

Abstract
P-cadherin is normally expressed in the basal layer of squamous epithelia and absent from the healthy intestine and colon. We have previously shown it to be expressed in all inflamed, hyperplastic and dysplastic intestinal and colonic mucosa. This study aimed to better understand the mechanisms controlling the expression of P-cadherin and the biological effects of its ectopic presence in the intestine and colon. We investigated the CpG methylation status of the P-cadherin (CDH3) promoter and P-cadherin mRNA and protein expression in cases of familial and sporadic colorectal cancer. The CDH3 promoter was hypomethylated in colonic aberrant crypt foci, colorectal cancer and occasionally in the normal epithelium adjacent to cancer, demonstrating a potential “field effect” of cancerisation. The hypomethylation was associated with induction of P-cadherin expression in the neoplastic colon (p<0.0001). We then created transgenic mice that over-expressed P-cadherin specifically in the intestinal and colonic epithelium under the liver fatty acid binding protein (L-FABP) promoter. Forced ectopic expression of P-cadherin accompanied by Indomethacin-induced inflammation resulted in a three-fold higher crypt fission rate within the small and large intestines in the homozygous mice compared to the wild-type animals (p<0.02). We conclude that epigenetic demethylation of the P-cadherin promoter in the human intestine permits its ectopic expression very early in the colorectal adenoma-carcinoma sequence and persists during invasive cancer. Induced P-cadherin expression, especially in mucosal damage, leads to an increased rate of crypt fission, a common feature of clonal expansion in gastrointestinal dysplasia.
Keywords: colorectal cancer, P-cadherin, stem cells, hypomethylation, crypt bifurcation
INTRODUCTION
Colorectal cancer (CRC) is currently the fourth most common malignancy and the second most common cause of death from cancer in US and UK (www.cancerresearchuk.org). It is typified by the adenoma to carcinoma sequence of tumour progression, with mutations in the Adenomatous Polyposis Coli (APC) gene believed to be one of the earliest genetic aberrations and present in up to 80% of sporadic colorectal cancers(1, 2). A heritable form of CRC, characterised by formation of multiple polyp adenomas, Familial Adenomatous Polyposis (FAP) also arises from APC gene mutations. APC binds to β-catenin and modulates its intracellular levels by marking it for degradation. Beta-catenin is a potent regulator of oncogene transcription and mediator of the Wnt signalling pathway, but has an additional role in cell-cell contact and adhesion through binding to cadherins(3).
Cadherins, as key cell adhesion molecules involved in organ development and morphogenesis, display tissue-specific distribution, identifying epithelial (E), neuronal (N) and placental (P) cadherin isoforms (reviewed in(4)). Through the interaction with β-catenin, cadherins are able to integrate cellular adhesion and tissue morphology with cell signalling and differentiation. P-cadherin is most strongly expressed in the stratified squamous epithelia, such as the esophagus in humans and placenta in mice(5). “Placentation” is characterised by physiological invasion and cellular division during gestation. In contrast to E-cadherin, which is present throughout the squamous epithelium and responsible for the maintenance of stratification and differentiation, the expression of P-cadherin is restricted to the basal cell layer, governing cell proliferation and glandular architecture(5). Altered expression of cadherins has been linked to changes in cellular behaviour and tumorigenesis(6). Downregulation of E-cadherin, often through epigenetic promoter silencing, has been associated with progression to breast(7), esophageal(8) and colorectal cancer(9), whilst upregulation of P-cadherin has been documented in esophageal, gastric, pancreatic, bladder and breast cancer(10-14). One study addressed the potential role of P-cadherin over-expression in tumorigenesis and observed changes in cell morphology, weakened cell-cell adhesion and increased cell motility in a pancreatic cancer cell line expressing P-cadherin. P-cadherin over-expression induced cytoplasmic accumulation of catenin p120ctn, a tyrosine kinase substrate involved in SRC oncogene-mediated cell transformation and cell signalling through growth factor receptors EGF and PDGF(15).
We have previously shown universal ectopic P-cadherin expression across different regenerative and hyperplastic lesions in the colon, such as colon injury, inflammation, restitution, hyperplasia and neoplasia(16, 17). In colorectal cancer, P-cadherin is present within the earliest morphologically recognised neoplastic lesion, the aberrant crypt focus (ACF). This is observed prior to, and independent of, disturbances in E-cadherin, catenins or APC expression, and persists into hyperplastic and adenomatous polyps(16). However, the mechanism underlying the ectopic P-cadherin expression in the ACF is unknown, as is the effect of P-cadherin on intestinal crypt biology. Gene-activating mutations and chromosomal amplifications of the P-cadherin chromosomal region 16q22.1 are not common in colorectal cancer (our unpublished observations) and regulation of P-cadherin expression in CRC by epigenetic alterations, such as CpG island methylation, has not been reported do date. Colorectal cancer is often coupled with epigenetic changes(18), although reports describing aberrant gene silencing through DNA hypermethylation outnumber examples of gene hypomethylation in CRC(19).
It has been shown that colorectal adenomas and hyperplastic polyps develop as a result of an increased rate of intestinal crypt proliferation, which occurs through the process of crypt fission, originating at the base of the crypt(20, 21). The majority of new crypts form during a short post-natal period, after which their numbers increase only gradually, following a cycle of around 3 months in mouse and 9-18 years in man(21). Adenomatous polyps bearing APC mutations expand through increased rates of intestinal crypt fission, identifying APC as one of the key precursors of adenoma progression(21). However, other factors which affect catenin/cadherin biology might also deregulate crypt fission and therefore influence the progression as well as initiation of colorectal tumours.
Here we show ectopic expression of P-cadherin in the colon mucosa from early stages of colorectal cancer, which is likely to be the result of abnormal gene activation through CDH3 gene promoter hypomethylation. Further, we describe a new in vivo model of induced P-cadherin expression in the mouse intestine and assess its effect on intestinal cell and crypt proliferation.
MATERIALS AND METHODS
Collection and characterization of tissue samples
Internationally accepted criteria such as the Vienna classification were used to characterise normal and CRC tissue in patients. Samples of normal colon were collected from patients with gastrointestinal complaints undergoing colonoscopy upon which no lesions were found. Samples of colon mucosa containing aberrant crypt foci, phenotypically normal colon (from outside the tumour margins) and colorectal cancer were collected at time of colectomy of CRC patients and either fixed in 10% neutral buffered formalin for embedding in paraffin blocks or immediately frozen in liquid nitrogen. Ethical approval was given at Queen Elisabeth Hospital, Birmingham, UK (ethical approval reference REC 3568), Leicester Royal Hospital, Leicester, UK (ethical approval ref. UHL 8500) and St. Mark’s Hospital, London, (ethical approval ref. 05/Q1605/66). Written informed consent was obtained from all individuals.
Microarray analysis of gene expression
Haematoxylin-eosin stained sections from frozen colon biopsies from either normal or adenomatous epithelium (> 80% dysplastic cells) were bisected. DNA was extracted from one section using the DNA Mini Kit (Qiagen, UK) and total RNA extracted from the other section using the Total RNA Kit (Sigma-Aldrich, UK). Adenoma DNA was screened for protein-truncating somatic APC mutations using fluorescence-SSCP and ABI3100 sequencers(22). Adenomas from 5 patients with known APC mutations were compared with normal samples for gene expression using Affymetrix HG-U133A array. Data were imported into GeneSpring (Agilent Technologies, UK), processed using MAS5 and subsequently median centred. Reporter genes called as absent in all cases were removed from the analysis and each gene was scaled to its median value within the normal control group. Differentially expressed genes between the tumour and normal control groups were selected by applying a 0.05 false discovery rate threshold. Differential gene expression in adenoma samples was measured relative to the median signal of all normal samples.
