→C mutation). The presence of genetic markers of toxicity could alert the physician that certain patients are at increased risk of intolerance and may require closer follow-up and counselling. However, this does not obviate the need for careful surveillance of white blood cells or whole blood counts together with liver and pancreatic function testing, so as to detect the common forms of toxicity unrelated to these genetic markers or genotypes [24, 46]. In addition, the combination of genotyping with measurement of the cytotoxic metabolites, 6-TGNs, could be an attractive approach for optimizing 6-MP therapy. Measurement of the metabolic profile could prove useful to avoid 6-MP underdosing and to identify patients in whom adherence to therapy is a problem.
REFERENCES
- 1.Mcleod HL, Coulthard S, Thomas AE, Pritchard SC, King DJ, Richards SM, Eden OB, Hall AG, Gibson BE. Analysis of thiopurine methyltransferase variant alleles in childhood acute lymphoblastic leukaemia. Br J Haematol. 1999;105:696–700. doi: 10.1046/j.1365-2141.1999.01416.x. [DOI] [PubMed] [Google Scholar]
- 2.Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE. Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood. 1999;93:2817–23. [PubMed] [Google Scholar]
- 3.Fraser AG, Orchard TR, Jewell DP. The efficacy of azathioprine for the treatment of inflammatory bowel disease: A 30 year review. Gut. 2002;50:485–9. doi: 10.1136/gut.50.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Escher JC, Taminiau JA, Nieuwenhuis EE, Buller HA, Grand RJ. Treatment of inflammatory bowel disease in childhood: best available evidence. Inflamm Bowel Dis. 2003;9:34–58. doi: 10.1097/00054725-200301000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Louis E, Belaiche J. Optimizing treatment with thioguanine derivatives in inflammatory bowel disease. Best Pract Res Clin Gastroenterol. 2003;17:37–46. doi: 10.1053/bega.2002.0346. [DOI] [PubMed] [Google Scholar]
- 6.Lennard L. The clinical pharmacology of 6-mercaptopurine. Eur J Clin Pharmacol. 1992;43:329–39. doi: 10.1007/BF02220605. [DOI] [PubMed] [Google Scholar]
- 7.Deininger M, Szumlanski CL, Otterness DM, Van Loon J, Ferber W, Weinshilboum RM. Purine substrates for human thiopurine methyltransferase. Biochem Pharmacol. 1994;48:2135–8. doi: 10.1016/0006-2952(94)90515-0. [DOI] [PubMed] [Google Scholar]
- 8.Derijks LJ, Gilissen LP, Hooymans PM, Hommes DW. Review article: Thiopurines in inflammatory bowel disease. Aliment Pharmacol Ther. 2006;24:715–29. doi: 10.1111/j.1365-2036.2006.02980.x. [DOI] [PubMed] [Google Scholar]
- 9.Schaeffeler E, Fischer C, Brockmeier D, Wernet D, Moerike K, Eichelbaum M, Zanger UM, Schwab M. Comprehensive analysis of thiopurine S-methyltransferase phenotype–genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics. 2004;14:407–17. doi: 10.1097/01.fpc.0000114745.08559.db. [DOI] [PubMed] [Google Scholar]
- 10.Lennard L, Lilleyman JS, Van Loon J, Weinshilboum RM. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336:225–9. doi: 10.1016/0140-6736(90)91745-v. [DOI] [PubMed] [Google Scholar]
- 11.Schmiegelow K, Schroder H, Gustafsson G, Kristinsson J, Glomstein A, Salmi T, Wranne L. Risk of relapse in childhood acute lymphoblastic leukemia is related to RBC methotrexate and mercaptopurine metabolites during maintenance chemotherapy. Nordic Society for Pediatric Hematology and Oncology. J Clin Oncol. 1995;13:345–51. doi: 10.1200/JCO.1995.13.2.345. [DOI] [PubMed] [Google Scholar]
- 12.Balis FM, Adamson PC. Application of pharmacogenetics to optimization of mercaptopurine dosing. J Natl Cancer Inst. 1999;91:1983–5. doi: 10.1093/jnci/91.23.1983. [DOI] [PubMed] [Google Scholar]
- 13.Aplenc R, Lange B. Pharmacogenetic determinants of outcome in acute lymphoblastic leukaemia. Br J Haematol. 2004;125:421–34. doi: 10.1111/j.1365-2141.2004.04932.x. [DOI] [PubMed] [Google Scholar]
