

Abstract
The androgen receptor (AR) is a member of the nuclear steroid hormone receptor family and is thought to play an important role in the development of both androgen-dependent and -independent prostatic malignancy. Elucidating roles by which cofactors regulate AR transcriptional activity may provide therapeutic advancement for prostate cancer (PCa). The DEAD box RNA helicase p68 (Ddx5) was identified as a novel AR-interacting protein by yeast two-hybrid screening, and we sought to examine the involvement of p68 in AR signalling and PCa. The p68-AR interaction was verified by co-localisation of over-expressed protein by immunofluorescence and confirmed in vivo by co-immunoprecipitation in the PCa LNCaP cell line. Chromatin-immunoprecipitation (ChIP) in the same cell line showed AR and p68 recruitment to the promoter region of the androgen-responsive Prostate Specific Antigen (PSA) gene. Luciferase reporter, minigene splicing assays, and RNA interference (RNAi) were used to examine a functional role of p68 in AR-regulated gene expression whereby p68 targeted RNAi reduced AR-regulated PSA expression, and p68 enhanced AR-regulated repression of CD44 splicing (p = 0.008). Tyrosine phosphorylation of p68 was found to enhance co-activation of ligand-dependent transcription of AR-regulated luciferase reporters independent of ATP-binding. Finally we observe increased frequency and expression of p68 in PCa compared with benign tissue using a comprehensive prostate tissue microarray (TMA) (p = 0.003; p = 0.008). These findings implicate p68 as a novel AR transcriptional co-activator that is significantly over-expressed in PCa with a possible role in progression to hormone-refractory disease.
Keywords: p68 DEAD Box RNA helicase, Androgen Receptor, Prostate Cancer, Tyrosine Kinase c-Abl, Alternative Splicing
Introduction
Prostate cancer (PCa) is the second leading cause of male cancer deaths with more than 670,000 men diagnosed worldwide each year. PCa incident rates for USA, European Union and the UK are currently 125, 106.2 and 107.3 cases per 100,000 men, respectively (1, 2). The onset and progression of PCa is driven by the transcriptional function of the androgen receptor (AR), a member of the nuclear hormone receptor family of transcriptional factors. Ablation of androgens, the major AR ligand, is a therapeutic modality in the early stages of the disease. However, PCa can progress to a hormone-refractory phenotype (HRPCa) due to mechanisms that are largely undefined. The majority of HRPCa still express AR and AR-regulated genes, indicating that an active AR-signalling cascade is maintained despite castrate levels of androgens (3). A suggested mechanism for maintenance of a functionally-active AR is aberrant expression of AR cofactors leading to increased AR sensitivity or promiscuous activation of AR by other steroid hormones (4, 5). There are currently no curative therapeutic agents for HRPCa (6).
The p68 DEAD box RNA helicase (Ddx5) is a growth- and developmentally- regulated prototypic member of the DEAD box family of enzymes, and an established RNA helicase. p68 has been found to play a role in a wide range of cellular functions including pre-mRNA, rRNA and miRNA processing (7, 8), ribosome biogenesis and cell proliferation (9), transcription (reviewed in (10, 11) and transcriptional deactivation by promotion of mRNA transcript release in Drosophila (12). A role for p68 in transcription is underscored through its interaction with components of the transcription machinery and other transcription-related factors (RNA Polymerase II [RNAPII], CBP, p300, HDAC1 and Smad3) (10). There is now considerable evidence indicating that p68 is a potent transcriptional co-activator of Estrogen Receptor α (13-15); tumor suppressor p53 (16); MyoD (17) and β-catenin (18).
p68 has been shown to be over-expressed and polyubiquitylated in colorectal tumors (19), suggesting that post-translational changes or modification of p68 expression may play a role in tumour development. In addition, modification of p68 by the small ubiquitin-like modifier, SUMO-2, was found to modulate the activity of p68 as a transcriptional regulator, favouring an action as a repressor (20, 21). Furthermore, tyrosine phosphorylation on Y593 of p68 by c-Abl was shown to be associated with cellular transformation and epithelial-mesenchymal transition in colon cancer (22-24) (although controversy exists as to whether this is through p68 nuclear translocation of β-catenin (25)), and tyrosine phosphorylation of p68 at this residue has also been shown to be important in PDGF-induced cell proliferation through the up-regulation of cyclin D1 and c-Myc expression (24).
Together, these findings indicate that the DEAD box RNA helicase p68 plays an important role in transcriptional regulation and post-translational modifications of the helicase may be important in tumor development. In this study we identify p68 as a novel AR co-activator and explore the relationship between p68 with AR-signalling in PCa, and suggest a role for p68 in cancer progression.
Materials and Methods
Yeast 2-Hybrid Assay
p68 was identified from a prostate library (Clontech) in a yeast two-hybrid screen using the AR bait clones pGBKT7-ARDS (containing the AR DNA and steroid binding domains but lacking the N-terminal domain) and pGBT9-AGA (with a substituted AR-DNA binding domain for a GAL4-binding domain), as previously described (26).
Cell Culture
All cells were grown at 37°C in 5% CO2. LNCaP cells were cultured in RPMI-1640, COS-7 and HEK 293 cells in Dulbecco’s Modified Eagle Media (D-MEM), plus 1% L-glutamine (Invitrogen) supplemented with 10% fetal calf serum (FCS) (Invitrogen) or 10% Dextran Charcoal Stripped FCS (Hyclone), to produce steroid-depleted media as detailed in figure legends. Where indicated, cells were treated with 10 nM R1881 (methyltrienolone).
Tissue Microarray and Immunohistochemistry
Immunohistochemistry was performed using a tissue microarray (TMA) of benign and malignant prostate biopsies derived from transrectal biopsy, transurethral resection and radical prostatectomy as previously described (27). All material was used in accordance with approval granted by the Northumberland, Tyne and Wear NHS Strategic Health Authority Research Ethics Committee (Ref: 2003/11; The Freeman Hospital, Newcastle-upon-Tyne, UK). The average core loss was 28 % and the final study included 147 cancer biopsies and 44 benign biopsies. Antigen retrieval was achieved by immersion in 10 mM Citric acid buffer (pH 6.0), followed by microwaving for 15 min (at 1000 W) in a pressure cooker. Sections were immunostained with PAb204 (mouse monoclonal antibody against p68, Upstate) on a DAKO autostainer using Vectastain® ABC kits (Vector Labs), according to the manufacturer’s protocol. Sections known to stain positively were included in each batch and negative controls were prepared by replacing the primary antibody with TBS buffer. p68 expression was scored blindly by intensity of staining in each biopsy core as being absent (0), weak (+), moderate (++) or strong (+++). Sections were viewed on a Nikon Eclipse 600 microscope and images captured using a Nikon OXM1200 camera with Eclipse net software.