Immunohistochemistry
Sections (5 μm) of paraffin embedded tissue were stained as previously described(16). Mouse anti-human P-cadherin Ab (clone 56, BD Biosciences, UK) was used at 1:100 dilution followed by biotinylated goat anti-mouse/rabbit IgG (Dako, UK) at 1:100 dilution and ABC/HRP (Dako) diluted 1: 1000. Goat anti-mouse P-cadherin primary antibody (AF761, R&D Systems, UK) was used at 1:100 dilution and secondary rabbit anti-goat biotinylated Ab (Dako, UK) at 1:400 dilution, followed by ABC/HRP (Dako, UK) diluted 1: 1000. Primary rabbit anti-mouse Rac1/Cdc42 polyclonal Ab and delta 1 Catenin phospho Y228 (p120ctn) monoclonal Abs (Abcam) were used at 1:50 and 1:75 dilution, respectively, each followed by secondary goat anti-rabbit biotinylated Ab (Dako, UK) at 1:300 dilution and ABC/HRP (Dako, UK) diluted 1:1000. The sections were analysed on BH-2 light microscope (Olympus, UK).
DNA extraction for methylation analysis
DNA was extracted from frozen samples of colon mucosa with aberrant crypt foci (n=15), normal colon mucosa (n=19), phenotypically normal mucosa adjacent to cancer (n=20) and colorectal cancer specimens (n=15) using the GenElute Mammalian Genomic DNA kit (Sigma-Aldrich, UK) by homogenising 20mg of tissue in the following lysis buffer: 50 mM KCl, 10 mM Tris HCl, 2.5 mM MgCl2, 0.01% gelatin, 0.5% NP40 and 0.5% Tween 20) containing 200 μg/ml Proteinase K (Sigma-Aldrich, UK).
Methylation specific PCR
The methylation status of a 149 bp fragment upstream of the P-cadherin exon 1 was analysed using bisulphite modification, as previously published(23). Briefly, NaOH-denatured DNA was treated with 5.5 M sodium bisulfite in the presence of hydroquinone for 4 h at 55°C. Modified DNA was desalted using the Wizard DNA Purification Resin (Promega, UK) and amplified with oligonucleotides specific for unmethylated and methylated DNA. The primers used for detection of unmethylated DNA (product size 149 bp): 5′-TTGTGAGGGGGTGGGATTTTGTGGT-3′ (forward), 5′-ATAAAACAACTACCACAACAACAAACCAA-3′ (reverse) and methylated DNA (141 bp): 5′-CGAGGGGGCGGGATTTCGTGGC-3′ (forward), 5′-ACAACTACCGCGACGACGAACCGA-3′ (reverse). PCR conditions used for unmethylated DNA: 5 min. at 95°C, followed by 35 cycles of 95°C (1 min), 61°C (1 min), 72°C (1 min) and final extension at 72°C for 7min. Conditions used for methylated DNA were the same apart from 64°C annealing temperature.
Sequencing of the methylation-specific PCR products
The products of methylation specific PCR were sequenced with reference to a previously published method(24). The PCR products were purified using Wizard PCR Preps DNA Purification System (Promega, UK) and inserted into vector pGEM-T using the LigaFast DNA Ligation System (Promega, UK); 50 ng vector DNA was used in each ligation. The quantity of insert DNA used in ligation reactions was calculated by using the equation: mass insert (ng)= mass vector (ng) x size of insert (kb)/size of vector (kb) x molar ratio of insert:vector. Vector and insert DNA were mixed with DNA ligation buffer and ligase and incubated at room temperature for 15 minutes before being placed on ice. Cloned inserts were sequenced with T7 oligonucleotide as a primer (5′-TAATACGACTCACTATAGGG 3′).
RT-PCR of human P-cadherin
The cDNA was synthesised from RNA using the Reverse Transcription System (Promega, UK). RNA at 1 mg/ml was added to the reverse transcription mix containing random hexamer oligonucleodites and heated to 19°C (10 min), 42°C (50 min), 99°C (5 min), then placed on ice. Primers used for amplifying the P-cadherin cDNA were: Forward: 5′-ACGGCTCCACCACCACGG-3′ and Reverse: 5′-TGGCTGTGGAGGTTGGGAG-3′ using the following PCR conditions: 3 min at 95°C, followed by 35 cycles of 95°C (1 min), 56°C (1 min), 72°C (1 min) and final extension at 72°C for 7min.
Generation of P-cadherin transgenic mice
Genetic modification of animals and all animal procedures were approved by the appropriate local and national authorities. P-cadherin mouse transgene construct was created by inserting mouse P-cadherin cDNA flanked by EcoRV sites (gift from Dr. S. Hirohashi(25)) into pL596hGHΔB2 plasmid (gift from Dr. J Gordon(26)). This construct contains nucleotides -596 to +21 of the promoter for the rat fatty acid binding protein gene (L-FABP), expressed in the liver and the intestine in order to induce intestine-specific expression of the transgene. The L-FABP/mP-cad construct was introduced into F1xF1 embryos (F1 from CBAxB6 cross) by pronuclear injection and mice maintained on this background by breeding to F1 mice. Two lines of experimental wild-type, hetero and homozygous animals were generated after four generations of breeding from two founder females. All animals were fed a standard chow diet ad libitum.
PCR of the mP-cadherin transgene
Transgenic animals were screened for the presence of the P-cadherin transgene using whole tail lysis-PCR and the following primers: 5′-CCCATTCTGATTTTGATTTTTATCGTT-3′ (forward), and 5′-CTCGGAGACCACGCTGCGTAG-3′ reverse, resulting in a 250 bp amplicon. PCR conditions used were: 5 min. at 95°C, followed by 35 cycles of 95°C (1min), 60°C (1 min), 72°C (1 min) and final extension at 72°C for 7 min.
Microdissection of intestinal crypts from P-cadherin transgenic mice and assessment of cell proliferation and crypt fission
Wild-type, homozygous and heterozygous mice were injected intra-peritonealy with vincristine-sulphate (David Bull Laboratories, UK) at 1mg per kg body weight, which causes cell arrest in metaphase. Mice were sacrificed 2 hours later in a CO2 chamber followed by cervical dislocation. The Indomethacin-treated mice were injected with Indomethacin (85 mg per kg body weight in fresh 5% bicarbonate) or vehicle (sham controls) 24 hours before sacrifice. The entire gastrointestinal tract was isolated, rinsed in cold PBS, fixed in Carnoy’s fixative for 3 h, transferred to 75% ethanol and stored until micro-dissection. For immunohistochemistry, gut preparations from ethanol were rolled up into a ‘Swiss-roll’ and embedded in paraffin.
