

Abstract
The expressions of proteins in the cell are carefully regulated by transcription factors that interact with their downstream targets in specific signal transduction cascades. Our understanding of the regulation of functional genes responsive to stress signals is still nascent. Plants like Arabidopsis thaliana, are convenient model systems to study fundamental questions related to regulation of the stress transcriptome in response to stress challenges. Microarray results of the Arabidopsis transcriptome indicate that several genes could be upregulated during multiple stresses, such as cold, salinity, drought etc. Experimental biochemical validations have proved the involvement of several transcription factors could be involved in the upregulation of these stress responsive genes. In order to follow the intricate and complicated networks of transcription factors and genes that respond to stress situations in plants, we have developed a computer algorithm that can identify key transcription factor binding sites upstream of a gene of interest. Hidden Markov models of the transcription factor binding sites enable the identification of predicted sites upstream of plant stress genes. The search algorithm, STIF, performs very well, with more than 90% sensitivity, when tested on experimentally validated positions of transcription factor binding sites on a dataset of 60 stress upregulated genes.
Availability
Supplementary data is available at http://caps.ncbs.res.in/download/stif.
Keywords: transcription factor binding site prediction, gene regulation, stress genes, Arabidopsis thaliana, HMM based algorithm
Background
The interactions between regulatory proteins and DNA control many important processes and responses to environmental stresses, and defects in these interactions can contribute to inefficient stress responses. Plants, like Arabidopsis thaliana, are very simple and good model systems to understand fundamental processes such as protein-DNA interactions that happen in response to environmental stresses [1,2]. Numerous studies have shown that transcription factors are important in regulating plant responses to stress. One important step in the control of stress responses is the transcriptional activation or repression of genes. Databases, such as ATHAMAP [3], offer information about the chromosomal positions of genes of interest and possible location of their transcription factors and binding sites. Multiple signalling pathways regulate the stress responses of plants and there is significant overlap between the patterns of gene expression that are induced in plants in response to different stresses [4]. Many genes induced by stress challenges, including those encoding transcription factors, have been identified and some of them have been shown to be essential for stress tolerance. Many studies have also revealed some of the complexity and overlap in the responses to different stresses, and are likely to lead to new ways to enhance crop tolerance to disease and environmental stress. The binding specificities of only a small number of transcription factors (TFs) are well characterized. Transcription-factor binding sites (TFBSs) are usually short (around 5-15 base-pairs (bp)) and they frequently contain degenerate sequence motifs. The sequence degeneracy of TFBSs has been selected through evolution and is beneficial, because it confers different levels of activity upon different promoters. Much of the information on TF binding specificity has been determined using traditional methodologies, such as foot-printing methods, (that identify the region of DNA protected by a bound protein), nitrocellulose binding assays, gel-shift analysis (that monitors the change in mobility when DNA and protein bind), South-western blotting (of both DNA and protein) or reporter constructs. These methods are generally quite time-consuming and are not readily scalable to a whole genome [5]. One of the interesting problems is to identify the cis-acting elements by computational techniques at a whole genome level so as to choose promising targets for detailed experimental investigation. Well-known eukaryotic transcription factors and their binding sites are recorded in TRANSFAC database [6]. There are computational tools to facilitate the retrieval of information from TRANSFAC database, but for the human genome [7]. There have also been algorithms that employ position-specific profiles and scoring schemes to recognize putative TFBS [8,9] or probabilistic models [10]. These servers and algorithms are largely for eukaryotic general-purpose transcription factors and not specific for plant stress induced genes. There are other computational algorithms to search for possible genes that are downstream of classical TFBS, where the binding site data are encoded as HMMs and searched all around the genome of interest. These methods are called as ‘targeted gene finding’ since they begin from known TFBS [11]. However, this approach is complicated for plant stress genes since stress TF-binding site signatures could potentially be upstream of constitutive genes as well and there could also be overlap in various TFBS. We have collected data of well-known stress specific transcription factors and generated Hidden Markov Method (HMM) of known TFBS. This knowledge-based approach, by building HMM models through well-known abiotic stress cis-elements, has been tested extensively to standardize thresholds for scores.
Methodology
Program input
In Arabidopsis thaliana, we have examined 10 families of transcription factors known to be involved in responses to abiotic stress (Table 1 under supplementary material).