Plasmids, Antibodies and Drugs
The following plasmids have been described previously, p(ARE)3 Luc, pCMV-β-gal, pcDNA3-ARWT (26), GFP-ARWT (28), pcDNA3-p68WT, pcDNA3-p72WT (16, 29), pCMV-c-Abl (30), pGFP3-Sam68 (31), pcDNA3-p68GLT and pSG5-p68Y593F were generated by site directed mutagenesis of p68WT and ligated into appropriate vectors. The YFP-p68 construct, CDM and CD44v5 minigenes were kind gifts from Angus Lamond (University of Dundee, UK), Bert O’Malley (Baylor College of Medicine, USA) and Stefan Stamm (University of Kentucky, USA), respectively.
The following antibodies p68 goat polyclonal, AR rabbit polyclonal C-19 and PSA goat polyclonal (Santa Cruz), p68 mouse monoclonal (Upstate), AR mouse monoclonal (BD Pharmingen), TATA Binding Protein mouse monoclonal (TBP-Abcam), α-tubulin mouse monoclonal (Sigma), c-Abl mouse monoclonal (Chemicon), P-Tyr-100 and myc mouse monoclonal (Cell Signalling), were used as stated in figure legends.
The tyrosine kinase inhibitor Imatinib was obtained from the Pharmacy department of the Royal Victoria Infirmary (RVI) Hospital (Newcastle Upon Tyne, UK).
Immunocytochemistry
Immunocytochemistry was performed on HEK 293 cells transfected with 1μg of GFP-AR and/or YFP-p68 construct using Superfect (Qiagen) as described previously (28). Transfected cells were placed in steroid depleted media for 24 hrs and treated with 10 nM R1881 for a further 16 hrs prior to fixing in methanol. Slides were visualised using laser scanning confocal microscopy (Leica).
Luciferase Reporter and Minigene Splicing Assays
Luciferase reporter assays were performed on COS-7 cells co-transfected in triplicate with 0.1 μg of p(ARE)3Luc, 0.1g of pcDNA3-ARWT, 0.1 μg of pCMV-β-gal and p68WT/Y593F, p72 and c-Abl pcDNA3 fusion constructs using Superfect (Qiagen) as described previously (30). The range of plasmid levels indicated in figures (+, ++, +++ and ++++), corresponds to 25, 50, 75 and 100ng respectively. Luciferase activity was corrected for the corresponding β-gal activity to give a relative activity.
Minigene splicing assays were performed in HEK 293 cells in 6-well plates as previously described (31, 32). Transfected cells were placed in steroid depleted media for 24 hrs and treated with 10 nM R1881, as detailed in figure legends. RNA was extracted using Trizol (Invitrogen) and RT-PCR performed using the One-Step RT-PCR kit (Qiagen). Densitometric band quantification was performed as previously described (33). The identities and sizes of bands were verified by sequencing.
All reporter and minigene assays shown are the mean of at least three independent experiments ± standard error.
Cell lysate extraction and co-immunoprecipitation
LNCaP cells were cultured in steroid depleted media for 48 hrs prior to 10nM R1881 treatment for 8 hrs. Cells were nuclear extracted using CelLytic™ NuCLEAR™ Extraction kit (Sigma, UK) with an extraction buffer salt concentration of 330mM, in the presence of PhosStop phosphatase inhibitors (Roche, UK) and RNase A (100μg/ml - Sigma). AR and p68 co-immunoprecipitation experiments were performed as described previously (34).
RNA interference (RNAi) and Quantitative real-time PCR (QPCR)
LNCaP cells seeded in 6-well plates were transfected with p68 or non-silencing (NS) siRNA using RNAifect (Qiagen), according to manufacturer’s instructions. The p68 and NS siRNA oligonucleotides have been described previously (16, 35). Transfected cells and non-treated controls (C) were grown in steroid-deplete media for 24 hrs and treated with 10 nM R1881 for a further 16 hrs. Cells were harvested in either SDS-sample buffer (protein) or TRIzol reagent (Invitrogen, followed by RT-PCR), to study mRNA expression by QPCR relative to GAPDH expression as previously described (35). The following oligonucleotides were used to determine p68 (CACATCAATCATCAGCCATTCCT and CAAACAAATAGGCCCATCGC) and AR (GGACTTGTGCATGCGGTACTCA and CCTGGCTTCGGCAACTTACAC) mRNA expression. The PSA and GAPDH oligonucleotides have been described previously (34).
Chromatin-Immunoprecipitation Assays
Chromatin-Immunoprecipitation (ChIP) assays were performed in LNCaP cells as described previously (35) in the presence of RNase A (100μg/ml). p68 and AR bound chromatin was immunoprecipitated using 2μg of p68 goat or AR rabbit polyclonal C19 antibody respectively. QPCR was performed on inputs and recovered material to assess the co-occupancy of AR and p68 to the proximal ARE I within the PSA promoter and distal ARE III within the PSA enhancer as described previously (34). The results of all ChIP assays shown are the mean of at least three independent experiments ± standard error.
Statistical Analysis
The independent sample T-test was used to compare differences in the ratio of exon skipping/inclusion products between groups in the minigene assays. For immunohistochemistry, differences in the frequency and expression of p68 between groups were examined using Fisher’s Exact and Mann-Whitney U-test respectively. Correlations with Gleason Grade (GG) were undertaken using the Kruskal-Wallis test. Patient survival was analysed using the Kaplan-Meier method with log rank testing, and multivariate analysis was performed using the Cox regression model. All tests were undertaken using SPSS version 11.0 computer software (SPSS, Inc). All tests were two-sided and a p-value of <0.05 was taken to indicate statistical significance.