Assessment of proliferation and fission throughout the gut was performed using the “crypt micro-dissection” method, as previously described(21, 27). Briefly, representative samples of tissue from the proximal, mid and distal small intestine and colon were hydrated, hydrolysed and stained with the Feulgen reaction, as described before(28). For studying metaphase arrest, the mucosal crypts were gently teased apart with needles and gently compressed with a cover slip. The number of metaphases per crypt was counted in 20 crypts per animal using a light microscope at x160 magnification. The number of crypt fission events per 200 crypts per animal was determined under a dissection microscope at x45 magnification(29). All samples were counted in a blinded fashion.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism software and p-values constrained with a 95% confidence interval. For the statistical analyses of metaphase and crypt fission, individual two-sided, non-parametric t-tests were performed, followed by Bonferroni correction for multiple comparisons.
RESULTS
P-cadherin is ectopically expressed in both sporadic and familial colorectal cancer
We have previously demonstrated a lack of P-cadherin (CDH3) expression in the normal colon mucosa and its over-expression in sporadic colorectal adenomas(16) as well as in the injured and regenerating colon(17). The over-expression of P-cadherin is first noted in the aberrant crypt foci, the earliest phenotypic precursors of colorectal cancer(16). Here we investigated the presence of P-cadherin protein and mRNA expression in samples of normal colon mucosa from patients with no evidence of colorectal inflammation or dysplasia, cancer samples from patients with sporadic CRC and from phenotypically normal mucosa adjacent to the cancer as well as samples from patients with Familial Adenomatous Polyposis.
As expected, all samples of normal colon mucosa from non-CRC subjects were negative for P-cadherin (n=19, Figure 1A). However, 1 out of 20 samples of phenotypically normal but adjacent to cancer mucosa, from sporadic CRC patients, showed aberrant P-cadherin expression, as did all of the cancer samples (n=15). The staining was confined to the epithelial cells (columnar and goblet). Positive staining for P-cadherin was also noted in all FAP samples (n=5). No staining was observed in samples from patients with a mild inflammation of the colon - diverticular colitis (data not shown). Normal esophageal squamous tissue which constitutively expresses P-cadherin in the basal layer was used as positive tissue control and, without the primary antibody, as the negative experimental control (Figure 1A).
Figure 1.
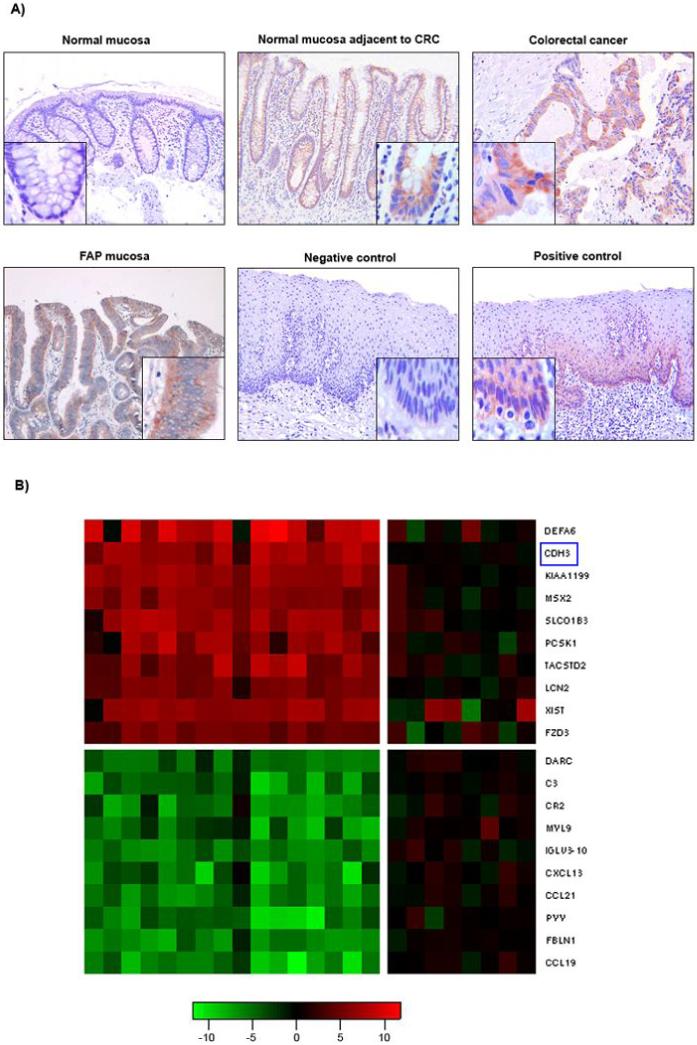
A) Immunohistochemical analysis of P-cadherin expression in normal and colorectal colon cancer mucosa. Images were taken at 100x and 250x magnification (inserts). Upper row: normal colon mucosa (no evidence of colorectal neoplasia) showing absence of P-cadherin; phenotypically normal colon mucosa adjacent to colorectal cancer from a patient with sporadic CRC (P-cadherin expression was found in 1 sample out of 20); colon mucosa from sporadic colorectal cancer showing P-cadherin expression.
Lower row: colon mucosa from a patient with Familial Adenomatous Polyposis (FAP) showing P-cadherin expression; negative control (esophageal epithelium, no primary Ab); positive control (esophageal epithelium, constitutive P-cadherin expression within the basal layer).
B) Expression heat-map for the top 10 up- and downregulated genes in FAP adenomas. Colorectal polyp adenomas and matched normal control tissue from five FAP patients with known germline APC mutations (total of 16 adenoma and 8 normal samples), were analysed for differential gene expression on a microarray of 14,500 genes. The red/green signals (columns 1-16) represent the adenoma samples relative to the median of all normal samples. Negative controls (columns 1-8) are the normal samples relative to the median of all normal samples, showing low variability among the normal samples. The genes are presented in decreasing order of expression: top (DEFA6) = most upregulated, bottom (CCL19) = most downregulated. P-cadherin (CDH3, marked by blue rectangle) was the second highest upregulated gene, present in polyp adenomas in an excess of 29-fold as compared to normal colon mucosa. Log2 ratio scale is shown below the map.
In order to investigate the timing and significance of P-cadherin expression in relation to APC aberration, a known early change in colorectal cancer, we used samples from patients with Familial Adenomatous Polyposis. We studied the expression of P-cadherin in colorectal polyp adenomas from FAP patients using microarray gene expression analysis (Figure 1B). Biopsies of colorectal adenomas (n=16) and normal tissue (n=8) were collected from five FAP patients with known germline mutations in the APC gene (S1190X, Q163X, 798FS, 1157FS and R213X). All biopsies were bisected for subsequent DNA and RNA extraction and snap-frozen. From each adenoma, the DNA extracted from half the biopsy was analysed for APC mutations. Biopsies with germline APC mutations along with the normal mucosa were subsequently analysed for gene expression using the Affymetrix HG-U133A gene array with 14,500 human genes.
We identified 205 significantly upregulated and 616 significantly downregulated genes in the analysed adenomas (Table 1). These comprised 15 previously identified Wnt targets (IL-8, MYB, CCND1, MMP7, CD44, DKK3, EPHB3, MSX1, MYC, SOX9, ENC1, EPHB2, ETS2, MET, MSX2). Remarkably, P-cadherin was the second highest upregulated gene (after Defensin), present in an excess of 29-fold over normal colon mucosa. A member of the Frizzled family of proteins (FZD3) which transmit Wnt signals through interaction with β-catenin was found to be upregulated 12-fold. Among the most significantly downregulated genes were several chemokine receptors and members of the complement pathway. A heat-map with a scale representing the log2 fold ratio for the 10 most upregulated and downregulated genes in the adenoma samples is shown in Figure 1B; the transcripts and their fold-changes are summarised in Table 1.
