

Introduction
Over the last ten years several international structural genomics initiatives have been funded [1;2] with diverse themes. Some are targeting specific biological and medical systems, whereas others, like the Protein Structure Initiative (PSI) in the United States, whilst incorporating some biological themes also aim to extend our ability to provide structural data to the increasing number of genomic sequences. By boosting the structural repertoire, it is hoped that we will be able to improve our understanding of fold space and how proteins evolve new functions. Consequently, the four major PSI Centres have targeted large sequence families that are most likely to adopt novel structures [1] (http://www.structuralgenomics.org), although these often have little or no functional annotation [3]. To maximise the biomedical benefit of PSI structures, recent reviews have proposed broadening selection criteria to explicitly focus on the relevance to human disease [4] or to provide structural characterisation of families with known functions [5].
Figure 1 shows that as the international genomics initiatives gather pace, both the number of sequences and protein families is still growing at an exponential rate, although the rate of expansion of protein families is substantially less. This trend is also observed among domain families, which are tenfold fewer (<10,000) than the number of protein families. By targeting these, the structural genomics initiatives can aim to characterise the major building blocks of whole proteins and since these domains recur in different combination in the genomes, it will be an important step towards understanding the complete structural repertoire in nature. Furthermore, the complement of molecular functions found within an organism is likely to be even fewer. For example, 97% of proteins in yeast can be assigned one or more of ~4000 unique GO terms. Therefore, target selection strategies that attempt to sample structurally uncharacterised domain families/subfamilies with distinct functions (known or predicted) will help to increase our knowledge of both structure and function space.
Figure 1.
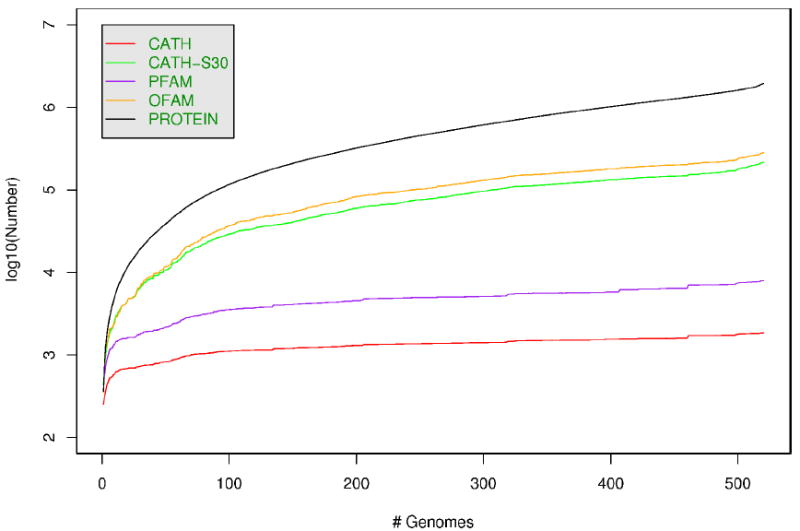
This figure shows the numbers (as a log scale) of genome sequences (PROTEIN), OFAMs (putative orthologous gene families), CATH-S30 (30% non-redundant sequence families), PFAM families, and CATH domain superfamilies, as genomes are added to the database in increasing order of size.
In this review, we first consider how approaches to defining the structural novelty of newly solved structures are changing and how this might impact on protein function. Similarly, we explore the ways in which analyses of diverse protein structural families have increased our understanding of how protein functions have evolved and how this knowledge can be exploited to improve methods for predicting function from structure. We then look at recent approaches to predicting function from sequence, focussing on those that are suitable for annotating genes at the whole genome scale, involving thousands or even tens of thousands of proteins. Such broad coverage of genomes with functional information is essential not only for interpreting functional genomics data, but also for dividing sequence space in a more biologically focussed way, allowing targeted selection of structurally uncharacterised functional families for structure determination.
How do we define structural novelty?
There have been several recent attempts to define a protein fold [6][Sippl] or question whether it is even realistic to seek to partition the protein universe at the fold level [7]. However, it appears that regardless of how structural novelty is defined, significant structural change often mediates divergence in functional properties, particularly in the very large, diverse superfamilies that tend to dominate the sequenced genomes [8]. Currently, the precise mechanisms by which this occurs are difficult to fathom given the sparsity of data. For example, in the largest of these diverse superfamilies, less than 10% of sequence diverse relatives (<30% sequence identity) have a close homologue with known structure [9].
The fact that homologues can change their fold has been commented on previously by Grishin [10] and others. The extent of this is becoming clearer as the new generation of powerful sequence profile (e.g. HMM-HMM) methods have detected some very remote homologues with different folds (see [11] and references therein). Furthermore, extreme examples of chameleon sequences that can adopt several alternative three-dimensional configurations [12], have complicated our view of the sequence to structure paradigm.
The traditional notion of a fold (as defined by SCOP and CATH) is to cluster domains based on a similar arrangement and connectivity of core secondary structures. However, these common elements may account for less than half of the domain size and in some fold groups relatives exhibit as much as a three fold size variation in terms of their numbers of secondary structures [8]. At what point these extra secondary structure elements can be thought to represent a change in fold is subjective and some have suggested that we can only view structure space as a continuum [7]. The lack of a clear definition of a protein fold becomes especially pertinent when assessing the power of structure prediction methods and the structural novelty that the PSI structures bring to the PDB.
In order to more objectively gauge the novelty of newly deposited structures and determine the extent to which structural similarities correlate with function, especially for the large structural families comprising hundreds of structures, we need fast and sensitive global structure comparison methods. A number of benchmark studies have been performed recently [13;14] which demonstrate the value of using normalised RMSD measures to assess structural similarity. Perhaps the most appropriate means of assessing global similarity, SIMAX [14], normalises by the number of aligned residues as a fraction of the number of resides in the larger protein, and thus decreases the significance of small common structural motifs.