Results
p68 is a novel nuclear Androgen Receptor-interacting protein in Prostate Cancer cells
The conserved helicase core (amino acids 203-353) of the p68 DEAD box RNA helicase was identified as a positive clone during a yeast two-hybrid screen for novel androgen receptor (AR) interacting partners. Subsequent immunoblot analysis demonstrated nuclear localisation of p68 in three different cell lines (Figure 1, A), that was unaffected by androgen treatment in the prostate cancer (PCa) LNCaP cell line (Figure S1). Accordingly, nuclear co-localisation of co-transfected p68WT-YFP and ARWT-GFP tagged constructs in HEK 293 cells grown in the presence of androgen was observed by confocal microscopy (Figure 1, B). Co-immunoprecipitation experiments with nuclear extracts of LNCaP cells further demonstrated that the endogenous p68 and AR interaction is enhanced in the presence of androgens (Figure 1, C). However, it is notable that the AR gene is itself androgen responsive and so the enhanced interaction is most likely due to an increase in AR levels and not due to a higher interaction affinity in the presence of androgens. No difference in the intensity of the AR protein band was seen between +/- RNase treated samples (in the presence of androgens), suggesting that the AR-p68 interaction was not RNA dependent. Together, these data provide evidence that p68 interacts with the AR in the nucleus of prostate cells and could thus function as a potential AR co-regulator.
Figure 1.

p68 is a nuclear protein and interacts with the AR. A. Cropped immunoblot images of p68 in nuclear (N) and cytosolic (C) cell lysates obtained from LNCaP, COS-7 and HEK 293 cells probed sequentially with p68, TATA Binding Protein (TBP) and α-tubulin antibody. B. Immunofluorescence images of HEK 293 cells transfected with ARWT-GFP or p68WT-YFP constructs in the presence of 10 nM R1881. Cells were nuclei stained with DAPI prior to confocal microscope imaging. C. Cropped immunoblot images of LNCaP nuclear lysates co-immunoprecipitated (IP) with either AR rabbit or p68 goat polyclonal antibody (+/- R1881 10nM), probed sequentially with p68 and AR mouse monoclonal antibodies. p68 co-immunoprecipitated AR +/- RNase treatment (100μg/ml), was in the presence of R1881 10nM. Control (Con) lanes contain LNCaP nuclear lysate and Protein G Sepharose (PGS) only.
Role of p68 as an Androgen Receptor co-activator
The LXXLL motif is observed in cofactors that interact with ligand-activated nuclear hormone receptors (36). We found that p68 contained an LXXLL motif at amino acid 146, which was a strong indicator that p68 would act as a transcriptional co-regulator of the AR. To further explore this, we undertook chromatin-immunoprecipitation (ChIP) assays to confirm p68 and AR co-occupancy at androgen responsive elements (ARE) within the promoter and enhancer regions of the androgen-responsive Prostate Specific Antigen (PSA) gene. Both p68 and AR were dynamically recruited to the ARE I and ARE III regions of the PSA promoter and enhancer respectively (Figure 2, A and B), with maximal observed recruitment to both regions at 80 min following androgen stimulation. These results clearly demonstrate the presence of p68 at the active promoter of an AR-regulated gene, suggesting a putative role for p68 in AR-dependent transcriptional initiation.
Figure 2.

p68 co-occupies the active PSA promoter at ARE regions and enhances AR transcriptional activity. QPCR analysis (n=3) of ChIP assays (n=3) normalised to GAPDH levels (+/- SE) demonstrate p68 (bar) and AR (line) recruitment to A. ARE I promoter and B. ARE III enhancer regions of the PSA gene over a time course of 100 mins after 10 nM R1881 treatment. COS-7 cells were co-transiently transfected in triplicate with 0.1 μg of p(ARE)3 luciferase reporter, 0.1 μg pcDNA3-AR, 0.1 μg of pCMV-β-gal and increasing concentrations of C. pcDNA3-p68WT and D. pcDNA3-p68GLT constructs (+/- 10nM R1881). Luciferase activity was corrected for the corresponding β-gal activity to give a relative activity. The range of plasmid levels (+, ++, +++ and ++++) corresponds to 25, 50, 75 and 100ng respectively. Data shows relative activity from at least three separate luciferase assay experiments (+/- SE).
To further examine whether p68 co-regulates AR activity, luciferase reporter assays driven by androgen-responsive fragments of the PSA promoter were used to monitor AR transcriptional activity. AR-mediated reporter activity was robustly stimulated by the addition of androgens and in the presence of p68; we observed a dose-dependent co-activation of AR-mediated activity above AR alone levels (Figure 2, C). p68 did not directly affect luciferase reporter activity in the absence of AR (Figure S2), illustrating that p68 is not acting via AR-independent pathways.
ATP dependent helicase enzymes such as p68 use the binding of ATP to increase the affinity and specificity of binding to RNA. Using ATPase mutants we sought to investigate whether the ATP binding/hydrolysis activity of p68 had a functional role in AR co-activation. An ATPase A (p68GLT) mutant maintained AR co-activation above levels of AR alone (Figure 2, D), similar to findings with a mutant directed to the ATPase B (p68NEAD) domain of p68 (data not shown), confirming that neither a conformational change upon p68 ATP binding nor helicase activity was required for p68 co-activation of the AR. This supports previous findings with other p68 interacting partners that demonstrated ATPase/RNA helicase activity was not required for transcriptional function (13, 16, 17).
In order to determine whether post-translational modifications were necessary for AR co-activation we used a p68Y593F mutant that was not tyrosine-phosphorylated at Y593, in luciferase reporter assays. We found that the p68Y593F mutant was unable to increase the activation of AR over AR alone levels compared to p68WT, indicating that the phosphorylation of p68 at Y593 was important for co-activation of the AR (Figure 3, A). In addition, co-immunoprecipitation experiments demonstrated that p68 was tyrosine phosphorylated and associated with c-Abl in the PCa LNCaP cell line (Figure 3, B), although LNCaP cells transiently transfected with the p68Y593F mutant also showed an interaction with c-Abl, indicating that sites other than Y593 may be phosphorylated by c-Abl. Further luciferase reporter assays showed that increasing amounts of c-Abl in the presence of a low concentration of p68WT, enhanced co-activation of AR over p68WT alone (Figure 3, C). c-Abl had no enhanced effect on luciferase reporter activity in the presence of the p68Y593F mutant, or in the absence of AR and/or p68WT indicating that c-Abl was neither enhancing the effect of AR directly nor acting via AR-independent pathways. Furthermore, luciferase reporter assays demonstrated a decrease in co-activation of the AR by p68WT in the presence of the tyrosine kinase inhibitor Imatinib (10 μM) compared to untreated cells (Figure 3, D). Collectively, these data demonstrate that p68 is a co-activator of the AR, independent of the helicase function of p68 and c-Abl activity enhances co-activation through the tyrosine phosphorylation of p68 at Y593.