Table 1.
Summary of the 10 most up- and down-regulated transcripts and their fold-change relative to the median value of the normal samples
| Gene symbol | Gene name | Log2 ratio (fold change up-regulated) | Gene symbol | Gene name | Log2 ratio (fold change down-regulated) |
|---|---|---|---|---|---|
| DEFA6 | Defensin | 5.13 (35x) | DARC | Blood group-Duffy system | -4.03 (16x) |
| CDH3 | P-cadherin | 4.84 (29x) | C3 | Complement component 3 | -4.11 (17x) |
| KIAA1199 | Inner ear protein | 4.57 (24x) | CR2 | Complement component receptor 2 | -4.12 (17x) |
| MSX2 | Muscle segment homeobox 2 | 4.45 (22x) | MYL9 | Myosin light chain 9 | -4.30 (20x) |
| SLCO1B3 | 4.21 (19x) | IGLV3-10 | -4.32 (20x) | ||
| PCSK1 | Proprotein convertase 1 | 3.96 (16x) | CXCL13 | Chemokine ligand 13 | -4.34 (20x) |
| TACSTD2 | Tumour-associated Ca signal transducer 2 | 3.84 (14x) | CCL21 | Chemokine ligand 21 | -4.43 (22x) |
| LCN2 | Lipocalin 2 | 3.59 (12x) | PYY | Peptide YY | -4.62 (24x) |
| XIST | X inactivation-specific transcript | 3.58 (12x) | FBLN1 | Fibulin 1 | -4.63 (25x) |
| FZD3 | Frizzled homolog 3 | 3.55 (12x) | CCL19 | Chemokine ligand 19 | -5.33 (40x) |
Aberrant expression of P-cadherin in colon neoplasia is associated with promoter hypomethylation
Analysis of the coding region of the CDH3 gene using single strand conformation polymorphism assays and DNA sequencing did not reveal any mutations in patients with CRC (unpublished observations) and our further work focused on the P-cadherin promoter. The upstream region of the P-cadherin gene has a GC content of 75% and contains 14 CpG dinucleotides grouped into three CpG islands. The third CpG island spans a 300 bp region proximal to exon 1 and harbours the CDH3 transcription initiation site.
We hence investigated the methylation pattern of the P-cadherin promoter in samples of sporadic colorectal cancer. This is the common form of CRC and can provide a more complete spectrum of samples in different stages of cancer development. In contrast to sporadic CRC, patients with FAP generally undergo prophylactic colonic resections before cancer, reducing the availability and diversity of samples across the adenoma-carcinoma sequence. To analyse the P-cadherin promoter methylation status we used bisulphate DNA treatment followed by methylation-specific PCR for the CDH3 promoter (NCBI sequence accession no. X95824).
Methylation analysis was carried out on DNA isolated from sporadic CRC biopsies (n=15), samples of cancer-adjacent, phenotypically normal colon mucosa (n=20) and normal colon (n=19), as well as samples of aberrant crypt foci (n=15). Normal colon samples demonstrated presence of both methylated and unmethylated P-cadherin promoter DNA (Figure 2A) as did 16/20 samples of ‘normal’ mucosa adjacent to CRC, suggesting an inactive (silenced) promoter. However, in the remaining four samples only unmethylated PCR products were detected (Figure 2B). Sequencing of the methylated PCR products revealed either partial or complete methylation of all 14 CpG dinucleotides. In contrast to the normal mucosa, all cancer samples showed the presence of only unmethylated DNA (Figure 2C), indicating an active P-cadherin promoter. Likewise, the P-cadherin promoter was unmethylated in all of the ACF samples, indicating that this methylation change occurs early in the development of colorectal cancer (Figure 2D).
Figure 2.
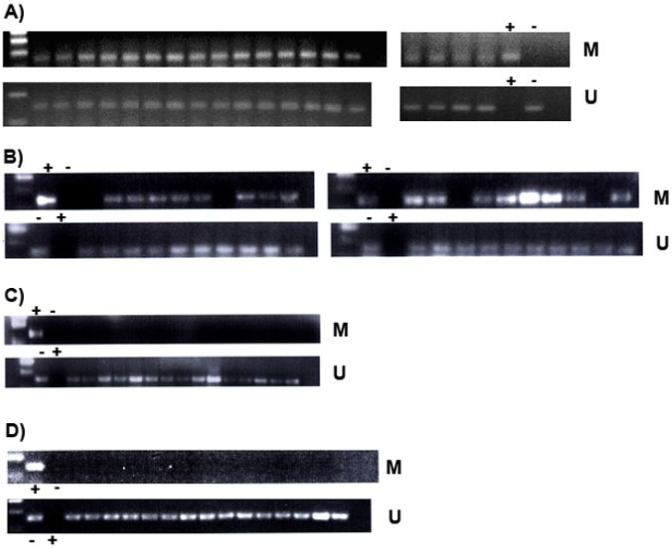
Analysis of the methylation status of the P-cadherin (CDH3) promoter in normal colon mucosa, phenotypically normal mucosa adjacent to CRC and CRC mucosa and aberrant crypt foci. DNA size standard is in the first lane of each panel except the right-hand panels in 2A.
A) Normal colon (n=19) shows presence of both methylated (141 bp, top row) and unmethylated (149 bp, bottom row) DNA.
B) Phenotypically normal colon tissue adjacent to cancer (n=20) shows both methylated and unmethylated PCR products, with the exception of four samples in which CDH3 is unmethylated (see text).
C) Colorectal cancer tissue (n=15) shows only unmethylated PCR products.
D) Mucosa from aberrant crypt foci (n=15) shows only unmethylated PCR products.
Bisulfite modified DNA from BJAB, a human lymphoma cell line with a fully methylated CDH3 promoter was used as PCR template for a positive methylation control (+) in all panels. Absence of methylation (-) was controlled for using the HT1080 human fibrosarcoma cell line, in which the CDH3 promoter is fully unmethylated.
In order to assess whether P-cadherin promoter methylation changes correlate with its transcription, the expression of P-cadherin mRNA was analysed in the cancer and cancer-adjacent normal colon mucosa samples using RT-PCR (Figure 3). Integrity of mRNA was assessed by amplification of β-actin (lower panel in Figures 3A and 3B). Consistent with the CDH3 promoter methylation patterns, cancer-adjacent normal colon mucosa (with the exception of one sample) showed absence of P-cadherin mRNA. The single sample expressing P-cadherin mRNA was one of the four samples that showed lack of promoter methylation and P-cadherin expression (Figures 2B and 1A). There was no detectable P-cadherin RNA in the remaining three unmethylated normal mucosa samples, giving an overall correlation coefficient of 0.84 (Pearson r, p<0.0001) between promoter methylation status and mRNA expression in the tissue adjacent to cancer. All of the cancer samples fully correlated with the unmethylated status of the promoter and were found to express P-cadherin mRNA (Figure 3B).
Figure 3.