- 14.Guerciolini R, Szumlanski C, Weinshilboum RM. Human liver xanthine oxidase: nature and extent of individual variation. Clin Pharmacol Ther. 1991;50:663–72. doi: 10.1038/clpt.1991.205. [DOI] [PubMed] [Google Scholar]
- 15.Lennard L. Therapeutic drug monitoring of antimetabolic cytotoxic drugs. Br J Clin Pharmacol. 1999;47:131–43. doi: 10.1046/j.1365-2125.1999.00884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marinaki AM, Ansari A, Duley JA, Arenas M, Sumi S, Lewis CM, Shobowale-Bakre E, Escuredo E, Fairbanks LD, Sanderson JD. Adverse drug reactions to azathioprine therapy are associated with polymorphism in the gene encoding inosine triphosphate pyrophosphatase (ITPase) Pharmacogenetics. 2004;14:181–7. doi: 10.1097/00008571-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Sumi S, Marinaki AM, Arenas M, Fairbanks L, Shobowale-Bakre M, Rees DC, Thein SL, Ansari A, Sanderson J, De Abreu RA, Simmonds HA, Duley JA. Genetic basis of inosine triphosphate pyrophosphohydrolase deficiency. Hum Genet. 2002;111:360–7. doi: 10.1007/s00439-002-0798-z. [DOI] [PubMed] [Google Scholar]
- 18.Armstrong VW, Oellerich M. New developments in the immunosuppressive drug monitoring of cyclosporine, tacrolimus, and azathioprine. Clin Biochem. 2001;34:9–16. doi: 10.1016/s0009-9120(00)00175-2. [DOI] [PubMed] [Google Scholar]
- 19.Weinshilboum R. Thiopurine pharmacogenetics: clinical and molecular studies of thiopurine methyltransferase. Drug Metab Dispos. 2001;29:601–5. [PubMed] [Google Scholar]
- 20.Lennard L. Therapeutic drug monitoring of cytotoxic drugs. Br J Clin Pharmacol. 2001;52(Suppl)(1):75S–87S. doi: 10.1046/j.1365-2125.2001.0520s1075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Armstrong VW, Shipkova M, von Ahsen N, Oellerich M. Analytic aspects of monitoring therapy with thiopurine medications. Ther Drug Monit. 2004;26:220–6. doi: 10.1097/00007691-200404000-00024. [DOI] [PubMed] [Google Scholar]
- 22.Gearry RB, Barclay ML. Azathioprine and 6-mercaptopurine pharmacogenetics and metabolite monitoring in inflammatory bowel disease. J Gastroenterol Hepatol. 2005;20:1149–57. doi: 10.1111/j.1440-1746.2005.03832.x. [DOI] [PubMed] [Google Scholar]
- 23.Gearry RB, Barclay ML, Roberts RL, Harraway J, Zhang M, Pike LS, George PM, Florkowski CM. Thiopurine methyltransferase and 6-thioguanine nucleotide measurement: early experience of use in clinical practice. Intern Med J. 2005;35:580–5. doi: 10.1111/j.1445-5994.2005.00904.x. [DOI] [PubMed] [Google Scholar]
- 24.Seidman EG. Clinical use and practical application of TPMT enzyme and 6-mercaptopurine metabolite monitoring in IBD. Rev Gastroenterol Disord. 2003;3(Suppl)(1):S30–8. [PubMed] [Google Scholar]
- 25.Baker DE. Pharmacogenomics of azathioprine and 6-mercaptopurine in gastroenterologic therapy. Rev Gastroenterol Disord. 2003;3:150–7. [PubMed] [Google Scholar]
- 26.Hawwa AF, Millership JS, Collier PS, McElnay JC. Improved HPLC methodology for the determination of mercaptopurine and its metabolites in plasma and red blood cells. J Pharm Pharmacol. 2006;58(Suppl)(1):A3. [Google Scholar]
- 27.Matthews JNS, Altman DG, Campbell MJ, Royston P. Analysis of serial measurements in medical research. BMJ. 1990;300:230–5. doi: 10.1136/bmj.300.6719.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lepage GA. Basic biochemical effects and mechanism of action of 6-thioguanine. Cancer Res. 1963;23:1202–6. [PubMed] [Google Scholar]
- 29.Warren DJ, Andersen A, Slordal L. Quantitation of 6-thioguanine residues in peripheral blood leukocyte DNA obtained from patients receiving 6-mercaptopurine-based maintenance therapy. Cancer Res. 1995;55:1670–4. [PubMed] [Google Scholar]
- 30.Yates CR, Krynetski EY, Loennechen T, Fessing MY, Tai HL, Pui CH, Relling MV, Evans WE. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med. 1997;126:608–14. doi: 10.7326/0003-4819-126-8-199704150-00003. [DOI] [PubMed] [Google Scholar]