Figure 3.

Co-activation of AR is enhanced by c-Abl mediated tyrosine phosphorylation of p68 at Y593. A. COS-7 cells were co-transiently transfected in triplicate with 0.1 μg of p(ARE)3 luciferase reporter, 0.1 μg pcDNA3-AR, 0.1 μg of pCMV-β-gal and increasing concentrations of pcDNA3-p68WT (bar) and pcDNA3-p68Y593F (line) constructs (+ 10 nM R1881). B. Cropped immunoblot images of LNCaP nuclear lysates (+/- 10 nM R1881) co-immunoprecipitated (IP) with p68 goat polyclonal antibody probed sequentially with P-Tyr-100, c-Abl and p68 mouse monoclonal antibodies. Nuclear lysates of LNCaP cells transfected with p68Y593F mutant (full media + 10 nM R1881) IP with c-Abl mouse monoclonal antibody and sequentially probed with myc (p68Y593F) or c-Abl mouse monoclonal antibody. C. (ARE)3 luciferase reporter plus 0.1 μg pcDNA3-AR and increasing concentrations of pcDNA3-p68WT, pcDNA3-p68Y593F and/or pcDNA3-c-AblWT (+ 10 nM R1881). D. (ARE)3 luciferase reporter plus 0.1 μg pcDNA3-AR and increasing concentrations of pcDNA3-p68WT +/- 10 μM Imatinib (+ 10 nM R1881). Luciferase activity was corrected for the corresponding β-gal activity to give a relative activity. The range of plasmid levels (+, ++, +++ and ++++) corresponds to 25, 50, 75 and 100ng respectively. Data is shown relative to AR activity alone from at least three separate luciferase assay experiments (+/- SE).
The highly-homologous p72 DEAD box RNA helicase (Ddx17) has been shown to interact with p68 and co-activate both Estrogen Receptor α and MyoD (14, 17). Therefore, we examined the effect of p72 on AR-mediated luciferase reporter activity. p72 did not enhance AR-regulated transcriptional activity above levels seen with AR alone (Figure S3). Moreover, increasing amounts of p72 had no effect on p68-mediated co-activation of AR transcriptional activity (at a low p68 concentration), and increasing p68 and p72 amounts (at the same concentration) did not enhance AR-mediated transcriptional activity over levels observed with p68 alone. These data demonstrate that p72 does not function as an AR transcriptional co-regulator either alone or in conjunction with p68.
p68 knockdown by RNAi reduces Androgen Receptor and Prostate Specific Antigen protein and mRNA expression
To determine a physiological role for p68 in AR transcriptional activity, we examined the effects of RNAi-mediated silencing of endogenous p68 gene expression on the expression of AR and the AR-regulated androgen responsive prostate specific antigen (PSA) gene in vivo in the LNCaP cell line. In the presence of androgens, p68 targeted RNAi (but not non-silencing (NS) RNAi), reduced the expression of p68 mRNA (>60%, Figure 4, B) and protein (Figure 4, A) compared to control (non RNAi transfected) LNCaP cells. Furthermore, a reduction in the expression of PSA and AR mRNA (∼40% and ∼60%, respectively, Figure 4, C and D) and protein (Figure 4, A) was also seen with p68 targeted RNAi compared to non-silencing controls. The AR is an androgen responsive gene itself and these findings clearly demonstrate that p68 targeted RNAi in PCa cells in the presence of androgens reduces protein expression of both the AR and the AR regulated PSA gene. These data are strong evidence that p68 is an important co-activator of AR mediated genes.
Figure 4.
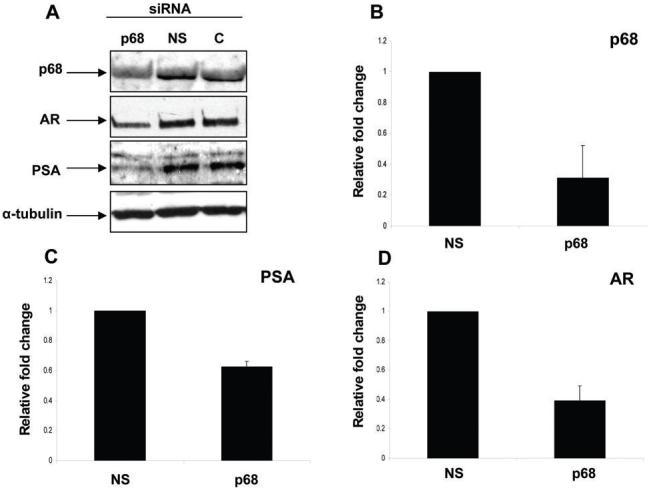
Silencing p68 protein expression by RNAi reduces AR and PSA mRNA and protein levels in LNCaP cells. A. Cropped immunoblot images of LNCaP lysates transfected with p68 and non-silencing (NS) targeted RNAi compared to control (C - non-transfected) cells (+ 10 nM R1881), probed sequentially with p68, AR, PSA and α-tubulin antibodies. N.B. NS and control protein levels are similar demonstrating no off target RNAi affects on protein expression. QPCR analysis of B. p68 C. PSA and D. AR mRNA expression in p68 targeted RNAi cells compared to non-silencing controls. QPCR data was normalised to GAPDH levels and fold change calculated to NS mRNA levels (set as 1). Figure shows the results from three independent RNAi experiments and at least three separate QPCR experiments (+/- SE).