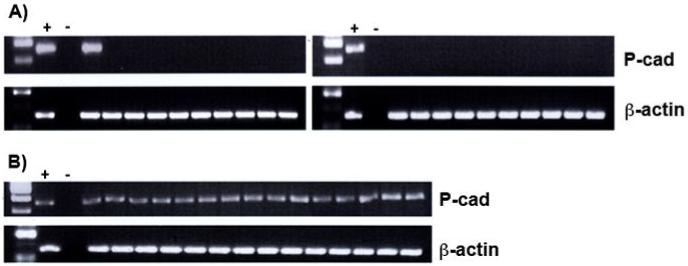
P-cadherin mRNA expression in normal and CRC colon mucosa, analysed using RT-PCR.
A) Normal colon mucosa adjacent to CRC; P-cadherin transcript (341 bp) is absent in the normal tissue (top panels) apart from one patient, as described in the text.
B) Samples of colorectal cancer all show abnormal ectopic expression of P-cadherin mRNA.
First two lanes following the size standard are the positive and negative control, respectively. Positive control: HaCaT human keratinocyte cell line known to express P-cadherin (gift from Dr Chris Tselepis); negative control: no template cDNA. Expression of β-actin (767 bp) was used as a positive control for mRNA integrity.
Phenotypic effects of induced mP-cadherin expression in the mouse intestine in vivo
To investigate the in vivo effects of ectopic P-cadherin expression alone, in the context of intact APC, we generated transgenic mice expressing P-cadherin in the intestine under the promoter for the liver fatty acid biding protein gene (L-FABP), which is expressed only in the mouse intestine and liver. This promoter has been extensively used to induce foreign gene expression in the mouse intestine(26). The plasmid construct, containing the mouse P-cadherin transgene (mP-cad) and L-FABP promoter, L-FABP/mP-cad, was introduced into wild-type mice by pronuclear injection (see Materials and Methods). The presence of the mP-cad transgene in the transgenic mice was confirmed by PCR of whole tail tissue lysate (Figure 4A) in all of the homozygous mice (total of 251 out of 505 genotyped mice). Tissue-specific expression of the P-cadherin transgene transcript in the homozygous mice was confirmed by RT-PCR and protein expression in the intestine and colon demonstrated using immunohistochemistry (Figure 4B). In order to confirm the functionality of the P-cadherin transgene, we investigated the events induced by P-cadherin over-expression, such as EGF/PDGF signalling evident through upregulation of Rac1 and Cdc42 and p120ctn translocation from the membrane into the cytoplasm and the nucleus(15) in four mice from each group. Wild-type mice showed very weak Rac1/Cdc42 expression and strictly membrane-associated p120ctn. In the intestines of homozygous mice, there was a notable upregulation of Rac1/Cdc42 and translocation of p120ctn from the membrane into the cytoplasm and, in some regions, the nuclei. Particularly strong upregulation of Rac1/Cdc42 coincided with the translocation of p120ctn into the nucleus (Figure 4C). The heterozygous mice had regions of increased Rac1/Cdc42 staining and some cytoplasmic but no nuclear localisation of p120ctn (data not shown).
Figure 4.
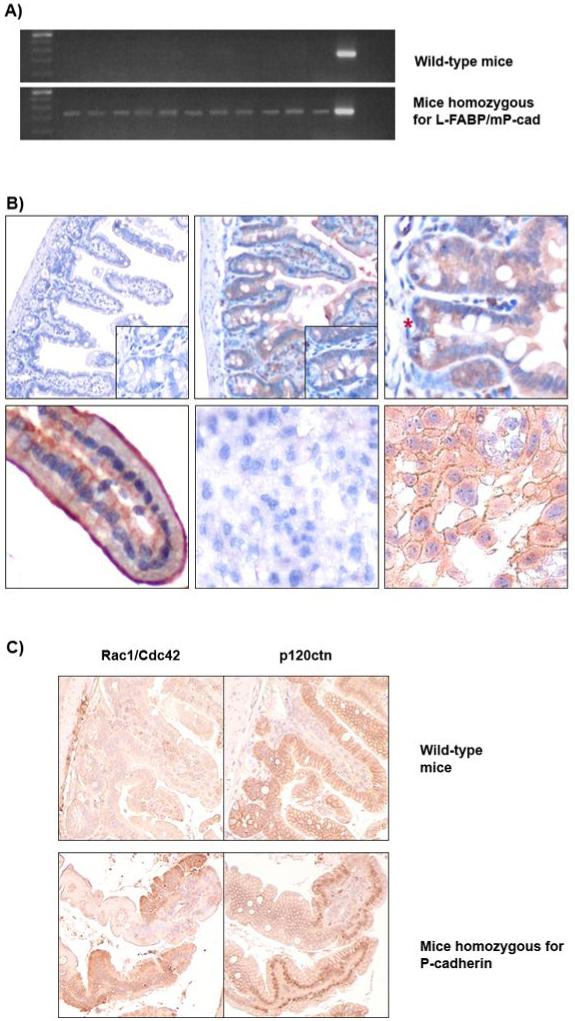
Analysis of the L-FABP/mP-cad transgenic mice for the presence of the mP-cad transgene.
A) Transgene-specific PCR of the tail tissue lysate. The wild-type mice were negative for the mP-cad transgene (250 bp) and all of the homozygous transgenic mice show the presence of mP-cad.
B) Immunohistochemical labelling of intestinal tissue from wild-type (n=4) and transgenic mice (n=4); magnification x100, x200 and x400. No transgene is detected in the small intestine of the wild-type mice (upper left) whereas the intestines of homozygous mice specifically express mP-cad (upper middle). Transgene mP-cad expression in the homozygous mice varied between animals, from medium intensity cytoplasmic to strong membranous. An intestinal crypt in fission is shown marked by a red asterisk (upper right) as well as an example of membranous staining within a villus (lower left). Primary antibody was omitted when staining mouse placenta as the negative control (lower middle), whilst the positive control (lower right) shows membranous and cytoplasmic expression of native P-cadherin in mouse placenta.
C) Demonstration of a functional P-cadherin transgene through Rac1/Cdc42 upregulation and p120ctn translocation. Upper row: wild-type mice (n=4), lower row homozygous mice (n=4) stained for Rac1/Cdc42 (left-hand panels) and p120ctn (right-hand panels), shown at 200x magnification. In the wild-type mice, Rac1/Cdc42 staining is low and correlates with membrane-bound p120ctn. In the presence of the mP-cad transgene (homozygous mice), upregulated Rac1/Cdc42 expression is associated with translocation of p120ctn into the nucleus as well as the cytoplasm.
The phenotypic effects of ectopic P-cadherin expression, specifically observing mucosal cell proliferation and intestinal crypt fission (as illustrated in Figure 4B), were assessed microscopically in the wild-type and homozygous animals (n=12 in each group). The degree of cell proliferation was evaluated as the number of cells arrested in metaphase by vincristine-sulphate, administered 2 h prior to sacrifice. The number of dividing cells per crypt, counted in 20 crypts per animal, was determined for proximal to distal segments along the small intestine (SI) and the colon (Figure 5A). Mice homozygous for L-FABP/mP-cad showed a marginally significant trend of fewer dividing cells per crypt (p=0.048), which decreased to p=0.053 following correction for multiple comparisons. In the heterozygous mice, the number of cells in metaphase was intermediate of those observed in wild-type and homozygous mice but there was no statistical significance. The intestinal crypt fission rate was calculated as the number of dividing crypts per 200 crypts per animal, which has been demonstrated as a method superior to the more traditional approaches using Bromodeoxyuridine incorporation or proliferating cell nuclear antigen (PCNA) staining(29). A non-significant increase in the crypt fission was found in homozygous mice (uncorrected p=0.053, Figure 5B). Among the heterozygous mice, the rate of crypt fission varied, again being intermediate at three of the analysed sites along the small and large intestine (see Supplementary Figure 2).