Applying this approach to compare structures classified in CATH (version 3.1) and assuming that two structures have “similar folds” or belong to structurally similar groups (SSGs) if they superpose with a SIMAX score <5Å (see Figure 2a), we find that there are approximately 3000 different SSGs classified to date. This contrasts with the ~1000 folds currently identified by SCOP and CATH using rather subjective criteria. In fact, Figure 3a shows that the largest 300 CATH superfamilies that are highly recurrent in the genomes [8;9], contain nearly 40% of SSGs in CATH and some contain more than 10 different SSGs (see Figure 3b). Analyses of these very large, diverse families in CATH [8] have revealed that relatives with significant divergence in structure or fold tend to have different functional roles and protein interactions (Figure 2b and Figure 4). Hence, by solving additional structures from these very large superfamilies, structural genomics could enrich our understanding of structure-function relationships within these superfamilies, most of which are structurally under-represented and highly recurrent in the genomes – although CATH comprises 2100 superfamilies the largest 300 superfamilies account for ~35% of predicted structural domains in genome sequences.
Figure 2.
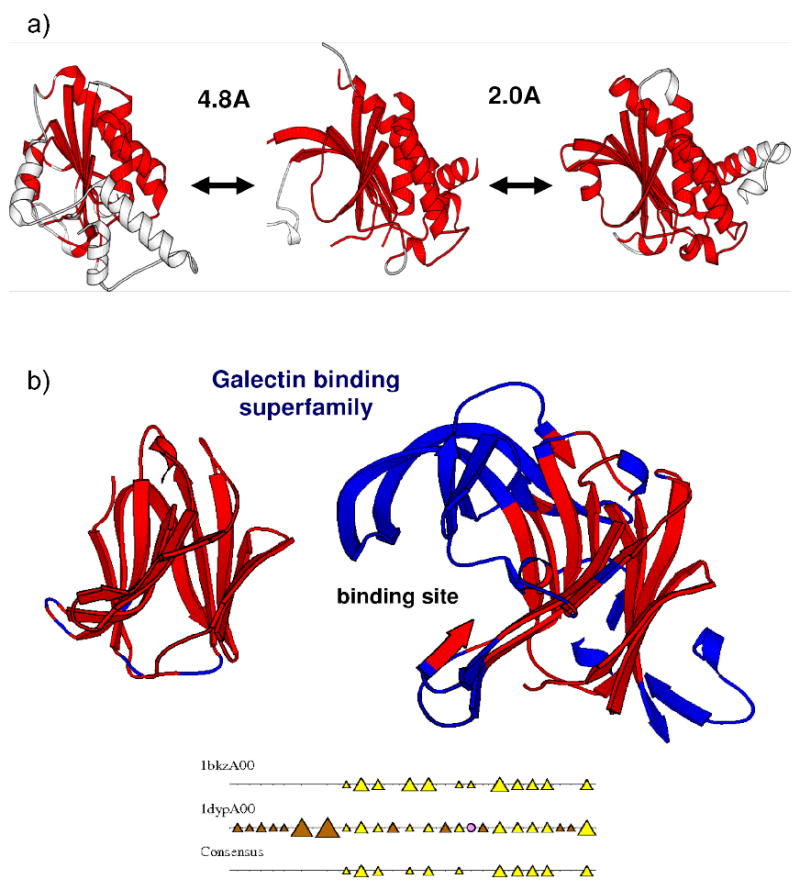
a) Two domains which differ by less than 5Å (SIMAX) can vary structurally but share the same fold. b) Two domains from the galectin-type carbohydrate recognition domain superfamily. The domains are coloured so that residues having the same α-helix or β-sheet conformation in 75% of domains appear red. Domains 1bkzA0 and 1dypA0 are in the similar orientations so that secondary structure embellishments to the core of 1dypA0 can be seen. 1dypA00 has a significant number of extra secondary structure elements, many of which are located at the binding site and are modifying the geometry of the active site.
Figure 3.
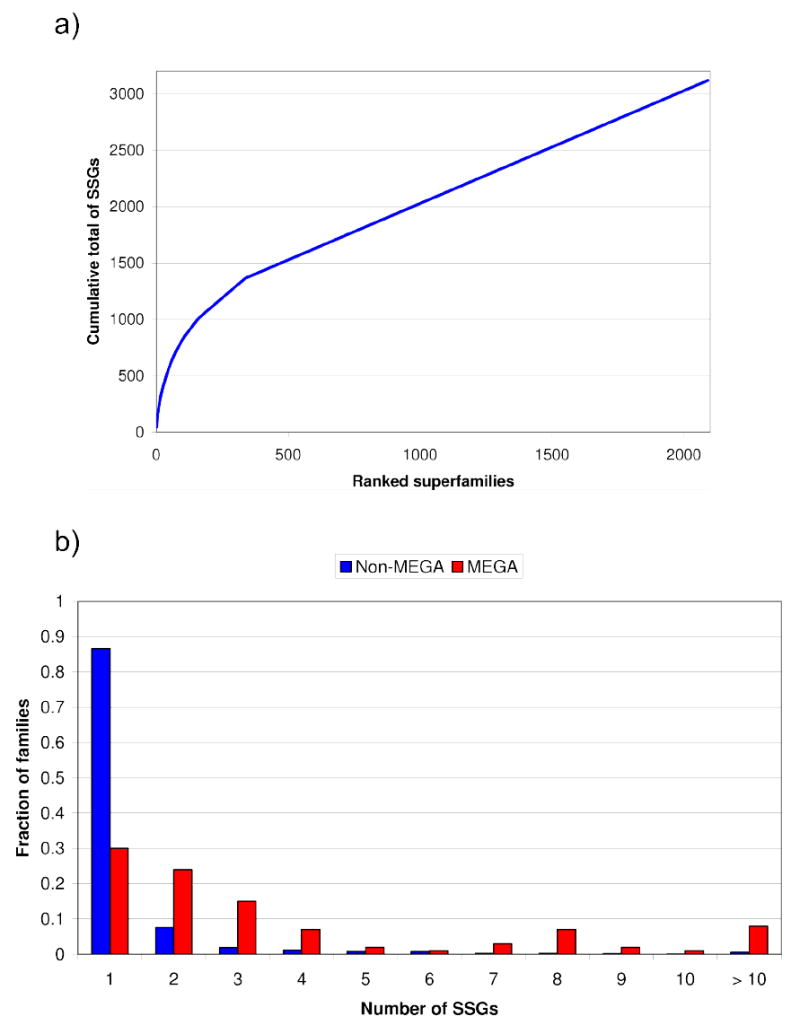
a) This figure shows a cumulative plot of the percentage of structurally similar groups (SSGs) for all superfamilies in CATH, when ranked by largest to smallest. It can be seen that the top 300 CATH superfamilies (MEGA) account for ~40% of the fold groups and are hence extremely structurally diverse. b) This histogram shows the fraction of SSGs in MEGA and NON-MEGA superfamilies in CATH. It can be seen that the MEGA superfamilies have a disproportionate number of SSGs.