p68 enhances Androgen Receptor-dependent repression of splicing of a hormone responsive CD44 variable exon minigene
Although p68 plays key roles in alternative splicing in some contexts - e.g. in splice site stability, spliceosome assembly, and H-ras splicing (reviewed in (10)); it is unable to directly affect CD44 variable exon inclusion (37) (Figure S4). However, transcriptional co-regulators of nuclear hormone receptors have been shown to affect splicing events (32, 38, 39). The AR has recently been shown to repress splicing of a CD44 variable exon minigene downstream of a hormone-responsive promoter (31, 39). Since p68 is recruited to the PSA promoter and co-activates AR-dependent transcription, we considered the possibility that p68 may also modulate AR-dependent splicing. We used a CD44 variable exon v4 and v5 minigene under the transcriptional control of the hormone-responsive MMTV promoter (CDM) transfected into HEK 293 cells with expression constructs encoding AR and/or p68 (Figure 5, A). Reverse Transcriptase (RT)-PCR and band densitometry (Figure 5, B and C) to study alternatively-spliced transcripts, showed that p68 enhanced AR-mediated exon skipping in the presence of androgens (compare the ratio of exon skipping/inclusion product in lanes 1 and 2 with lanes 5 and 6) (p = 0.008).
Figure 5.
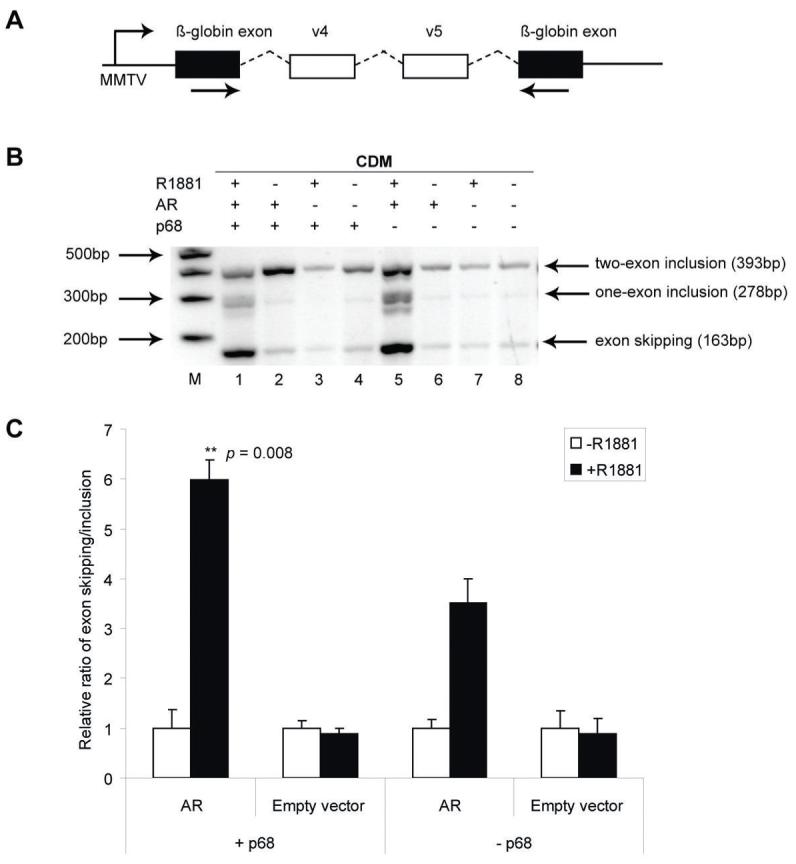
p68 enhances AR-dependent repression of a CD44 variable exon minigene. A. Minigene CDM contains variable exons v4 and v5 cloned downstream of steroid-responsive MMTV promoter. Arrows show location of primers used for RT-PCR. B. HEK 293 cells cultured in steroid-depleted media prior to transfection with the CDM minigene, pcDNA3-AR and pcDNA3-p68WT (+/- 10 nM R1881). Arrows indicate RT-PCR two exon inclusion (393bp), one exon inclusion (278bp) and exon skipped (163bp) products. Image is representative of at least three independent experiments. Marker (M): 1Kb plus DNA ladder (Invitrogen). C. Densitometric assessment from gel images to obtain mean relative ratios of exon skipped/inclusion product (+/- SE).
p68 protein expression in clinical prostate biopsies
Since other AR transcriptional co-regulators have been shown to be over-expressed in PCa (40), we decided to examine p68 protein expression by immunostaining of a prostate tissue microarray (TMA) containing 147 cancer and 44 benign biopsies. The incidence of p68 expressing biopsies was significantly greater in the PCa cohort (66/147; 45%) as compared with benign prostatic hyperplasia (BPH) (11/44; 25%) (p = 0.003; two-sided Fisher’s Exact test) (Figure 6, A). In addition, an increasing frequency of p68 expression was observed with increasing Gleason Grade (GG) tumors: GG3=14/38 (37%), GG4 = 23/51 (45%), GG5 = 26/55 (47%), although the trend did not reach statistical significance (p = 0.3; Kruskal-Wallis test) (Figure 6, B and C). Up-regulation in the level of p68 expression by nearly 3-fold was seen in PCa biopsies compared with BPH (p=0.008, Mann-Whitney test) (Figure 6, D). No statistical correlation with local stage, PSA, metastases, time treatment failure or survival was seen with p68 expression in either multivariate or univariate analyses.
Figure 6.

p68 protein expression levels in clinical prostate biopsies. A. Frequency of p68 immunostaining expression in PCa (66/147; 45%, Gleason Grade [GG] 3, 4 and 5) compared with BPH (11/44; 25%) biopsies. Fisher’s exact test was used for statistical significance. B. Representative images of p68 immunostaining in BPH, GG3, GG4 and GG5 PCa biopsy cores. Magnification, x 5. C. Increasing p68 immunostaining expression with increasing GG of PCa. Kruskall-Wallis test was used for statistical significance. D. Average p68 immunostaining expression in PCa compared with BPH. Mann-Whitney test was used for statistical significance.