Figure 5.
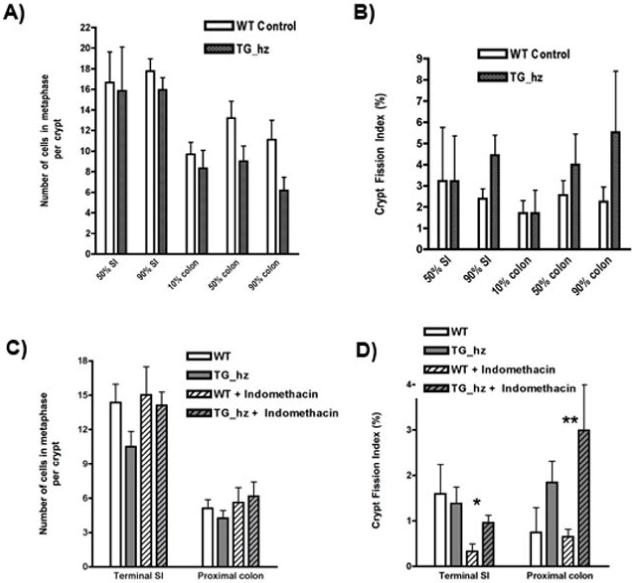
A and B: Cell division (metaphases per crypt) and intestinal crypt bifurcation analysis in the small intestine and colon of the wild-type mice (WT, n=12) and the mice homozygous for the mP-cad transgene (TG_hz, n=12). Data are expressed as mean ± SEM.
A) Number of dividing cells per crypt (number of metaphases per 20 crypts per animal) shows non-significant reduction in the number of dividing cells in the transgenic mice compared to wild-type.
B) Crypt fission index (number of dividing crypts per 200 crypts per animal) shows an increase in rate of crypt fission in the transgenic mice (non-significant).
C and D: Cell division and crypt bifurcation in wild-type and mP-cad transgenic mice injected with Indomethacin (85 mg per kg of body weight) to induce intestinal inflammation. Animals analysed: wild-type (WT, n=8), transgenic homozygous (TG_hz, n=6), WT+Indomethacin (n=9) and TG_hz + Indomethacin (n=7).
C) Number of dividing cells per crypt shows no significant difference between the wild-type and the homozygous animals with or without Indomethacin injection.
D) Intestinal crypt division rate was higher in the proximal colon of the homozygous mice as compared to the wild-type. Statistically significant difference was found between the wild-type and the homozygous animals treated with Indomethacin in both terminal small intestine (*p=0.04) and proximal colon (**p=0.02).
We have previously shown in the jejunum of transgenic mice that other genes regulating mucosal physiology may be biologically neutral unless epithelial integrity is compromised (30). We therefore investigated the phenotypic effects of the transgene in the context of proximal to middle gut intestinal inflammation and ulceration induced by subcutaneous administration of Indomethacin (85 mg per kg body weight) 24 h prior to animal sacrifice (Figure 5C). Within the sites available for analysis, terminal SI and proximal colon, the previously observed downward trend in the number of metaphases per crypt in homozygous L-FABP/mP-cad animals was reproduced but remained non-significant. Following Indomethacin injury this effect was not observed. Furthermore, the increased crypt fission rate within the intestine of homozygous L-FABP/mP-cad animals was significantly higher following Indomethacin injection as compared to wild-type mice (Figure 5D; p=0.04 for terminal SI and p=0.02 for proximal colon, following Bonferroni correction). Indomethacin injury alone had no effect on either cell division or crypt fission, as treated wild-type animals did not differ significantly from the untreated ones. These results strongly suggest that ectopic expression of P-cadherin in the intestine, exacerbated by mucosal damage, can lead to increased crypt fission.
DISCUSSION
The expression of P-cadherin within tissues is strictly programmed, due to its role in the development of normal stratified squamous epithelium(5). This study demonstrates that in colorectal cancer the control of P-cadherin expression is disrupted through abnormal promoter methylation, induced gene transcription and ectopic presence of P-cadherin from the earliest stages of carcinoma development. Increased P-cadherin expression was observed in both sporadic cancer and heritable colorectal adenomas and gene expression analysis showed high upregulation of P-cadherin in familial adenomatous polyps. Forced expression of P-cadherin accompanied by inflammation in the mouse intestine in vivo resulted in an increased rate of intestinal crypt fission.
Over-expression through promoter hypomethylation of P-cadherin, a gene not classically associated with imprinting or a defined proto-oncogene, has been reported previously in a proportion of breast cancer cases(31). Furthermore, in inflamed premalignant gastrointestinal mucosa such as Barrett’s metaplasia and ulcerative colitis similar de novo expression of P-cadherin occurs early in carcinogenesis(32), implying that these changes could have similar epigenetic control. Our study shows that a CpG island in the P-cadherin (CDH3) promoter is methylated in the normal colon mucosa and demethylated in colorectal cancer. Importantly, we demonstrate an early disruption of normal P-cadherin promoter methylation within aberrant crypt foci, potential precursors of CRC, indicating a putative role of P-cadherin in the initiation of colorectal cancer. Hypomethylation of the P-cadherin promoter in CRC appears to be gene-specific and unlikely due to genome-wide demethylation since P-cadherin maps adjacent to E-cadherin whose promoter is hypermethylated in a proportion of colorectal cancers(9).
We have previously reported early ectopic P-cadherin expression, both transient and sustained, in the reparative colon mucosa and hyperplastic crypts and benign adenomatous tumours(16, 17). This study demonstrates that the aberrant expression of P-cadherin persists in sporadic colorectal cancer. We show that the ectopic expression of P-cadherin in the colon is not limited to the neoplastic region but can extend into the adjacent, phenotypically normal, colon mucosa. This finding resembles the previously described “field effect” phenomenon which suggests that precancerous cells neighbouring cancer have some but not all of the genetic alterations of the fully developed tumour(33, 34). This concept has been illustrated with hypermethylation in colorectal cancer(35), but has not been documented in the context of methylation loss until now. The observed field effect indicates that demethylation of P-cadherin might be an early event in the colorectal mucosa, prior to any morphological changes. It is possible that this early epigenetic change triggers downstream events, which, in an inflamed gut niche, can lead to bifurcation, clonal expansion and ultimately neoplastic changes. One of the candidate downstream pathways could be the upregulation of Rac1 and nuclear translocation of p120ctn, as observed in the mice homozygous for P-cadherin transgene. Upregulation of Rac1 has been shown to induce nuclear localisation of p120ctn(36), which has been associated with early stages of lobular breast cancer(37).