Figure 4.
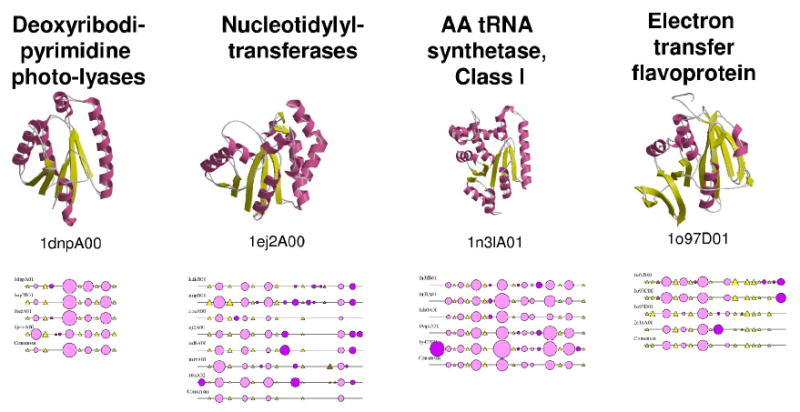
Function and structure diversity in the HUP domain superfamily (CATH code 3.40.50.620). Four major functions in the superfamily are represented on this figure, together with one protein of yet unknown function. For each of the functional groups, a representative structure is displayed in cartoons with alpha-helices coloured pink and beta-strands coloured yellow. CATH domain identifiers of the displayed structures are specified on the figure. 2DSEC plots [8] showing the conservation of secondary structure elements within each functional groups are also shown to illustrate that the different functional groups tend to be characterised by specific secondary structure patterns. These 2DSEC plots represent conserved helices as pink circles and conserved strands as yellow triangles. This figure illustrates the diversity of function that can be found within a homologous superfamily, and the correlation between function and structure divergence.
How does function evolve within superfamilies?
It is clear that the relationship between the evolution of the function of proteins with respect to the evolution of their sequence and structure is complex [15]. Very close homologues can differ in function, and there are many known instances of a given protein being reused for entirely unrelated functions, depending on the context in which they are found, in a phenomenon known as “recruitment” or “moonlighting” [16;17]. A classic example of this is that of duck eye lens crystallins, which are identical in sequence to liver enolase and lactate dehydrogenase [18].
Given the complexity of the relationship between sequence, structure and function divergence in proteins sensu lato, a sensible approach is to study these relationships in the limited context of individual families, superfamilies and structural folds (for several recent examples of such studies, see [19-23]). In fact, functional diversity within a given fold or superfamily can span a wide variety of different molecular activities and biological processes, and studies of large and diverse superfamilies are particularly relevant given the recent evidence that they are more likely to contain proteins with essential functions [24]. For example, protein domains in the HUP-domain superfamily [25] illustrate the diversity of structures and functions that can be observed amongst homologous protein domains (Figure 4).
A detailed analysis of limited sets of superfamilies recently culminated in the development of a new resource for the study of structure-function relationships within enzyme superfamilies, namely the Structure Function Linkage Database (SFLD) [26]. Divergence of function amongst homologues can result from several mechanisms such as substitutions in the active sites [27], changes in residues that determine the specificity of interactions (see [28;29] for recent methods that exploit this principle), or variation in environmental context such as the presence of other potential protein interaction partners [23]. However, one conclusion derived from the analysis of the data in the SFLD is that many functionally divergent enzyme superfamilies seem to conserve a common partial reaction or other chemical capability [26]. This is further supported by structural comparisons of adenine-binding proteins using a new generation of structure-function comparison method[30]. This approach detected ligand binding similarities between domains in different SCOP superfamilies, suggesting remote homologies between these proteins, where the ability to bind adenine-containing ligands is maintained as other functional properties diverge.
The development of other databases – EzCatDB [31] and MACiE [32] – are aimed at the description of catalytic reactions from a mechanistic point-of-view. These should help in deciphering further relationships between the catalytic mechanisms of homologous enzymes, which might be overlooked when describing reactions using EC numbers alone. Recent studies using these catalytic mechanism databases have already identified multiple cases of the convergence on a common catalytic mechanism in unrelated enzymes [33].
Figure 4 shows that each functional sub-group in the HUP-domain superfamily can be characterised by distinct secondary structure features. Recent studies have shown that successive indels in a particular structural region were a common phenomenon and that such “nested indels” could result in the occurrence of new domains and/or new functions [34]. In particular, it was demonstrated in a set of diverse CATH superfamilies that such embellishments often occur in regions close to the active site of enzymes, or at the surface of proteins thus providing alternative interfaces for interaction with other proteins and domains [8]. It was also observed that some structural architectures, and hence the superfamilies therein, accommodate structural embellishments more easily thanks to their higher intrinsic stability or to their specific features (e.g. large central beta-sheets to which peripheral beta-strands can be added relatively easily), and that these embellishments in turn allow exploration of new functions in superfamilies. Other recent results suggest that in addition, another structural measure, namely the structural designability (i.e. mutational plasticity), correlates with functional flexibility amongst homologous proteins [35] and that the size of permitted indels depends on the specific function [36].