Discussion
A number of androgen receptor (AR) transcriptional co-regulators have been identified by our group and others that play a role in prostate cancer (PCa) (40). In this study, we reveal a novel role for the DEAD box RNA helicase p68 as an AR co-activator in PCa. Our results show that p68 is an AR-interacting protein and is associated with the AR at the promoter region of an androgen-responsive gene (PSA). By over-expression of p68 in luciferase reporter assays, we demonstrate a functional role for p68 as an AR transcriptional co-activator, dependent on Y593 phosphorylation, but independent of ATP-binding. Furthermore, p68 co-activator function was verified in vivo in PCa cells by p68 targeted RNAi demonstrating reduced expression of androgen responsive endogenous protein (AR and PSA).
The requirement of ATP dependency in p68-mediated transcription is conflicting. A recent study suggested that ATP binding was required for the transcriptional activation of cyclin D1 by tyrosine phosphorylated p68 (24). However, helicase inactive ATPase mutants have been shown to co-regulate transcription as efficiently as wild type for several other transcription factors (13, 14, 16, 17, 29), suggesting that the function of p68 as a transcriptional co-regulator is independent of its RNA helicase activity. In agreement with these findings, we also demonstrate p68 ATPase mutants maintain co-activator function in ARE luciferase reporter assays demonstrating that neither ATPase activity nor a conformational change upon p68 ATP binding is required for AR co-activation. At present, it is unclear why the ATP-dependent activity of tyrosine phosphorylated p68 is specifically required for the transcription of the cyclin D1 promoter (24), but it may reflect differences in the way p68 functions as a direct transcriptional activator and as a co-regulator.
The C-terminal domain of p68 can bind single stranded RNA and has been shown to be a target for protein kinase C (41). Phosphorylation of the C-terminal domain of p68 consequently abolished RNA binding and it was hypothesised that phosphorylated p68 may act as a molecular switch between transcriptional functions and splicing (42). In this study, we demonstrate that tyrosine phosphorylation of p68 on Y593 by c-Abl increased transcriptional co-activation of the AR. The Y593 phosphorylation of p68 has been implicated in promoting epithelial-mesenchymal transition via PDGF and c-Abl-dependent pathways (23). Paracrine signalling and the role of stromal-epithelial interactions leading to loss of cell-cell adhesion in PCa has been well documented (43). Hence, a model where p68 may directly co-activate AR transcriptional activity and promote epithelial-mesenchymal transition-driven PCa progression would be of notable interest.
Recent evidence suggests that p68 may function as an ‘adaptor’ or ‘coupling’ protein that coordinates the tightly integrated processes of transcription and mRNA processing (10, 44, 45). p68 has key roles in U1snRNP-5′ splice site complex stability, spliceosome assembly, pre-mRNA splicing (10) and mRNA transcript clearance (12). In this study, we show that p68 potentiates AR-regulated repression of splicing of a CD44 variable exon minigene. In contrast, the homologous DEAD box RNA helicase p72 promotes CD44 exon inclusion downstream of a hormone-responsive promoter (32). These results suggest that the effect of p68 on AR-dependent splicing may be via transcriptional co-regulatory mechanisms, possibly regulated by the post-translational status of p68. Alternatively, recruitment of p68 to endogenous AR-responsive genes may facilitate splicesome assembly and increase the rate of RNA polymerase II (RNAPII) elongation, affecting splice site recognition and promotion of exon-skipping in nascent transcripts (46). AR is thought to enhance RNAPII elongation by interacting with Transcription Factor IIH (TFIIH) and Positive Transcription Elongation Factor b (P-TEFb) (47) which phosphorylate the C-terminal domain (CTD) of RNAPII switching from a non-processive to a processive form (48-50). In this study we demonstrate p68 is involved in both AR transcriptional co-activation and AR mediated repression of splicing of an androgen responsive gene. Therefore, p68 may serve as a common link between transcription and splicing machinery. RNA processing activities are thought to be strongly connected to efficient transcriptional co-regulation and this study provides evidence for p68 as part of a ‘molecular link’ that involves the coupling of transcriptional initiation to mRNA processing.
Finally, we show that p68 is over-expressed in PCa biopsies as compared with benign prostatic hyperplasia controls. There is an increase in the incidence of p68 expression by two-fold and the level of expression increases by 3-fold (p = 0.008), highlighting the clinical significance of p68 in PCa. Not all PCa samples express p68 and this may represent a specific mechanism of disease progression in a discrete cohort. Although, there was limited correlation with other clinicopathological parameters (grade, stage, PSA, and prognosis), a putative role of p68 in PCa progression is demonstrated by the functional data shown and the increased levels of expression in clinical samples.
Identifying cofactors that influence the development of hormone escape of PCa will be important in the development of effective therapies and prevention of hormone refractory PCa. This study provides strong evidence of the up-regulation of a novel AR co-activator in PCa which may play a significant role in progression to androgen independence. As well as a role in epithelial mesenchymal transition, activation of c-Abl pathways may enhance p68 function as a co-activator of AR-dependent gene expression in PCa. Hence, tyrosine kinase inhibitors such as Imatinib may be of therapeutic benefit if post-translational modifications are shown to be important in modulating p68 function and affect disease phenotypes.
Acknowledgments
We thank Susan Bray and the Tayside Tissue bank (University of Dundee, UK) for the immunostaining of the prostate TMA, Professor Angus Lamond (University of Dundee, UK) for the p68-YFP expression construct and Professors Bert O’Malley (Baylor College of Medicine, USA) and Stefan Stamm (University of Kentucky, USA) for their kind gifts of minigene constructs. The funding bodies had no role in the design of the study, the collection, analysis and interpretation of the data, the decision to submit the manuscript for publication, or the writing of the manuscript.
