

Abstract

A new method for the synthesis of nitriles is described. As a complement to the classic cyanation of aryl halides using cyanide sources and a transition metal catalyst, the palladium-catalyzed cross-coupling of thiocyanates with boronic acids in the presence of copper(I) thiophene-2-carboxylate (CuTC) affords nitriles in good to excellent yields.
Aryl nitriles are an important class of compounds. They are key components of pharmaceuticals and natural products and appear as basic constituents of dyes and herbicides.1 A variety of processes for the introduction of cyanide functionality into aromatic compounds have been described. 2 – 3 The Rosenmund-von Braun reaction, a traditional method for preparing aryl nitriles, involves the direct reaction of copper (I) cyanide with aryl halides.4 The reaction usually proceeds at high temperature and with prolonged times. More recently, transition-metalcatalyzed cyanations of aryl halides have been developed as a useful alternative for the preparation of aryl nitriles. Unfortunately, various problems limit the efficiency of this process. For example, mechanistic studies revealed that excess cyanide significantly inhibits the transition metal catalyst by formation of inactive complexes.5 This problem can be circumvented by careful control of the concentration of dissolved cyanide ions. Thus, nonpolar solvents such as toluene or xylene were used to maintain low concentrations of cyanide salts like KCN, Zn(CN)2 in solution.6 Another novel way to overcome this problem was achieved by employing a slow dosage of liquid cyanide precursors such as TMSCN or acetone cyanohydrin to maintain a low concentration of cyanide, but these sources are prone to liberate hydrogen cyanide.7 Recently, potassium hexacyanoferrate (K4[Fe(CN)6]) was introduced as a nontoxic cyanide source for metalcatalyzed cyanations. 8 Other developments are represented by the cyanation of aryl bromides through a copper-catalyzed domino halide exchange and the use of specific amines (TMEDA or 1,1'-methylenedipiperidine) as co-catalysts. 9 – 10 Herein is reported the cyanide-free cyanation of boronic acids with organic thiocyanates as a complementary method for the synthesis of nitriles. This mild and general protocol is an extension of a growing family of thioorganic/boronic acid cross-couplings discovered in this laboratory11 and extended in others.12
Initially, the coupling of boronic acids with several thiocyanates was investigated (Scheme 1). Both catalytic palladium and stoichiometric copper(I) carboxylate are required for the cross-coupling of boronic acids and organic thiocyanates, just as with other thioorganic-boronic acid cross-couplings.11,12 Among the systems studied for the cyanative cross-coupling the Pd(PPh3)4/CuTC system gave the best results and 1.5 equiv of boronic acids were used to optimize the cross-coupling yields. In all cases, the cyanation products were obtained in good to excellent yields (70–92%) in THF at 50 °C.
Scheme 1.

Variation of Thiocyanates for the Cyanation of Boronic Acids
As probed by the examples depicted in Scheme 1, the nature of the organic thiocyanate did not seem critical to the reaction, so commercially available benzyl thiocyanate was employed as the cyanide source in subsequent experiments and served as an excellent cyanide source in boronic acid cross-coupling reactions.13
With these results in hand, the scope and the possible limitations of this new protocol were studied. As shown in Table 1, a variety of different boronic acids were tested in the copper-mediated, palladium-catalyzed cyanation reaction. Very efficient transformations of unactivated (electron-neutral) (entries 1–3) and activated (electron– rich) aryl boronic acids (entries 4–6) were observed under the above conditions (1.5 equiv CuTC, THF/50 °C). However, deactivated (electron-deficient) aryl and heteroaryl boronic acids were generally less reactive and led to incomplete conversion under the same conditions. To overcome this problem, a variety of Pd catalysts and ligand systems were screened, but the survey did not lead to improved results. Fortunately, the use of an excess of CuTC (2–3 equiv) at a higher reaction temperature (100 °C) in 1,4-dioxane afforded much higher yields. Other solvents tested (DMF, DMA, NMP, toluene) were not as effective. Under such conditions all but the most electron-deficient aryl boronic acids gave the corresponding benzonitriles in good yields. Sterically congested substrates (entries 5–7) undergo cyanation efficiently. The cyanation of heteroaryl boronic acids is also possible. 3-Thiopheneboronic acid and 2-methoxy-5-pyridineboronic acid gave corresponding nitriles in 86% and 84% yields, respectively (entries 13–14). With the exception of 3-nitrophenylboronic acid (entry 12), 14 electron-deficient substrates can be effectively cyanated (entries 8, 9, and 11). It is worth noting that alkenyl nitriles, as well, can also be obtained in good yields by this new method (entries 15–16). Unlike the Suzuki-Miyaura cross-coupling of organoboron reagents, where the presence of a base is essential, 15 the CuTC-mediated reaction proceeds efficiently under non-basic conditions. This offers the possibility of the synthesis of base-sensitive compounds. Finally, in contrast to boronic acids, neither boronate esters nor boroxines (boronic acid anhydrides) were effective in this new cyanative coupling.