How can we predict function from structure?
Although the majority of structural data are associated with additional experimental data regarding a protein’s function, this is not the case for a large proportion of PSI structures [3]. For these proteins it is necessary to attempt to predict the function from the structure. Furthermore, to better understand the structure-function relationship, it is useful to sub-classify structures within superfamilies according to their functions.
Significant global structural similarity can be used to transfer functional annotations where close homology can often be identified in the absence of high similarity at the sequence level, using methods such as CE [37], STRUCTAL [13], DALI [38], CATHEDRAL [14], FATCAT [39] and SSM [40]. Recently, methods such as ProKnow [41], Annolite [42] and PHUNCTIONER [43] have provided interfaces for assigning confidence values to GO functional annotations transferred from putative relatives using global structure comparison, structural motif and sequence methods.
Where global structural similarity provides insufficient evidence for functional annotation, methods which detect binding or catalytic site similarity can provide additional clues. PDBSITE and MSDSITE permit the detection of known functional sites annotated by the authors of a structure. The PROFUNC [44] and TEMPURA interfaces allows the user to scan a novel structure against hand-curated catalytic residues in the Catalytic Site Atlas[45].
To manually generate structural templates based on known functional residues is hugely time-consuming and hence the literature contains numerous examples of methods that automatically generate these templates to classify new structures. The reverse template method available as part of the PROFUNC suite decomposes the query structure into tri-peptide fragments (putative functional residues) which are then matched against a non-redundant set of PDB structures using JESS [46]. Each hit is then compared to the query according to the sequence similarity of the local environment of the template. A recent extension of the Evolutionary Trace method for binding site prediction was to generate structural templates based on predicted functional residues[47]. SiteEngines [48] produces templates by comparing the physico-chemical properties of residues in binding site clefts. As well as atom or residue-level templates, other approaches seek to compare the electrostatic properties of binding sites (ef-Site, [49], SURF’s UP [50]. For enzymes, pvSOAR (CASTp) [51] compares surface accessible clefts, which often co-locate with the active site.
The GASP method [52] instead uses a genetic algorithm to construct templates based on their ability to discriminate between different protein families against a background of representatives from the SCOP database. Similarly, DRESPAT [53] implements a graph theoretical approach to discover structural patterns associated with a given family of protein templates. At a coarser level, Doig et al. [54], developed a Support Vector Machine (SVM) to predict enzyme class based purely on general properties, such as secondary structure content, solvent accessibility and amino acid composition.
One of the inherent problems with using PDB structures to transfer annotations between enzymes is the binding state in which it is crystallised. These vary between structures crystallised with non-cognate ligands, transition state structures or apo-enzymes. Hence, precise geometric matching in the active site region can be problematic. The SOIPPA method [30;55] addresses this by introducing the concept of a “geometric potential” to characterise the shape formed by a given set of C-alpha atoms, which accounts for both local and global relationships between residues across the protein structure. This is used to compare ligand binding sites and was able to detect distant evolutionary relationships between proteins binding a range of adenine-containing ligands.
Arguably, the most difficult problem facing prediction methods is the complexity of protein function. Errors in annotation databases mean it can be difficult to generate a gold standard data set to test methods, although the above-mentioned SFLD [26] continues to expand its database of manually curated families, creating a useful benchmark.
Exploring functional diversity within superfamilies from a sequence perspective – which subfamiles should be targeted for structural determination?
For most organisms, the level of direct experimental characterisation is low (<30% of all proteins) [56]. As previously discussed, better characterisation of the molecular functions of proteins and the interaction networks in which they participate will illuminate functional evolution and lead to more powerful prediction methods. In general, the relationship between sequence and function divergence is subject to much debate, and several contradictory conclusions have been published on the level of sequence similarity needed to confidently transfer functional annotations between homologues [17;27;57-59].
Identifying orthology is perhaps the safest way of transferring functions between relatives and various approaches exist for recent reviews and resources (see [60-63]), yet the majority of relatives in the highly diverse superfamilies are paralogues – the result of different evolutionary history in different organisms. Whilst knowledge of their functions would be extremely valuable for understanding phenotypic diversity, it is important to bear in mind that functions can vary considerably between paralogues. However, as described above, studies of large superfamilies have shown that frequently some central aspect of function (e.g. catalytic chemistry), is shared between relatives and can provide useful clues for genomics initiatives in the context of other data. Various domain and protein family resources cluster relatives at different levels of sequence similarity reflecting higher likelihood of shared functions (see [62] for review of these resources).
Most of these family based resources have evolved through application of pair-wise or profile based algorithms established to recognise homologues but which have not been specifically optimised or benchmarked for capturing functional similarity. In the PANTHER database [63] which explicitly seeks to classify functional relatives, relationships are manually validated, so whilst less prone to error the resource is less comprehensive than some others.
More specific approaches which attempt to divide families into functional subfamilies generally mine information captured in a multiple sequence alignment of relatives. Phylogenetic trees derived from this alignment can be interrogated at different nodes to trace changes in the patterns of conserved residues necessary for different functions (see [62;64;64-67] for a review and recent approaches). Barton and co-workers recently compared a range of different conservation measures ranging from simple counts of identical positions through to entropy based approaches [68]. Since highly conserved residues are likely to be close to a functionally important site, the evolutionary trace method of Lichtarge and co-workers exploits structural data, where available, attaching significance to conserved residues clustering in 3D space. A more recent implementation has shown improvements using a refined entropy based scoring scheme [69]. The sequence harmony method of Heringa and co-workers also exploits an entropy-based approach to identify positions distinguishing specific functional subtypes [70]. Variations on this theme, developed in the Valencia group, include approaches which reflect the multi-factorial nature of function and overlay trees derived from other properties associated with function onto the phylogenetic tree to confirm functional groupings [71].