Dataset for validation
We have identified 60 stress responsive genes from six different microarray databases and these were collected on the basis of their consistent upregulation in response to abiotic stress signals in most of these microarray databases and across different microarray experiments. To compare the cis-elements both in stress on-off conditions, we also identified 60 constitutive genes from six different databases. Genes that get consistently upregulated under abiotic conditions were identified from these databases and used for the validation study.
RARGE
RARGE [12] presents Arabidopsis resource data (cDNAs, transposon mutants and microarray experiments) for all biology researchers. RARGE has data from 6 different abiotic stress experiments (i.e. cold, drought, salt, ABA, Light, dehydration stress) with expression levels at different time courses.
DRASTIC
DRASTIC [13] is a database of plant genes regulated in response to biotic and abiotic stress, developed and maintained by the Scottish Crop Research Institute.
StressLink
StressLink[14] is a database linking genome information to the primary literature on stress-related genes in Arabidopsis thaliana.
AtGenExpress
AtGenExpress [15] is a multinational effort designed to uncover the transcriptome of the multicellular model organism Arabidopsis thaliana [1]15].
DATF
The Database of Arabidopsis Transcription Factors (DATF) [16] collects all Arabidopsis transcription factors and classifies them into 64 families. It uses not only locus (gene), but also gene model (transcript, protein) and the detail information is for each gene model not for locus. It adds multiple alignment of the DNA- binding domain of each family.
TAIR
The Arabidopsis Information Resource (TAIR) [17] maintains a database of genetic and molecular biology data for the model higher plant Arabidopsis thaliana. Data available from TAIR includes the complete genome sequence along with gene structure, gene product information, metabolism, gene expression, DNA and seed stocks, genome maps, genetic and physical markers, publications, and information about the Arabidopsis research community.
Construction of a Hidden Markov Model
Hidden Markov Model (HMM) is a probabilistic method, which is used for TFBS detection. The consensus (S) of length (L) was taken from the literature and the probabilistic score (P(S)) and log-odd score were calculated by using equation (1) and (2) (see supplementary material) respectively.
As plant sequences are rich in GC content, we gave higher weight to AT than GC in log-odd score (please see Figure 1 for an example).
Figure 1.

Construction of a Hidden Markov Model of transcription factor binding sites given the experimentally observed nucleotide patterns.
STIF - TFBS search algorithm
The search program will accept nucleotide stretch that is upstream of an abiotic stress gene. A detailed flow-chart of the search algorithm is given in Figure 2. The TFBS search algorithm searches for cis-elements both in forward and reverse direction in the query sequence from the built models and the acquired hit gets a HMM score. The chromosomal position, UTR position, cis-element, orientation of the cis-element are also recorded and mentioned in the output.
Figure 2.
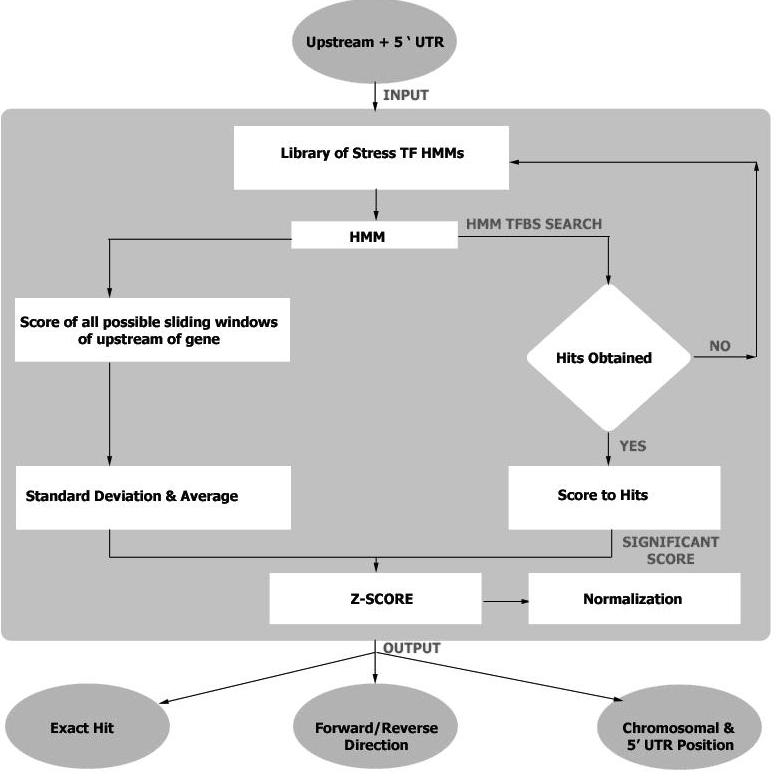
Flow chart diagram of STIF search algorithm.