Grant Support: Medical Research Council/Cancer Research UK/Department of Health - Prostate cancer Mechanisms of Progression and Treatment ‘ProMPT’ collaboration (G0100100/64424 to CNR); Association for International Cancer Research (06-613 to FVF-P); Association for International Cancer Research (06-705 to DJE and HYL)
References
- 1.Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boyle P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol. 2007;18:581–92. doi: 10.1093/annonc/mdl498. [DOI] [PubMed] [Google Scholar]
- 2.Gann PH. Interpreting recent trends in prostate cancer incidence and mortality. Epidemiology. 1997;8:117–20. [PubMed] [Google Scholar]
- 3.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nature Review Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 4.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nature Medicine. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 5.Chmelar R, Buchanan G, Need EF, Tilley W, Greenberg NM. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. International Journal of Cancer. 2007;120:719–33. doi: 10.1002/ijc.22365. [DOI] [PubMed] [Google Scholar]
- 6.Nieto M, Finn S, Loda M, Hahn WC. Prostate cancer: Re-focusing on androgen receptor signaling. Int J Biochem Cell Biol. 2007;39:1562–8. doi: 10.1016/j.biocel.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukuda T, Yamagata K, Fujiyama S, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nature Cell Biology. 2007;9:604–11. doi: 10.1038/ncb1577. [DOI] [PubMed] [Google Scholar]
- 8.Salzman DW, Shubert-Coleman J, Furneaux H. P68 RNA helicase unwinds the human let-7 microRNA precursor duplex and is required for let-7-directed silencing of gene expression. The Journal of Biological Chemistry. 2007;282:32773–9. doi: 10.1074/jbc.M705054200. [DOI] [PubMed] [Google Scholar]
- 9.Jalal C, Uhlmann-Schiffler H, Stahl H. Redundant role of DEAD box proteins p68 (Ddx5) and p72/p82 (Ddx17) in ribosome biogenesis and cell proliferation. Nucleic Acids Research. 2007;35:3590–601. doi: 10.1093/nar/gkm058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuller-Pace FV. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Research. 2006;34:4206–15. doi: 10.1093/nar/gkl460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caretti G, Lei EP, Sartorelli V. The DEAD-box p68/p72 proteins and the noncoding RNA steroid receptor activator SRA: eclectic regulators of disparate biological functions. Cell Cycle. 2007;6:1172–6. doi: 10.4161/cc.6.10.4228. [DOI] [PubMed] [Google Scholar]
- 12.Buszczak M, Spradling AC. The Drosophila P68 RNA helicase regulates transcriptional deactivation by promoting RNA release from chromatin. Genes and Development. 2006;20:977–89. doi: 10.1101/gad.1396306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Endoh H, Maruyama K, Masuhiro Y, et al. Purification and identification of p68 RNA helicase acting as a transcriptional coactivator specific for the activation function 1 of human estrogen receptor alpha. Molecular and Cellular Biology. 1999;19:5363–72. doi: 10.1128/mcb.19.8.5363. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Watanabe M, Yanagisawa J, Kitagawa H, et al. A subfamily of RNA-binding DEAD-box proteins acts as an estrogen receptor alpha coactivator through the N-terminal activation domain (AF-1) with an RNA coactivator, SRA. The EMBO Journal. 2001;20:1341–52. doi: 10.1093/emboj/20.6.1341. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Metivier R, Penot G, Hubner MR, et al. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–63. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 16.Bates GJ, Nicol SM, Wilson BJ, et al. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. The EMBO Journal. 2005;24:543–53. doi: 10.1038/sj.emboj.7600550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caretti G, Schiltz RL, Dilworth FJ, et al. The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Developmental Cell. 2006;11:547–60. doi: 10.1016/j.devcel.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Shin S, Rossow KL, Grande JP, Janknecht R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Research. 2007;67:7572–8. doi: 10.1158/0008-5472.CAN-06-4652. [DOI] [PubMed] [Google Scholar]
- 19.Causevic M, Hislop RG, Kernohan NM, et al. Overexpression and poly-ubiquitylation of the DEAD-box RNA helicase p68 in colorectal tumours. Oncogene. 2001;20:7734–43. doi: 10.1038/sj.onc.1204976. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs AM, Nicol SM, Hislop RG, Jaffray EG, Hay RT, Fuller-Pace FV. SUMO modification of the DEAD box protein p68 modulates its transcriptional activity and promotes its interaction with HDAC1. Oncogene. 2007;30:5866–76. doi: 10.1038/sj.onc.1210387. [DOI] [PubMed] [Google Scholar]
- 21.Fuller-Pace FV, Jacobs AM, Nicol SM. Modulation of transcriptional activity of the DEAD-box family of RNA helicases, p68 (Ddx5) and DP103 (Ddx20), by SUMO modification. Biochemical Society Transactions. 2007;35:1427–9. doi: 10.1042/BST0351427. [DOI] [PubMed] [Google Scholar]
- 22.Yang L, Lin C, Liu ZR. Signaling to the DEAD box--regulation of DEAD-box p68 RNA helicase by protein phosphorylations. Cellular Signalling. 2005;17:1495–504. doi: 10.1016/j.cellsig.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Yang L, Lin C, Liu ZR. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell. 2006;127:139–55. doi: 10.1016/j.cell.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 24.Yang L, Lin C, Zhao S, Wang H, Liu ZR. Phosphorylation of p68 RNA Helicase Plays a Role in Platelet-derived Growth Factor-induced Cell Proliferation by Up-regulating Cyclin D1 and c-Myc Expression. The Journal of Biological Chemistry. 2007;282:16811–9. doi: 10.1074/jbc.M610488200. [DOI] [PubMed] [Google Scholar]
- 25.Stucke VM, Gorses D, Hofmann F. DEAD-box RNA helicase p68 is not required for nuclear transloction of beta-catenin in colon cancer cells. Cell Cycle. 2008;7:830–32. doi: 10.4161/cc.7.6.5614. [DOI] [PubMed] [Google Scholar]
- 26.Brady ME, Ozanne DM, Gaughan L, et al. Tip60 is a nuclear hormone receptor coactivator. The Journal of Biological Chemistry. 1999;274:17599–604. doi: 10.1074/jbc.274.25.17599. [DOI] [PubMed] [Google Scholar]