Whilst many of these approaches appear powerful with promising performance for selected superfamiles, few have been benchmarked on large sets of families nor applied on a genome wide scale. The need to predict functions on a larger scale, to assist both structural and functional genomics initiatives have prompted the recent development of some new strategies that can be applied to larger datasets or entire family resources (e.g. Pfam) [64;68;72]. Some attempt to bolster confidence in functional assignments through homology by examining the similarity of functions from multiple relatives identified by BLAST[73;74] or apply ‘phylogenomics’ using Bayesian approaches to combine information in close branches of a phylogenetic tree [75].
Others apply sensitive profile-profile based comparison strategies with residue conservation scores finely tuned to recognising functional homologues [64-66]. One of the challenges in developing methods for large scale application is the lack of any large datasets to optimise thresholds for safe functional inheritance. In this context, the SFLD resource [26] described above, provides highly valuable data. The SCI-PHY method of the Sjolander group explores phylogenetic trees by using iterative HMM-HMM alignment and entropy based scoring to detect putative functional subgroups and has been benchmarked on 12 curated families (5 from the SFLD) and more than 500 Pfam families. Performance was assessed using informative measures based both on the purity of the subgroups and ability of the method to capture diverse relatives within the subgroups.
Another similar approach which has been applied to recognise shifts in function across superfamilies and has been applied to a large dataset of superfamilies is the FunShift method of Sonnhammer et al. [64]. Entropy based methods can also be applied to iteratively characterise and progressively merge related functional sub-clusters within a family, avoiding the need for a phylogenetic tree [28].
Many of the methods described above attempt to predict the molecular function of proteins, yet it is clearly also important to consider the functional complexes and biological processes in which they participate. There are increasing number of resources providing data on protein networks and also methods for predicting protein interactions (see [62] for reviews). Thus the structural genomics initiatives also target structurally uncharacterised proteins predicted to participate in networks of biological and medical interest (e.g. cancer). Recent interest in protein families highly enriched in gut microbiomes would also benefit from targeting proteins in key metabolic pathways.
Summary
Over the next five years, the worldwide structural genomics initiatives may add a further 2000-3000 structures to the PDB. Some structure genomics initiatives already explore the relationship between structure and function [26;76]. The PSI initiative is currently depositing more structures in the PDB per annum than all other initiatives combined [ref]. Many of these structures are novel - using the 5Å threshold defined above for fold similarity). By refining target selection to include all available functional annotations (both experimental and predicted) this initiative could enrich our understanding of structure-function space and help to reveal structural mechanisms by which functions are modified. We will need further developments in methods for classifying sequence space, in order to recognise and target distinct functional groups. However, to assess the extent to which these initiatives succeed, we will also need better methods for recognising function from structure. Expanding the repertoire of structure-function groups will give valuable insights into protein evolution, which are not only likely to be fascinating but will also ultimately improve the power and accuracy of function prediction methods.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chandonia JM, Brenner SE. The impact of structural genomics: expectations and outcomes. Science. 2006;311:347–351. doi: 10.1126/science.1121018. [DOI] [PubMed] [Google Scholar]
- 2.Todd AE, Marsden RL, Thornton JM, Orengo CA. Progress of structural genomics initiatives: an analysis of solved target structures. J Mol Biol. 2005;348:1235–1260. doi: 10.1016/j.jmb.2005.03.037. [DOI] [PubMed] [Google Scholar]
- 3.Watson JD, Sanderson S, Ezersky A, Savchenko A, Edwards A, Orengo C, Joachimiak A, Laskowski RA, Thornton JM. Towards fully automated structure-based function prediction in structural genomics: a case study. J Mol Biol. 2007;367:1511–1522. doi: 10.1016/j.jmb.2007.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie L, Bourne PE. Functional coverage of the human genome by existing structures, structural genomics targets, and homology models. PLoS Comput Biol. 2005;1:e31. doi: 10.1371/journal.pcbi.0010031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedberg I, Godzik A. Functional differentiation of proteins: implications for structural genomics. Structure. 2007;15:405–415. doi: 10.1016/j.str.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Taylor WR. Evolutionary transitions in protein fold space. Curr Opin Struct Biol. 2007;17:354–361. doi: 10.1016/j.sbi.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Kolodny R, Petrey D, Honig B. Protein structure comparison: implications for the nature of ‘fold space’, and structure and function prediction. Curr Opin Struct Biol. 2006;16:393–398. doi: 10.1016/j.sbi.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 8.Reeves GA, Dallman TJ, Redfern OC, Akpor A, Orengo CA. Structural diversity of domain superfamilies in the CATH database. J Mol Biol. 2006;360:725–741. doi: 10.1016/j.jmb.2006.05.035. [DOI] [PubMed] [Google Scholar]
- 9.Marsden RL, Lee D, Maibaum M, Yeats C, Orengo CA. Comprehensive genome analysis of 203 genomes provides structural genomics with new insights into protein family space. Nucleic Acids Res. 2006;34:1066–1080. doi: 10.1093/nar/gkj494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kinch LN, Grishin NV. Evolution of protein structures and functions. Curr Opin Struct Biol. 2002;12:400–408. doi: 10.1016/s0959-440x(02)00338-x. [DOI] [PubMed] [Google Scholar]