Validation
Validation was performed using a leave-one-out approach (Jackknifing method). The threshold and maximum scores of each HMM model was further decided by this statistical test of jackknifing (Figure 1). The parameters (coverage, sensitivity and specificity) chosen for the statistical tests were calculated using the equations (3), (4) and (5) given under supplementary material.
Implementation
We have used Perl based programs exclusively developed for STIF algorithm to perform HMM related computation, searching, calculation of statistics and input - output parsing. Scripts for parsing, searching, statistics and other calculations like Z-Score and normalization are coded for the STIF algorithm in Perl as in equation (6) and (7) respectively (under supplementary material). The source code is available from the corresponding author upon request.
Discussion
A computational method, STIF, has been developed to search for potential transcription factor binding sites of stress-specific transcription factors, starting from Hidden Markov Models of nucleotide binding site patterns of cis-elements that are well-known to respond during stress situations in plants. The 19 models of cis-elements, based on abiotic stress transcription factor families, were built as Hidden Markov Models and were validated using Jackknifing method. We had applied our HMM-based search algorithm, STIF, to search 100 base pairs upstream of the gene with its 5′UTR. We identified 60 abiotic stress genes from well-known microarray databases based on the high stress-induced expression profiles. These genes were known to be upregulated during stress and their validated TFBS information is also clearly available. To evaluate the method further, we also searched against 1000 base pairs with its 5′UTR.
In our validation set, at a Z-score of 2.0 when searched 100 base pairs with 5′UTR, the sensitivity of the method is very high, since we identify 18 out of 20 hits (95% coverage) with only two false negatives Table 1 (see supplementary material). We therefore propose that a Z-score of 2.0 or more could be effective in not missing out the associated TFBS when searched for 100 base pairs with 5′UTR. In several instances, more than one transcription factor has been recorded for a stress gene of interest (for instance, COR15a has both DREB_AP2_EREBP and G_ABRE_bZIP (Figure 3a and Table 1 in supplementary material). The 60 stress genes that we have considered for validation are known to be upregulated during different types of stress - such as cold, dehydration, salinity etc. It is possible that, during a particular type of stress, any one of these transcription factors would selectively respond by binding upstream of the gene of interest.
Figure 3.

(a) The validation set of 11 stress responsive genes when searched for 100 base pairs with its 5′UTR with 11 stress responsive genes. The total number of false positives obtained during the search is compared against the total number of false negatives for various Z-score thresholds applied for the statistical tests. (b) Same as Figure 3a but for a validation set of 29 stress genes where search for TFBS was performed 1000 base pairs with its 5′UTR.
We also noticed that there are very few ‘validated’ TFBS which are mapped 100 base pairs upstream of stress genes. Therefore, we extended this validation to searches 1000 base pairs upstream of the gene and likewise a Z-score threshold of 1.5 is appropriate for 1000 base pairs with 5′UTR (Figure 3b and Table 2 under supplementary material). 90% sensitivity is achieved in STIF, where 71 out of 78 hits could be correctly identified with Z-scores above the threshold. As with most other algorithms, we notice that the method is not highly specific and can generate false positives. The specificities for searches in the validation set, by searching 100 base pairs and 1000 base pairs, is 57 and 18.6 (for Z-score threshold o1.5) and 54 and 20.4 (for Z-score threshold of 2.0), respectively. The difficulty in obtaining high specificities has been due to simple and short nucleotide patterns that describe some of the transcription factors like bHLH. Such TFs, would respond frequently and that too with very good match with HMM and are reflected as high scores. We have proposed an alternate normalized score for these frequently responding TFs. STIF employs Hidden Markov Models of binding site information of well-known plant transcription factors in stress. Microarray results of key stress upregulated genes in plants have shown that a large number of these genes are upregulated in response to a variety of genes generating redundancy in the dataset of stress upregulated plant genes. Further, the experimentally ‘validated’ results also indicate that more than one transcription factor can turn ON the stress genes in our dataset. The scoring schemes and thresholds established should be useful for dealing with redundancy and occurrence of multiple true positives.
We have built each HMM model and provided a stringent threshold for the scoring schemes. STIF algorithm, along with its database, is highly specific for plant stress cis-elements. However, this can be easily applied and extended to general systems after updating the HMM library and carefully standardizing the scoring scheme thresholds. The availability of such sensitive and specialized search algorithms can be very useful for addressing particular biological problems.