Table 1.
Pd-Catalyzed Cyanation of Boronic Acids Mediated by CuTC16
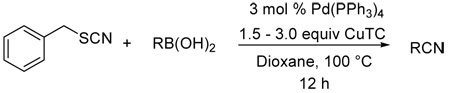 | |||
|---|---|---|---|
| entry | R | RCN | yield (%)d |
| 1a | phenyl | 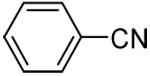 |
90 |
| 2a | 2-naphthyl | 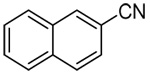 |
90 |
| 3a | 1-naphthyl |  |
82 |
| 4a | 4-(N,Ndimethylamino) | 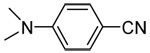 |
95 |
| 5a | Phenyl | 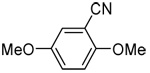 |
90 |
| 6a | 2,5-dimethoxyphenyl | 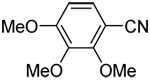 |
85 |
| 7c | 2,3,4-trimethoxyphenyl |  |
76 |
| 8b | 3-(acetyl)phenyl | 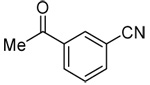 |
86 |
| 9b | 4-(methoxycarbonyl)phenyl | 83 | |
| 10c | 4-chlorophenyl | 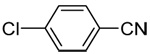 |
80 |
| 11c | 4-cyanophenyl | 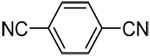 |
80 |
| 12c | 3-nitrophenyl | 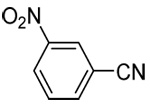 |
tracee |
| 13c | 3-thienyl | 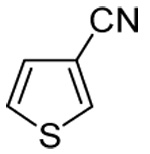 |
83 |
| 14c | 2-methoxy-5-pyridinyl | 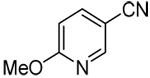 |
84 |
| 15c | E-β-styryl | 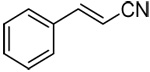 |
82 |
| 16c | 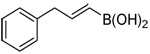 |
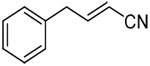 |
75 |
1.5 equiv CuTC.
2.0 equiv CuTC.
3.0 equiv CuTC.
Isolated yield.
Detected by GC-MS.
From a mechanistic perspective, the coupling may start with an oxidative addition of the thiocyanate to Pd0,17 followed by a transmetalation from boron to palladium (Scheme 2). Reductive elimination would afford the expected nitrile and regenerate a catalytically active Pd(0). On the basis of previous observations, it is suggested that the copper(I) carboxylate functions as a unique, dual activator; the soft Cu(I) ion acts as a thiophilic agent to help polarize the palladium thiolate bond, while simultaneously providing borophilic activation by coordination of the carboxylate to the trivalent boron atom.11a The much greater reactivity of boronic acids compared to that of boronate esters and boroxines is consistent with the proposal of the hydrogen bonded, ternary complex depicted in Scheme 2 as the reactive intermediate.
Scheme 2.