Crypt fission is the dominant mechanism of early adenoma development in the intestine, whereas top-down growth into adjacent crypts is prevalent in advanced dysplasia later in carcinogenesis(21, 38). Our present findings further support a potential role of P-cadherin in early adenoma formation and neoplastic development. The enhanced (albeit statistically non-significant) rate of intestinal crypt fission induced by the transgenic P-cadherin in our murine model was further increased following the injection of Indomethacin. Indomethacin, administered as a single injection, induces acute epithelial inflammation and mucosal injury, which reach maximum one day post-injection and resolve completely after 3-7 days(39). Our previous work shows upregulation of P-cadherin in the colon mucosa following severe injury (radiation proctitis)(17). In the current study, both injury and P-cadherin transgene maximally resulted in crypt fission. This might indicate that prior endogenous P-cadherin expression is required to maximally exploit the exogenous inflammatory stimuli through activation of downstream factors, the ‘fertile soil’ theory(40) or that a threshold of P-cadherin level necessary for a significant increase in rate of crypt proliferation was reached by a subtle upregulation of the native P-cadherin due to Indomethacin-induced injury combined with the transgene expression.
In sporadic colorectal adenomas, ectopic P-cadherin is functional with respect to β-catenin binding, and, importantly, expressed in the absence of disturbances in the APC gene(16), which are viewed as a key initiating event of malignant transformation in the colon (2, 41, 42). In addition, other members of the Wnt signalling pathway are known to play critical roles in colorectal tumorigenesis in the presence of wild-type APC and β-catenin(43, 44). Mutations in APC and β-catenin have been shown to give rise to CRC independently(45), and an APC-independent mode of β-catenin degradation has also been reported(46). Another report showed that increased β-catenin signalling can lead to upregulation of P-cadherin expression(47), while a recent study further highlighted the complexities of the Wnt pathway by uncovering that one of its targets, C-myc oncogene, can rescue the perturbed cell differentiation and proliferation phenotype induced by APC loss in the mouse intestine(48). We show here that in vivo expression of P-cadherin, in the context of an intestinal inflammation and intact APC and β-catenin, is sufficient to trigger an increased rate of crypt proliferation. In this regard, P-cadherin might be implicated in governing the niche of stem cells as has been demonstrated for other cadherins at other epithelial sites(49, 50).
In conclusion, we show here that ectopic expression of P-cadherin in the mouse intestine can lead to increased crypt fission under conditions of intestinal damage and provide another example of gene promoter hypomethylation associated with ectopic protein expression very early in colorectal cancer development.
Acknowledgements
We thank Dr. Ian Rosewell for mice breeding and maintenance, Mr. Neil Bailey for technical advice, Dr. Paul Atherfold for technical help, Mr Dion Morton, Birmingham and Prof. Theresa Pretlow, Ohio for help with selecting tissue samples and Sir Walter Bodmer, Oxford for cell lines and helpful comments on our data.
Financial support: This work was funded by Cancer Research, UK, University Hospitals of Leicester and The Wellcome Trust, UK.
Abbreviations
- ACF
aberrant crypt foci
- APC
adenomatous polyposis coli
- CRC
colorectal cancer
- FAP
familial adenomatous polyposis
- IHC
immunhistochemistry
- L-FABP
liver fatty acid binding protein
- P-cad
P-cadherin
- RT-PCR
reverse transcriptase PCR
- SI
small intestine
Footnotes
There are no conflicts of interest to declare.
References
- 1.Powell SM, Zilz N, Beazer-Barclay Y, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359(6392):235–7. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 2.Takayama T, Ohi M, Hayashi T, et al. Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology. 2001;121(3):599–611. doi: 10.1053/gast.2001.27203. [DOI] [PubMed] [Google Scholar]
- 3.Jamora C, Fuchs E. Intercellular adhesion, signalling and the cytoskeleton. Nat Cell Biol. 2002;4(4):E101–8. doi: 10.1038/ncb0402-e101. [DOI] [PubMed] [Google Scholar]
- 4.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6(8):622–34. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 5.Shimoyama Y, Hirohashi S, Hirano S, et al. Cadherin cell-adhesion molecules in human epithelial tissues and carcinomas. Cancer Res. 1989;49(8):2128–33. [PubMed] [Google Scholar]
- 6.Wheelock MJ, Johnson KR. Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol. 2003;19:207–35. doi: 10.1146/annurev.cellbio.19.011102.111135. [DOI] [PubMed] [Google Scholar]
- 7.Berx G, Van Roy F. The E-cadherin/catenin complex: an important gatekeeper in breast cancer tumorigenesis and malignant progression. Breast Cancer Res. 2001;3(5):289–93. doi: 10.1186/bcr309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corn PG, Heath EI, Heitmiller R, et al. Frequent hypermethylation of the 5′ CpG island of E-cadherin in esophageal adenocarcinoma. Clin Cancer Res. 2001;7(9):2765–9. [PubMed] [Google Scholar]
- 9.Wheeler JM, Kim HC, Efstathiou JA, Ilyas M, Mortensen NJ, Bodmer WF. Hypermethylation of the promoter region of the E-cadherin gene (CDH1) in sporadic and ulcerative colitis associated colorectal cancer. Gut. 2001;48(3):367–71. doi: 10.1136/gut.48.3.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanders DS, Bruton R, Darnton SJ, et al. Sequential changes in cadherincatenin expression associated with the progression and heterogeneity of primary oesophageal squamous carcinoma. Int J Cancer. 1998;79(6):573–9. doi: 10.1002/(sici)1097-0215(19981218)79:6<573::aid-ijc4>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 11.Shimoyama Y, Hirohashi S. Expression of E- and P-cadherin in gastric carcinomas. Cancer Res. 1991;51(8):2185–92. [PubMed] [Google Scholar]
- 12.Palacios J, Benito N, Pizarro A, et al. Anomalous expression of P-cadherin in breast carcinoma. Correlation with E-cadherin expression and pathological features. Am J Pathol. 1995;146(3):605–12. [PMC free article] [PubMed] [Google Scholar]
- 13.Bryan R, Atherfold P, Yeo Y, et al. Cadherin switching dictates the biology of transitional cell carcinoma of the bladder: ex vivo and in vitro studies. J Pathol. 2008;215(2):184–94. doi: 10.1002/path.2346. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura T, Furukawa Y, Nakagawa H, et al. Genome-wide cDNA microarray analysis of gene expression profiles in pancreatic cancers using populations of tumor cells and normal ductal epithelial cells selected for purity by laser microdissection. Oncogene. 2004;23(13):2385–400. doi: 10.1038/sj.onc.1207392. [DOI] [PubMed] [Google Scholar]
- 15.Taniuchi K, Nakagawa H, Hosokawa M, et al. Overexpressed P-cadherin/CDH3 promotes motility of pancreatic cancer cells by interacting with p120ctn and activating rho-family GTPases. Cancer Res. 2005;65(8):3092–9. doi: 10.1158/0008.5472.CAN-04-3646. [DOI] [PubMed] [Google Scholar]