Conclusion
Computational Transcription Factor Binding Site (TFBS) prediction is a mature domain in the field of Bioinformatics. Various algorithms, stand-alone software and web servers are available for the effective prediction of transcription start from sequence information using knowledge based and motif based methods [5,18]. A wide array of TFBS prediction programs are available based on different biological contexts. For example a novel method for prokaryotic promoter prediction based on DNA stability that utilises structural properties of DNA is developed and analysed across different organisms [19], time-delay neural network based method (NNPP), is available specifically for the analysis of Drosophila melanogaster promoter regions [20]. An HMM based method based on markov chain optimization is available for the identification of hepatocyte nuclear factor 4-alpha in human genome [21]. Due to availability of such generic as well as specific TFBS prediction algorithms specific algorithms for prediction of transcription factor binding sites, users are recommended to use multiple programs to obtain a consensus result. STIF algorithm explained in this manuscript which uses HMM models of known Abiotic stress factors will be useful for further analysis and understanding of stress gene regulation in the plant model system Arabidopsis thaliana. Since no bioinformatics tool provides a complete solution for the transcription factor identification problem, it is always better to analyse the promoter regions with more than one algorithm or program that based on the biological context.
Supplementary material
Acknowledgments
The project is funded by a grant from the Department of Biotechnology, India. Susan Mary Varghese acknowledges the CSIR for the Senior Research Fellowship during the course of this research work. We thank UAS and NCBS (TIFR) for infrastructural support.
References
- 1.The Arabidopsis genome initiative Nature. 2000;408:796. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 2.Lander E, et al. Nature. 2001;409:860. [Google Scholar]
- 3.Steffens NO, et al. Nucleic Acids Res. 2004;1:D368. doi: 10.1093/nar/gkh017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh K. Curr Opin Plant Biol. 2002;5:430. doi: 10.1016/s1369-5266(02)00289-3. [DOI] [PubMed] [Google Scholar]
- 5.Bulyk ML, et al. Genome Biol. 2003;5:201. doi: 10.1186/gb-2003-5-1-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wingender E, et al. Nucleic Acids Res. 2001;29:316. [Google Scholar]
- 7.Zhang H, et al. Nucleic Acids Res. 2002;30:e121. doi: 10.1093/nar/gnf120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sandelin A, et al. Nucleic Acids Res. 2004;1:D91. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schones DE, et al. Bioinformatics. 2003;21:307. doi: 10.1093/bioinformatics/bth480. [DOI] [PubMed] [Google Scholar]
- 10.Pudimat R, et al. Bioinformatics. 2005;21:3082. doi: 10.1093/bioinformatics/bti477. [DOI] [PubMed] [Google Scholar]
- 11.Zhang W, et al. Bioinformatics. 2005;21:3074. doi: 10.1093/bioinformatics/bti490. [DOI] [PubMed] [Google Scholar]
- 12.Sakurai T, et al. Nucleic Acids Res. 2005;1:D368. [Google Scholar]
- 13.Button DK, et al. Nucleic Acids Res. 2006;1:D712. doi: 10.1093/nar/gkj136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warren GJ. Curr Biol. 1998;8:R514. doi: 10.1016/s0960-9822(07)00335-1. [DOI] [PubMed] [Google Scholar]
- 15.Schmid M. Nat Genetics. 2005;37:501. doi: 10.1038/ng1543. [DOI] [PubMed] [Google Scholar]
- 16.Guo A, et al. Bioinformatics. 2005;21:2568. doi: 10.1093/bioinformatics/bti334. [DOI] [PubMed] [Google Scholar]
- 17.Rhee SY, et al. Nucleic Acids Res. 2006;31:224. doi: 10.1093/nar/gkg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tompa M, et al. Nat Biotechnol. 2005;23:137. doi: 10.1038/nbt1140. [DOI] [PubMed] [Google Scholar]
- 19.Kanhere A, Bhansal M. BMC Bioinformatics. 2005;6:1. doi: 10.1186/1471-2105-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reese MG. Comput Chem. 2001;26:51. doi: 10.1016/s0097-8485(01)00099-7. [DOI] [PubMed] [Google Scholar]
- 21.Ellrott K, et al. Bioinformatics. 2002;18:S100. doi: 10.1093/bioinformatics/18.suppl_2.s100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.