Proposed Mechanism for Thiocyanate / Boronic Acid Coupling
In summary, a new cyanide-free cyanation of boronic acids has been developed through the use of a palladium-catalyzed, copper(I)-mediated coupling of benzylthiocyanate with boronic acids. Using this protocol a variety of nitriles can be easily synthesized in high yields. The method is a useful complement to the traditional synthesis of nitriles through transition metal-catalyzed cross-coupling of aryl halides/sulfonates with cyanide sources.
Supplementary Material
Complete description of experimental details and product characterization (7 pages). This material is available free of charge via Internet at http://pubs.acs.org.
Acknowledgment
The National Institutes of General Medical Sciences, DHHS supported this investigation through grant No. GM066153.
References
- 1.Sundermeier M, Zapf A, Beller M, Sans S. Tetrahedron Lett. 2001;42:6707. [Google Scholar]
- 2.Sundermeier M, Zapf A, Beller M.Eur. J. Inorg. Chem 20033513 and references therein.Ellis GP, Romney-Alexander TM. Chem. Rev. 1987;87:779.
- 3.(a) Tschaen DM, Desmond R, King AO, Fortin MC, Pipik B, King S, Verhoeven TR. Synth. Commun. 1994;24:887. [Google Scholar]; (b) Maligres PE, Waters MS, Flietz F, Askin D. Tetrahedron Lett. 1999;40:8193. [Google Scholar]; (c) Anderson BA, Bell EC, Ginah FO, Harn NK, Pagh LM, Wepsiec JP. J. Org. Chem. 1998;63:8224. [Google Scholar]; (d) Sundermeier M, Zapf A, Beller M. Angew. Chem., Int. Ed. 2003;42:1661. doi: 10.1002/anie.200250778. [DOI] [PubMed] [Google Scholar]; (e) Okano T, Kiji J, Toyooka Y. Chem Lett. 1998:425. [Google Scholar]; (f) Chidambaram R. Tetrahedron Lett. 2004;45:1441. [Google Scholar]
- 4.(a) Mowry DT. Chem. Rev. 1948;42:189. doi: 10.1021/cr60132a001. [DOI] [PubMed] [Google Scholar]; (b) Friedman L, Shechter H. J. Org. Chem. 1961;26:2522. [Google Scholar]; (c) Maligres PE, Waters MS, Fleitz F, Askin D. Tetrahedron Lett. 1999;40:8193. [Google Scholar]
- 5.(a) Sekiya A, Ishikawa N. Chem. Lett. 1975:277. [Google Scholar]; (b) Takagi K, Okamoto T, Sakakibara Y, Ohno A, Oka S, Hayama N. Bull. Chem. Soc. Jpn. 1972;49:3177. [Google Scholar]
- 6.(a) Okano T, Kiji J, Toyooka Y. Chem. Lett. 1998:425. [Google Scholar]; (b) Maligres PE, Waters MS, Fleitz F, Askin D. Tetrahedron Lett. 1999;40:8193. [Google Scholar]; (c) Veauthier JM, Carlson CN, Collis GE, Kiplinger JL, John KD. Synthesis. 2005:2683. [Google Scholar]
- 7.(a) Cassar L, Ferrara S, Foa M. Adv. Chem. Ser. 1974;132:252. [Google Scholar]; (b) Sundermeier M, Zapf A, Beller M. Angew. Chem., Int. Ed. 2003;42:1661. doi: 10.1002/anie.200250778. [DOI] [PubMed] [Google Scholar]
- 8.(a) Schareina T, Zapf A, Beller M. Chem. Comm. 2004:1388–1389. doi: 10.1039/b400562g. [DOI] [PubMed] [Google Scholar]; (b) Weissman SA, Zewge D, Chen C. J. Org. Chem. 2005;70:1508–1510. doi: 10.1021/jo0481250. [DOI] [PubMed] [Google Scholar]; (c) Grossman O, Gelman D. Org. Lett. 2006;8:1189. doi: 10.1021/ol0601038. [DOI] [PubMed] [Google Scholar]
- 9.Zanon J, Klapars A, Buchwald SL. J. Am. Chem. Soc. 2003;125:2890. doi: 10.1021/ja0299708. [DOI] [PubMed] [Google Scholar]