- 11.Reid AJ, Yeats C, Orengo CA. Methods of remote homology detection can be combined to increase coverage by 10% in the midnight zone. Bioinformatics. 2007;23:2353–2360. doi: 10.1093/bioinformatics/btm355. [DOI] [PubMed] [Google Scholar]
- 12.Andreeva A, Murzin AG. Evolution of protein fold in the presence of functional constraints. Curr Opin Struct Biol. 2006;16:399–408. doi: 10.1016/j.sbi.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 13.Kolodny R, Koehl P, Levitt M. Comprehensive evaluation of protein structure alignment methods: scoring by geometric measures. J Mol Biol. 2005;346:1173–1188. doi: 10.1016/j.jmb.2004.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Redfern OC, Harrison A, Dallman T, Pearl FM, Orengo CA. CATHEDRAL: a fast and effective algorithm to predict folds and domain boundaries from multidomain protein structures. PLoS Comput Biol. 2007;3:232e. doi: 10.1371/journal.pcbi.0030232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whisstock JC, Lesk AM. Prediction of protein function from protein sequence and structure. Q Rev Biophys. 2003;36:307–340. doi: 10.1017/s0033583503003901. [DOI] [PubMed] [Google Scholar]
- 16.Jeffery CJ. Moonlighting proteins: old proteins learning new tricks. Trends Genet. 2003;19:415–417. doi: 10.1016/S0168-9525(03)00167-7. [DOI] [PubMed] [Google Scholar]
- 17.Sangar V, Blankenberg DJ, Altman N, Lesk AM. Quantitative sequence-function relationships in proteins based on gene ontology. BMC Bioinformatics. 2007;8:294. doi: 10.1186/1471-2105-8-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piatigorsky J, Kantorow M, Gopal-Srivastava R, Tomarev SI. Recruitment of enzymes and stress proteins as lens crystallins. EXS. 1994;71:241–250. doi: 10.1007/978-3-0348-7330-7_24. [DOI] [PubMed] [Google Scholar]
- 19.Balaji S, Aravind L. The RAGNYA fold: a novel fold with multiple topological variants found in functionally diverse nucleic acid, nucleotide and peptide-binding proteins. Nucleic Acids Res. 2007;35:5658–5671. doi: 10.1093/nar/gkm558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burroughs AM, Allen KN, Dunaway-Mariano D, Aravind L. Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol. 2006;361:1003–1034. doi: 10.1016/j.jmb.2006.06.049. [DOI] [PubMed] [Google Scholar]
- 21.Burroughs AM, Balaji S, Iyer LM, Aravind L. Small but versatile: the extraordinary functional and structural diversity of the beta-grasp fold. Biol Direct. 2007;2:18. doi: 10.1186/1745-6150-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Favia AD, Nobeli I, Glaser F, Thornton JM. Molecular docking for substrate identification: the short-chain dehydrogenases/reductases. J Mol Biol. 2008;375:855–874. doi: 10.1016/j.jmb.2007.10.065. [DOI] [PubMed] [Google Scholar]
- 23.Ojha S, Meng EC, Babbitt PC. Evolution of Function in the “Two Dinucleotide Binding Domains” Flavoproteins. PLoS Comput Biol. 2007;3:121e. doi: 10.1371/journal.pcbi.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shakhnovich BE. Relative contributions of structural designability and functional diversity in molecular evolution of duplicates. Bioinformatics. 2006;22:e440–e445. doi: 10.1093/bioinformatics/btl211. [DOI] [PubMed] [Google Scholar]
- 25.Aravind L, Anantharaman V, Koonin EV. Monophyly of class I aminoacyl tRNA synthetase, USPA, ETFP, photolyase, and PP-ATPase nucleotide-binding domains: implications for protein evolution in the RNA. Proteins. 2002;48:1–14. doi: 10.1002/prot.10064. [DOI] [PubMed] [Google Scholar]
- 26.Pegg SC, Brown SD, Ojha S, Seffernick J, Meng EC, Morris JH, Chang PJ, Huang CC, Ferrin TE, Babbitt PC. Leveraging enzyme structure-function relationships for functional inference and experimental design: the structure-function linkage database. Biochemistry. 2006;45:2545–2555. doi: 10.1021/bi052101l. [DOI] [PubMed] [Google Scholar]
- 27.Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol. 2001;307:1113–1143. doi: 10.1006/jmbi.2001.4513. [DOI] [PubMed] [Google Scholar]
- 28.Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8:R232. doi: 10.1186/gb-2007-8-11-r232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye K, Anton FK, Heringa J, Ijzerman AP, Marchiori E. Multi-RELIEF: a method to recognize specificity determining residues from multiple sequence alignments using a Machine-Learning approach for feature weighting. Bioinformatics. 2008;24:18–25. doi: 10.1093/bioinformatics/btm537. [DOI] [PubMed] [Google Scholar]
- 30.Xie L, Bourne PE. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments. Proc Natl Acad Sci U S A. 2008;105:5441–5446. doi: 10.1073/pnas.0704422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagano N. EzCatDB: the Enzyme Catalytic-mechanism Database. Nucleic Acids Res. 2005;33:D407–D412. doi: 10.1093/nar/gki080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holliday GL, Almonacid DE, Bartlett GJ, O’Boyle NM, Torrance JW, Murray-Rust P, Mitchell JB, Thornton JM. MACiE (Mechanism, Annotation and Classification in Enzymes): novel tools for searching catalytic mechanisms. Nucleic Acids Res. 2007;35:D515–D520. doi: 10.1093/nar/gkl774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Boyle NM, Holliday GL, Almonacid DE, Mitchell JB. Using reaction mechanism to measure enzyme similarity. J Mol Biol. 2007;368:1484–1499. doi: 10.1016/j.jmb.2007.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang H, Blouin C. Insertions and the emergence of novel protein structure: a structure-based phylogenetic study of insertions. BMC Bioinformatics. 2007;8:444. doi: 10.1186/1471-2105-8-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shakhnovich BE, Koonin EV. Origins and impact of constraints in evolution of gene families. Genome Res. 2006;16:1529–1536. doi: 10.1101/gr.5346206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf Y, Madej T, Babenko V, Shoemaker B, Panchenko AR. Long-term trends in evolution of indels in protein sequences. BMC Evol Biol. 2007;7:19. doi: 10.1186/1471-2148-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 38.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 39.Ye Y, Godzik A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics. 2003;19(Suppl 2):ii246–ii255. doi: 10.1093/bioinformatics/btg1086. [DOI] [PubMed] [Google Scholar]