- 16.Hardy RG, Tselepis C, Hoyland J, et al. Aberrant P-cadherin expression is an early event in hyperplastic and dysplastic transformation in the colon. Gut. 2002;50(4):513–9. doi: 10.1136/gut.50.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardy RG, Brown RM, Miller SJ, et al. Transient P-cadherin expression in radiation proctitis; a model of mucosal injury and repair. J Pathol. 2002;197(2):194–200. doi: 10.1002/path.1092. [DOI] [PubMed] [Google Scholar]
- 18.Wong JJ, Hawkins NJ, Ward RL. Colorectal cancer: a model for epigenetic tumorigenesis. Gut. 2007;56(1):140–8. doi: 10.1136/gut.2005.088799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Das PM, Singal R. DNA methylation and cancer. J Clin Oncol. 2004;22(22):4632–42. doi: 10.1200/JCO.2004.07.151. [DOI] [PubMed] [Google Scholar]
- 20.Araki K, Ogata T, Kobayashi M, Yatani R. A morphological study on the histogenesis of human colorectal hyperplastic polyps. Gastroenterology. 1995;109(5):1468–74. doi: 10.1016/0016-5085(95)90632-0. [DOI] [PubMed] [Google Scholar]
- 21.Wasan HS, Park HS, Liu KC, et al. APC in the regulation of intestinal crypt fission. J Pathol. 1998;185(3):246–55. doi: 10.1002/(SICI)1096-9896(199807)185:3<246::AID-PATH90>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 22.Groves C, Lamlum H, Crabtree M, et al. Mutation cluster region, association between germline and somatic mutations and genotype-phenotype correlation in upper gastrointestinal familial adenomatous polyposis. Am J Pathol. 2002;160(6):2055–61. doi: 10.1016/S0002-9440(10)61155-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93(18):9821–6. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29(13):E65–5. doi: 10.1093/nar/29.13.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimoyama Y, Yoshida T, Terada M, Shimosato Y, Abe O, Hirohashi S. Molecular cloning of a human Ca2+-dependent cell-cell adhesion molecule homologous to mouse placental cadherin: its low expression in human placental tissues. J Cell Biol. 1989;109(4 Pt 1):1787–94. doi: 10.1083/jcb.109.4.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong MH, Hermiston ML, Syder AJ, Gordon JI. Forced expression of the tumor suppressor adenomatosis polyposis coli protein induces disordered cell migration in the intestinal epithelium. Proc Natl Acad Sci U S A. 1996;93(18):9588–93. doi: 10.1073/pnas.93.18.9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bashir O, FitzGerald AJ, Goodlad RA. Both suboptimal and elevated vitamin intake increase intestinal neoplasia and alter crypt fission in the ApcMin/+ mouse. Carcinogenesis. 2004;25(8):1507–15. doi: 10.1093/carcin/bgh137. [DOI] [PubMed] [Google Scholar]
- 28.Park HS, Goodlad RA, Wright NA. Crypt fission in the small intestine and colon. A mechanism for the emergence of G6PD locus-mutated crypts after treatment with mutagens. Am J Pathol. 1995;147(5):1416–27. [PMC free article] [PubMed] [Google Scholar]
- 29.Alferez D, Goodlad RA. To best measure cell proliferation in samples from the intestine. Cell Prolif. 2007;40(2):231–40. doi: 10.1111/j.1365-2184.2007.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marchbank T, Cox HM, Goodlad RA, et al. Effect of ectopic expression of rat trefoil factor family 3 (intestinal trefoil factor) in the jejunum of transgenic mice. J Biol Chem. 2001;276(26):24088–96. doi: 10.1074/jbc.M101363200. [DOI] [PubMed] [Google Scholar]
- 31.Paredes J, Albergaria A, Oliveira JT, Jeronimo C, Milanezi F, Schmitt FC. P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with CDH3 promoter hypomethylation. Clin Cancer Res. 2005;11(16):5869–77. doi: 10.1158/1078-0432.CCR-05-0059. [DOI] [PubMed] [Google Scholar]
- 32.Bailey T, Biddlestone L, Shepherd N, Barr H, Warner P, Jankowski J. Altered cadherin and catenin complexes in the Barrett’s esophagus-dysplasia-adenocarcinoma sequence: correlation with disease progression and dedifferentiation. Am J Pathol. 1998;152(1):135–44. [PMC free article] [PubMed] [Google Scholar]
- 33.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6(5):963–8. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 34.Giovannucci E, Ogino S. DNA methylation, field effects, and colorectal cancer. J Natl Cancer Inst. 2005;97(18):1317–9. doi: 10.1093/jnci/dji305. [DOI] [PubMed] [Google Scholar]
- 35.Shen L, Kondo Y, Rosner GL, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97(18):1330–8. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 36.Lanning CC, Ruiz-Velasco R, Williams CL. Novel mechanism of the co-regulation of nuclear transport of SmgGDS and Rac1. J Biol Chem. 2003;278(14):12495–506. doi: 10.1074/jbc.M211286200. [DOI] [PubMed] [Google Scholar]
- 37.Sarrio D, Perez-Mies B, Hardisson D, et al. Cytoplasmic localization of p120ctn and E-cadherin loss characterize lobular breast carcinoma from preinvasive to metastatic lesions. Oncogene. 2004;23(19):3272–83. doi: 10.1038/sj.onc.1207439. [DOI] [PubMed] [Google Scholar]
- 38.Preston SL, Wong WM, Chan AO, et al. Bottom-up histogenesis of colorectal adenomas: origin in the monocryptal adenoma and initial expansion by crypt fission. Cancer Res. 2003;63(13):3819–25. [PubMed] [Google Scholar]
- 39.Yamada T, Deitch E, Specian RD, Perry MA, Sartor RB, Grisham MB. Mechanisms of acute and chronic intestinal inflammation induced by indomethacin. Inflammation. 1993;17(6):641–62. doi: 10.1007/BF00920471. [DOI] [PubMed] [Google Scholar]
- 40.Urbanski SJ, Haber G, Hartwick W, Kortan P, Marcon N, Miceli P. Mucosal changes associated with adenomatous colonic polyps. Am J Pathol. 1986;124(1):34–8. [PMC free article] [PubMed] [Google Scholar]
- 41.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 42.Korinek V, Barker N, Morin PJ, et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275(5307):1784–7. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- 43.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58(6):1130–4. [PubMed] [Google Scholar]
- 44.Suzuki H, Watkins DN, Jair KW, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36(4):417–22. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 45.Morin PJ, Sparks AB, Korinek V, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275(5307):1787–90. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 46.Xiao JH, Ghosn C, Hinchman C, et al. Adenomatous polyposis coli (APC)-independent regulation of beta-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem. 2003;278(32):29954–62. doi: 10.1074/jbc.M304761200. [DOI] [PubMed] [Google Scholar]
- 47.Faraldo MM, Teuliere J, Deugnier MA, et al. beta-Catenin regulates P-cadherin expression in mammary basal epithelial cells. FEBS Lett. 2007;581(5):831–6. doi: 10.1016/j.febslet.2007.01.053. [DOI] [PubMed] [Google Scholar]
- 48.Sansom OJ, Meniel VS, Muncan V, et al. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446(7136):676–9. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- 49.Hayashi R, Yamato M, Sugiyama H, et al. N-Cadherin is expressed by putative stem/progenitor cells and melanocytes in the human limbal epithelial stem cell niche. Stem Cells. 2007;25(2):289–96. doi: 10.1634/stemcells.2006-0167. [DOI] [PubMed] [Google Scholar]
- 50.Lee DM, Kiener HP, Agarwal SK, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315(5814):1006–10. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]