- 31.Krynetski EY, Evans WE. Pharmacogenetics of cancer therapy: getting personal. Am J Hum Genet. 1998;63:11–6. doi: 10.1086/301941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bostrom B, Erdmann G. Cellular pharmacology of 6-mercaptopurine in acute lymphoblastic leukemia. Am J Pediatr Hematol Oncol. 1993;15:80–6. [PubMed] [Google Scholar]
- 33.Zimm S, Collins JM, Riccardi R, O'Neill D, Narang PK, Chabner B, Poplack DG. Variable bioavailability of oral mercaptopurine. Is maintenance chemotherapy in acute lymphoblastic leukemia being optimally delivered? N Engl J Med. 1983;308:1005–9. doi: 10.1056/NEJM198304283081705. [DOI] [PubMed] [Google Scholar]
- 34.Krynetski EY, Krynetskaia NF, Yanishevski Y, Evans WE. Methylation of mercaptopurine, thioguanine, and their nucleotide metabolites by heterologously expressed human thiopurine S-methyltransferase. Mol Pharmacol. 1995;47:1141–7. [PubMed] [Google Scholar]
- 35.Lewis AS, Murphy L, McCalla C, Fleary M, Purcell S. Inhibition of mammalian xanthine oxidase by folate compounds and amethopterin. J Biol Chem. 1984;259:12–5. [PubMed] [Google Scholar]
- 36.McLeod HL, Miller DR, Evans WE. Azathioprine-induced myelosuppression in thiopurine methyltransferase deficient heart transplant recipient. Lancet. 1993;341:1151. doi: 10.1016/0140-6736(93)93168-z. [DOI] [PubMed] [Google Scholar]
- 37.McLeod HL, Relling MV, Liu Q, Pui CH, Evans WE. Polymorphic thiopurine methyltransferase in erythrocytes is indicative of activity in leukemic blasts from children with acute lymphoblastic leukemia. Blood. 1995;85:1897–902. [PubMed] [Google Scholar]
- 38.Black AJ, McLeod HL, Capell HA, Powrie RH, Matowe LK, Pritchard SC, Collie-Duguid ES, Reid DM. Thiopurine methyltransferase genotype predicts therapy-limiting severe toxicity from azathioprine. Ann Intern Med. 1998;129:716–8. doi: 10.7326/0003-4819-129-9-199811010-00007. [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: insights, challenges and future directions. Oncogene. 2006;25:1629–38. doi: 10.1038/sj.onc.1209372. [DOI] [PubMed] [Google Scholar]
- 40.Nebert DW, Jorge-Nebert L, Vesell ES. Pharmacogenomics and ‘individualized drug therapy’: high expectations and disappointing achievements. Am J Pharmacogenomics. 2003;3:361–70. doi: 10.2165/00129785-200303060-00002. [DOI] [PubMed] [Google Scholar]
- 41.Lennard L. TPMT in the treatment of Crohn's disease with azathioprine. Gut. 2002;51:143–6. doi: 10.1136/gut.51.2.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Derijks LJ, Gilissen LP, Engels LG, Bos LP, Bus PJ, Lohman JJ, Curvers WL, Van Deventer SJ, Hommes DW, Hooymans PM. Pharmacokinetics of 6-mercaptopurine in patients with inflammatory bowel disease: implications for therapy. Ther Drug Monit. 2004;26:311–8. doi: 10.1097/00007691-200406000-00016. [DOI] [PubMed] [Google Scholar]
- 43.Marinaki AM, Duley JA, Arenas M, Ansari A, Sumi S, Lewis CM, Shobowale-Bakre M, Fairbanks LD, Sanderson J. Mutation in the ITPA gene predicts intolerance to azathioprine. Nucleosides Nucleotides Nucleic Acids. 2004;23:1393–7. doi: 10.1081/NCN-200027639. [DOI] [PubMed] [Google Scholar]
- 44.Allorge D, Hamdan R, Broly F, Libersa C, Colombel JF. ITPA genotyping test does not improve detection of Crohn's disease patients at risk of azathioprine/6-mercaptopurine induced myelosuppression. Gut. 2005;54:565. doi: 10.1136/gut.2004.055947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D'Halluin PN, Tribut O, Branger B, Lebreton C, Bretagne JF, Bentue-Ferrer D, Heresbach D. RBC 6-TGN and hematological parameters in patients with Crohn's disease treated by azathioprine. Gastroenterol Clin Biol. 2005;29:1264–9. doi: 10.1016/s0399-8320(05)82219-5. [DOI] [PubMed] [Google Scholar]
- 46.Colombel J, Ferrari N, Debuysere H, Marteau P, Gendre J, Bonaz B, Soulé J, Modigliani R, Touze Y, Catala P, Libersa C, Broly F. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn's disease and severe myelosuppression during azathioprine therapy. Gastroenterology. 2000;118:1025–30. doi: 10.1016/s0016-5085(00)70354-4. [DOI] [PubMed] [Google Scholar]