- 40.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 41.Pal D, Eisenberg D. Inference of protein function from protein structure. Structure. 2005;13:121–130. doi: 10.1016/j.str.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 42.Marti-Renom MA, Rossi A, Al-Shahrour F, Davis FP, Pieper U, Dopazo J, Sali A. The AnnoLite and AnnoLyze programs for comparative annotation of protein structures. BMC Bioinformatics. 2007;8(Suppl 4):S4. doi: 10.1186/1471-2105-8-S4-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pazos F, Sternberg MJ. Automated prediction of protein function and detection of functional sites from structure. Proc Natl Acad Sci U S A. 2004;101:14754–14759. doi: 10.1073/pnas.0404569101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laskowski RA, Watson JD, Thornton JM. ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Res. 2005;33:W89–W93. doi: 10.1093/nar/gki414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Porter CT, Bartlett GJ, Thornton JM. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004;32:D129–D133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laskowski RA, Watson JD, Thornton JM. Protein function prediction using local 3D templates. J Mol Biol. 2005;351:614–626. doi: 10.1016/j.jmb.2005.05.067. [DOI] [PubMed] [Google Scholar]
- 47.Kristensen DM, Ward RM, Lisewski AM, Erdin S, Chen BY, Fofanov VY, Kimmel M, Kavraki LE, Lichtarge O. Prediction of enzyme function based on 3D templates of evolutionarily important amino acids. BMC Bioinformatics. 2008;9:17. doi: 10.1186/1471-2105-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shulman-Peleg A, Nussinov R, Wolfson HJ. SiteEngines: recognition and comparison of binding sites and protein-protein interfaces. Nucleic Acids Res. 2005;33:W337–W341. doi: 10.1093/nar/gki482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kinoshita K, Nakamura H. eF-site and PDBjViewer: database and viewer for protein functional sites. Bioinformatics. 2004;20:1329–1330. doi: 10.1093/bioinformatics/bth073. [DOI] [PubMed] [Google Scholar]
- 50.Sasin JM, Godzik A, Bujnicki JM. SURF’S UP! - protein classification by surface comparisons. J Biosci. 2007;32:97–100. doi: 10.1007/s12038-007-0009-0. [DOI] [PubMed] [Google Scholar]
- 51.Binkowski TA, Joachimiak A, Liang J. Protein surface analysis for function annotation in high-throughput structural genomics pipeline. Protein Sci. 2005;14:2972–2981. doi: 10.1110/ps.051759005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Polacco BJ, Babbitt PC. Automated discovery of 3D motifs for protein function annotation. Bioinformatics. 2006;22:723–730. doi: 10.1093/bioinformatics/btk038. [DOI] [PubMed] [Google Scholar]
- 53.Wangikar PP, Tendulkar AV, Ramya S, Mali DN, Sarawagi S. Functional sites in protein families uncovered via an objective and automated graph theoretic approach. J Mol Biol. 2003;326:955–978. doi: 10.1016/s0022-2836(02)01384-0. [DOI] [PubMed] [Google Scholar]
- 54.Dobson PD, Doig AJ. Predicting enzyme class from protein structure without alignments. J Mol Biol. 2005;345:187–199. doi: 10.1016/j.jmb.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 55.Xie L, Bourne PE. A robust and efficient algorithm for the shape description of protein structures and its application in predicting ligand binding sites. BMC Bioinformatics. 2007;8(Suppl 4):S9. doi: 10.1186/1471-2105-8-S4-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karp PD, Keseler IM, Shearer A, Latendresse M, Krummenacker M, Paley SM, Paulsen I, Collado-Vides J, Gama-Castro S, Peralta-Gil M, Santos-Zavaleta A, Penaloza-Spinola MI, Bonavides-Martinez C, Ingraham J. Multidimensional annotation of the Escherichia coli K-12 genome. Nucleic Acids Res. 2007;35:7577–7590. doi: 10.1093/nar/gkm740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hegyi H, Gerstein M. Annotation transfer for genomics: measuring functional divergence in multi-domain proteins. Genome Res. 2001;11:1632–1640. doi: 10.1101/gr.183801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rost B. Enzyme function less conserved than anticipated. J Mol Biol. 2002;318:595–608. doi: 10.1016/S0022-2836(02)00016-5. [DOI] [PubMed] [Google Scholar]
- 59.Tian W, Skolnick J. How well is enzyme function conserved as a function of pairwise sequence identity? J Mol Biol. 2003;333:863–882. doi: 10.1016/j.jmb.2003.08.057. [DOI] [PubMed] [Google Scholar]
- 60.Jensen LJ, Julien P, Kuhn M, von MC, Muller J, Doerks T, Bork P. eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008;36:D250–D254. doi: 10.1093/nar/gkm796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Brien KP, Remm M, Sonnhammer EL. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005;33:D476–D480. doi: 10.1093/nar/gki107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat Rev Mol Cell Biol. 2007;8:995–1005. doi: 10.1038/nrm2281. [DOI] [PubMed] [Google Scholar]
- 63.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abhiman S, Sonnhammer EL. FunShift: a database of function shift analysis on protein subfamilies. Nucleic Acids Res. 2005;33:D197–D200. doi: 10.1093/nar/gki067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown DP, Krishnamurthy N, Sjolander K. Automated protein subfamily identification and classification. PLoS Comput Biol. 2007;3:e160. doi: 10.1371/journal.pcbi.0030160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marttinen P, Corander J, Toronen P, Holm L. Bayesian search of functionally divergent protein subgroups and their function specific residues. Bioinformatics. 2006;22:2466–2474. doi: 10.1093/bioinformatics/btl411. [DOI] [PubMed] [Google Scholar]
- 67.del Sol MA, Pazos F, Valencia A. Automatic methods for predicting functionally important residues. J Mol Biol. 2003;326:1289–1302. doi: 10.1016/s0022-2836(02)01451-1. [DOI] [PubMed] [Google Scholar]
- 68.Manning JR, Jefferson ER, Barton GJ. The contrasting properties of conservation and correlated phylogeny in protein functional residue prediction. BMC Bioinformatics. 2008;9:51. doi: 10.1186/1471-2105-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mihalek I, Res I, Lichtarge O. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J Mol Biol. 2004;336:1265–1282. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 70.Pirovano W, Feenstra KA, Heringa J. Sequence comparison by sequence harmony identifies subtype-specific functional sites. Nucleic Acids Res. 2006;34:6540–6548. doi: 10.1093/nar/gkl901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pazos F, Rausell A, Valencia A. Phylogeny-independent detection of functional residues. Bioinformatics. 2006;22:1440–1448. doi: 10.1093/bioinformatics/btl104. [DOI] [PubMed] [Google Scholar]