- 27.Sahadevan K, Darby S, Leung HY, Mathers ME, Robson CN, Gnanapragasam VJ. Selective over-expression of fibroblast growth factor receptors 1 and 4 in clinical prostate cancer. The Journal of Pathology. 2007;213:82–90. doi: 10.1002/path.2205. [DOI] [PubMed] [Google Scholar]
- 28.Rigas AC, Ozanne DM, Neal DE, Robson CN. The scaffolding protein RACK1 interacts with androgen receptor and promotes cross-talk through a protein kinase C signaling pathway. The Journal of Biological Chemistry. 2003;278:46087–93. doi: 10.1074/jbc.M306219200. [DOI] [PubMed] [Google Scholar]
- 29.Wilson BJ, Bates GJ, Nicol SM, Gregory DJ, Perkins ND, Fuller-Pace FV. The p68 and p72 DEAD box RNA helicases interact with HDAC1 and repress transcription in a promoter-specific manner. BMC Molecular Biology. 2004;5:11. doi: 10.1186/1471-2199-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaughan L, Logan IR, Neal DE, Robson CN. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic Acids Research. 2005;33:13–26. doi: 10.1093/nar/gki141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajan P, Gaughan L, Dalgliesh C, et al. The RNA-binding and adaptor protein Sam68 modulates signal-dependent splicing and transcriptional activity of the androgen receptor. The Journal of Pathology. 2008;215:67–77. doi: 10.1002/path.2324. [DOI] [PubMed] [Google Scholar]
- 32.Auboeuf D, Honig A, Berget SM, O’Malley BW. Coordinate regulation of transcription and splicing by steroid receptor coregulators. Science. 2002;298:416–9. doi: 10.1126/science.1073734. [DOI] [PubMed] [Google Scholar]
- 33.Venables JP, Bourgeois CF, Dalgliesh C, Kister L, Stevenin J, Elliott DJ. Up-regulation of the ubiquitous alternative splicing factor Tra2beta causes inclusion of a germ cell-specific exon. Human Molecular Genetics. 2005;14:2289–303. doi: 10.1093/hmg/ddi233. [DOI] [PubMed] [Google Scholar]
- 34.Gaughan L, Logan IR, Cook S, Neal DE, Robson CN. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. The Journal of Biological Chemistry. 2002;277:25904–13. doi: 10.1074/jbc.M203423200. [DOI] [PubMed] [Google Scholar]
- 35.Logan IR, Gaughan L, McCracken SR, Sapountzi V, Leung HY, Robson CN. Human PIRH2 enhances androgen receptor signaling through inhibition of histone deacetylase 1 and is overexpressed in prostate cancer. Molecular and Cellular Biology. 2006;26:6502–10. doi: 10.1128/MCB.00147-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plevin MJ, Mills MM, Ikura M. The LxxLL motif: a multifunctional binding sequence in transcriptional regulation. Trends in Biochemical Sciences. 2005;30:66–9. doi: 10.1016/j.tibs.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Honig A, Auboeuf D, Parker MM, O’Malley BW, Berget SM. Regulation of alternative splicing by the ATP-dependent DEAD-box RNA helicase p72. Molecular and Cellular Biology. 2002;22:5698–707. doi: 10.1128/MCB.22.16.5698-5707.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Auboeuf D, Batsche E, Dutertre M, Muchardt C, O’Malley BW. Coregulators: transducing signal from transcription to alternative splicing. Trends in Endocrinology and Metabolism. 2007;18:122–9. doi: 10.1016/j.tem.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Sun J, Blair AL, Aiyar SE, Li R. Cofactor of BRCA1 modulates androgen-dependent transcription and alternative splicing. The Journal of Steroid Biochemistry and Molecular Biology. 2007;107:131–9. doi: 10.1016/j.jsbmb.2007.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heemers HV, Tindall DJ. Androgen Receptor (AR) Coregulators: A Diversity of Functions Converging on and Regulating the AR Transcriptional Complex. Endocrine Reviews. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 41.Buelt MK, Glidden BJ, Storm DR. Regulation of p68 RNA helicase by calmodulin and protein kinase C. The Journal of Biological Chemistry. 1994;269:29367–70. [PubMed] [Google Scholar]
- 42.Yang L, Yang J, Huang Y, Liu ZR. Phosphorylation of p68 RNA helicase regulates RNA binding by the C-terminal domain of the protein. Biochemical and Biophysical Research Communications. 2004;314:622–30. doi: 10.1016/j.bbrc.2003.12.129. [DOI] [PubMed] [Google Scholar]
- 43.Chung LW, Baseman A, Assikis V, Zhau HE. Molecular insights into prostate cancer progression: the missing link of tumor microenvironment. The Journal of Urology. 2005;173:10–20. doi: 10.1097/01.ju.0000141582.15218.10. [DOI] [PubMed] [Google Scholar]
- 44.Auboeuf D, Dowhan DH, Dutertre M, Martin N, Berget SM, O’Malley BW. A subset of nuclear receptor coregulators act as coupling proteins during synthesis and maturation of RNA transcripts. Molecular and Cellular Biology. 2005;25:5307–16. doi: 10.1128/MCB.25.13.5307-5316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ares M, Jr., Proudfoot NJ. The spanish connection: transcription and mRNA processing get even closer. Cell. 2005;120:163–6. doi: 10.1016/j.cell.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 46.Clark EL, Fuller-Pace FV, Elliott DJ, Robson CN. Coupling transcription to RNA processing via the p68 DEAD box RNA helicase androgen receptor co-activator in prostate cancer. Biochemical Society Transactions. 2008;36:546–7. doi: 10.1042/BST0360546. [DOI] [PubMed] [Google Scholar]
- 47.Lee DK, Duan HO, Chang C. Androgen receptor interacts with the positive elongation factor P-TEFb and enhances the efficiency of transcriptional elongation. The Journal of Biological Chemistry. 2001;276:9978–84. doi: 10.1074/jbc.M002285200. [DOI] [PubMed] [Google Scholar]
- 48.Fong YW, Zhou Q. Stimulatory effect of splicing factors on transcriptional elongation. Nature. 2001;414:929–33. doi: 10.1038/414929a. [DOI] [PubMed] [Google Scholar]
- 49.Ni Z, Schwartz BE, Werner J, Suarez JR, Lis JT. Coordination of transcription, RNA processing, and surveillance by P-TEFb kinase on heat shock genes. Molecular Cell. 2004;13:55–65. doi: 10.1016/s1097-2765(03)00526-4. [DOI] [PubMed] [Google Scholar]
- 50.Nogues G, Kadener S, Cramer P, Bentley D, Kornblihtt AR. Transcriptional activators differ in their abilities to control alternative splicing. The Journal of Biological Chemistry. 2002;277:43110–4. doi: 10.1074/jbc.M208418200. [DOI] [PubMed] [Google Scholar]