- 10.Sundermeier M, Zapf A, Mutyala S, Baumann W, Sans S, Weiss S, Beller M. Chem. Eur. J. 2003;9:165. doi: 10.1002/chem.200390210. [DOI] [PubMed] [Google Scholar]
- 11.(a) Srogl J, Liebeskind LS. J. Am. Chem. Soc. 2000;122:11260. [Google Scholar]; (b) Savarin C, Srogl J, Liebeskind LS. Org. Lett. 2001;3:91. doi: 10.1021/ol006807d. [DOI] [PubMed] [Google Scholar]; (c) Savarin C, Liebeskind LS. Org. Lett. 2001;3:2149. doi: 10.1021/ol010060p. [DOI] [PubMed] [Google Scholar]; (d) Srogl J, Liebeskind LS. Org. Lett. 2002;4:979. doi: 10.1021/ol0200091. [DOI] [PubMed] [Google Scholar]; (e) Kusturin CL, Liebeskind LS, Neumann WL. Org. Lett. 2002;4:983. doi: 10.1021/ol025547s. [DOI] [PubMed] [Google Scholar]; (f) Liebeskind LS, Srogl J, Savarin C, Polanco C. Pure Appl. Chem. 2002;74:115. [Google Scholar]; (g) Kusturin C, Liebeskind LS, Rahman H, Sample K, Schweitzer B, Srogl J, Neumann WL. Org. Lett. 2003;5:4349. doi: 10.1021/ol035649y. [DOI] [PubMed] [Google Scholar]; (h) Lory P, Gilbertson SR. Chemtracts. 2005;18:569. [Google Scholar]
- 12.(a) Alphonse F-A, Suzenet F, Keromnes A, Lebret B, Guillaumet G. Org. Lett. 2003;5(6):803. doi: 10.1021/ol027453o. [DOI] [PubMed] [Google Scholar]; (b) Alphonse F-A, Suzenet F, Keromnes A, Lebret B, Guillaumet G. Synlett. 2002;3:447. doi: 10.1021/ol027453o. [DOI] [PubMed] [Google Scholar]; (c) Lengar A, Kappe CO. Org. Lett. 2004;6(5):771. doi: 10.1021/ol036496h. [DOI] [PubMed] [Google Scholar]; (d) Oumouch S, Bourotte M, Schmitt M, Bourguignon J-J. Synthesis. 2005:25. [Google Scholar]; (e) Morita A, Kuwahara S. Org. Lett. 2006;8:1613. doi: 10.1021/ol053122a. [DOI] [PubMed] [Google Scholar]
- 13.Ethyl- and benzyl thiocyanates are commercially available; phenylthiocyanate and p-nitrophenylthiocyanate were prepared from the corresponding thiol according to Still, Ian WJ, Watson Iain DG. Synth. Commun. 2001;31:1355.
- 14.The poor reactivity of 3-nitrophenylboronic acid, in contrast to the other electron-deficient substrates, is not understood at this time.
- 15.Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- 16.Typical experimental procedure: Benzylthiocyanate (1.0 equiv), boronic acid (1.5 equiv), CuTC (1.5-3.0 equiv), and Pd(PPh3)4 (3 mol %) were added to a flask that was then flushed with argon. Dry and degassed 1,4-dioxane (2 mL per 0.1 mmol) was added. The brown suspension was stirred under the protection of argon at 100 °C for 12 h. Then 10 mL Et2O was added and the reaction mixture was washed with satd. NH4Cl followed by satd. Na2CO3 and then brine. After evaporation of the solvent, the residue was purified by preparative plate silica gel chromatography (eluted with 4:1 hexanes/EtOAc) to give the desired product.
- 17.Kamiya I, Kawakami J-i, Yano S, Nomoto A, Ogawa A. Organometallics. 2006;25:3562. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Complete description of experimental details and product characterization (7 pages). This material is available free of charge via Internet at http://pubs.acs.org.