- 72.Krishnamurthy N, Brown DP, Kirshner D, Sjolander K. PhyloFacts: an online structural phylogenomic encyclopedia for protein functional and structural classification. Genome Biol. 2006;7:R83. doi: 10.1186/gb-2006-7-9-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin DM, Berriman M, Barton GJ. GOtcha: a new method for prediction of protein function assessed by the annotation of seven genomes. BMC Bioinformatics. 2004;5:178. doi: 10.1186/1471-2105-5-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hawkins T, Luban S, Kihara D. Enhanced automated function prediction using distantly related sequences and contextual association by PFP. Protein Sci. 2006;15:1550–1556. doi: 10.1110/ps.062153506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Engelhardt BE, Jordan MI, Muratore KE, Brenner SE. Protein molecular function prediction by Bayesian phylogenomics. PLoS Comput Biol. 2005;1:e45. doi: 10.1371/journal.pcbi.0010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Najmanovich RJ, lali-Hassani A, Morris RJ, Dombrovsky L, Pan PW, Vedadi M, Plotnikov AN, Edwards A, Arrowsmith C, Thornton JM. Analysis of binding site similarity, small-molecule similarity and experimental binding profiles in the human cytosolic sulfotransferase family. Bioinformatics. 2007;23:e104–e109. doi: 10.1093/bioinformatics/btl292. [DOI] [PubMed] [Google Scholar]
Selected References
- Kolodny R, Petrey D, Honig B. Protein structure comparison: implications for the nature of ’fold space’, and structure and function prediction. Curr Opin Struct Biol. 2006;16:393–398. doi: 10.1016/j.sbi.2006.04.007. This review challenges the idea that structure space can be partitioned into discrete fold groups and suggests that for the purposes of structure and function prediction, it might be more useful to recognise a continuum.
- Xie L, Bourne PE. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments. Proc Natl Acad Sci U S A. 2008;105:5441–5446. doi: 10.1073/pnas.0704422105. The authors report the application of their “geometric potential” to detecting similarities between ligand binding sites. The SOIPPA method they describe is a good example of the need for fast, robust algorithms for detecting functional similarities at the level of protein structure.
- Pal D, Eisenberg D. Inference of protein function from protein structure. Structure. 2005;13:121–130. doi: 10.1016/j.str.2004.10.015. A good example of a method which combines structure and sequence-based comparison approaches to predict functional annotations (in this case Gene Ontology terms) and assign confidence values based on a Bayesian probability model.
- Brown DP, Krishnamurthy N, Sjolander K. Automated protein subfamily identification and classification. PLoS Comput Biol. 2007;3:e160. doi: 10.1371/journal.pcbi.0030160. A robust automated method for identifying functional subfamilies in protein families. The approach can be applied on a large scale and has been benchmarked on both manually curated families and a very large dataset of Pfam families. It exploits a good scheme for assessing the purity and the coverage of the functional clusters. SCI-PHY has been used to provide functional classifications in the Phylofacts database.
- Reva B, Antipin Y, Sander C. Determinants of protein function revealed by combinatorial entropy optimization. Genome Biol. 2007;8:R232. doi: 10.1186/gb-2007-8-11-r232. This method aims to find an optimal division of a family into functional subfamilies using combinatorial entropy optimisation. The method returns a set of subfamilies and the specificity residues associated with them. It has also been applied to identify binding sites and validated on protein/peptide, protein/NA complexes classified in SCOP, with some success. Application of the method to subdivision of Pfam families into functional subfamilies can be examined on the Protein Keys web site http://proteinkeys.net.
- Engelhardt BE, Jordan MI, Muratore KE, Brenner SE. Protein molecular function prediction by Bayesian phylogenomics. PLoS Comput Biol. 2005;1:e45. doi: 10.1371/journal.pcbi.0010045. Statistical approach to function prediction based on phylogenomic principles. SIFTER uses a reconciled phylogenetic tree and derives a posterior probability for the functional annotation of each node. It allows a function to evolve from any other function and more rapidly after duplication than speciation events. It has been tested on 100 Pfam families with promising results.
- Abhiman S, Sonnhammer EL. Large-Scale Prediction of Function Shift in Protein Families with a Focus on Enzymatic Function. PROTEINS: Structure, Function and Bioinformatics. 2005;60:758–768. doi: 10.1002/prot.20550. Method to determine whether functions are likely to have shifted between subfamilies within a family. Applied and tested on a large number of families – enzyme families in Pfam. It could be used to identify structurally uncharacterised subfamilies having different functions to relatives of known structure and these would make good targets for the structural genomics initiatives.
- Burroughs AM, Allen KN, Dunaway-Mariano D, Aravind L. Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol. 2006;361:1003–1034. doi: 10.1016/j.jmb.2006.06.049. An excellent example of a detailed analysis of a structurally diverse superfamily.
- Pegg SC, Brown SD, Ojha S, Seffernick J, Meng EC, Morris JH, Chang PJ, Huang CC, Ferrin TE, Babbitt PC. Leveraging enzyme structure-function relationships for functional inference and experimental design: the structure-function linkage database. Biochemistry. 2006;45:2545–2555. doi: 10.1021/bi052101l. A description of the SFLD resource where large, diverse enzyme superfamilies have been manually classified by experts according to conservation of specific partial reactions or other chemical capabilities.