

Abstract
Manganese transport regulator (MntR) is a member of the diphtheria toxin repressor (DtxR) family of transcription factors that is responsible for manganese homeostasis in Bacillus subtilis. Prior biophysical studies have focused on the metal-mediated DNA binding of MntR [Lieser, S. A., Davis, T. C., Helmann, J. D., and Cohen, S. M. (2003) Biochemistry 42, 12634-12642], as well as metal stabilization of the MntR structure [Golynskiy, M. V., Davis, T. C., Helmann, J. D., and Cohen, S. M. (2005) Biochemistry 44, 3380-3389], but only limited data on the metal-binding affinities for MntR are available. Herein, the metal-binding affinities of MntR were determined by using electron paramagnetic resonance (EPR) spectroscopy, as well as competition experiments with the fluorimetric dyes Fura-2 and Mag-fura-2. MntR was not capable of competing with Fura-2 for the binding of transition metal ions. Therefore, the metal-binding affinities and stoichiometries of Mag-fura-2 for Mn2+, Co2+, Ni2+, Zn2+, and Cd2+ were determined and utilized in MntR/Mag-fura-2 competition experiments. The measured Kd values for MntR metal binding are comparable to those reported for DtxR metal binding [Kd from 10-7 to 10-4 M; D’Aquino, J. A., et al. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 18408-18413], AntR [a homologue from Bacillus anthracis; Sen, K. I. et al. (2006) Biochemistry 45, 4295-4303], and generally follow the Irving-Williams series. Direct detection of the dinuclear Mn2+ site in MntR with EPR spectroscopy is presented, and the exchange interaction was determined, J = -0.2 cm-1. This value is lower in magnitude than most known dinuclear Mn2+ sites in proteins and synthetic complexes and is consistent with a dinuclear Mn2+ site with a longer Mn···Mn distance (4.4 Å) observed in some of the available crystal structures. MntR is found to have a surprisingly low binding affinity (∼160 μM) for its cognate metal ion Mn2+. Moreover, the results of DNA binding studies in the presence of limiting metal ion concentrations were found to be consistent with the measured metal-binding constants. The metal-binding affinities of MntR reported here help to elucidate the regulatory mechanism of this metal-dependent transcription factor.
Bacteria handle the delicate issue of metal ion homeostasis using a class of transcription factors known as metalloregulatory proteins (metalloregulators). In some systems, these metal-sensing proteins are involved in mediating the removal of toxic metals, while in other systems they are central to maintaining the required levels of essential metals. To date, a large number of metalloregulatory proteins have been identified that respond to a variety of metal ions (1-3). The subject of how metal binding is translated into an ability to control transcription via a metalloregulator has become a prominent subject of investigation (4-10). Indeed, many metalloregulators are reported to bind several metal ions in vitro, while only eliciting a specific transcriptional response when they are bound to the cognate metal in vivo (5-7, 9). This observation naturally leads to the question: how does a metalloregulator selectively respond to its cognate metal ion as opposed to other available metal ion activators? The ability of a metalloregulator to respond selectively to a metal ion may depend on several factors, including the availability of the requisite metal ion, the binding affinity for the metal ion, the charge on the metal ion, and the coordination geometry/number assumed by the metal ion upon binding. Determining which of these factors are most important for a given metalloregulator is essential for gaining a better understanding of how these proteins elicit transcriptional control.
The manganese transport regulator MntR1 is found in Bacillus subtilis and is a member of the iron-responsive DtxR family of proteins (11). MntR is a 142-amino acid protein that binds its DNA-recognition sequence as a homodimer in the presence of activating metal ions (12, 13). Like other members of the DtxR family, MntR requires the binding of two metal ions per protein monomer and utilizes a helixturn-helix DNA-binding motif (12, 13). Despite these similarities, MntR differs from other members of the DtxR family in that it lacks a C-terminal SH3 domain and forms a metal-independent dimer. Another significant difference is that the two metal ions in MntR form a bridged, dinuclear metal active site (M···M distances ranging from 3.3 to 4.4 Å), which is distinct from the two mononuclear sites (M···M distance ∼9 Å) found in DtxR (13-15). Finally, DtxR is an iron responsive repressor (16), while MntR is responsive to manganese and cadmium in vivo (11).
Structural studies on MntR show that, with the exception of the metal site, the overall protein structure is the same for various metal isoforms (13, 15). As for the metal-binding site, three different arrangements have been observed for MntR: (i) a binuclear AB conformer with a 3.3 Å M···M distance, (ii) a binuclear AC conformer with a 4.4 Å M···M distance, and (iii) a mononuclear conformer with only the A site occupied (Figure 1). The AB and AC binuclear clusters utilize the same protein ligands, but the binding mode and side chain orientations in the two forms are different. The AC form of MntR has been observed upon reconstitution with Ca2+, Mn2+, and Cd2+, while the AB form has only been observed with Mn2+ (13, 15). With Mn2+, formation of the AB versus AC form is dependent upon conditions such as temperature and pH; from these studies, the AC form has been proposed as the more physiologically relevant conformer (15). The mononuclear A only site form has been observed with Co2+ (A. Glasfeld, personal communication) and Zn2+ (15). On the basis of the observation that the A site is conserved in all three structure types, it has been suggested that the A site serves as an “activation filter,” and that the formation of a binuclear site is essential for full activation of the protein (15).
FIGURE 1.
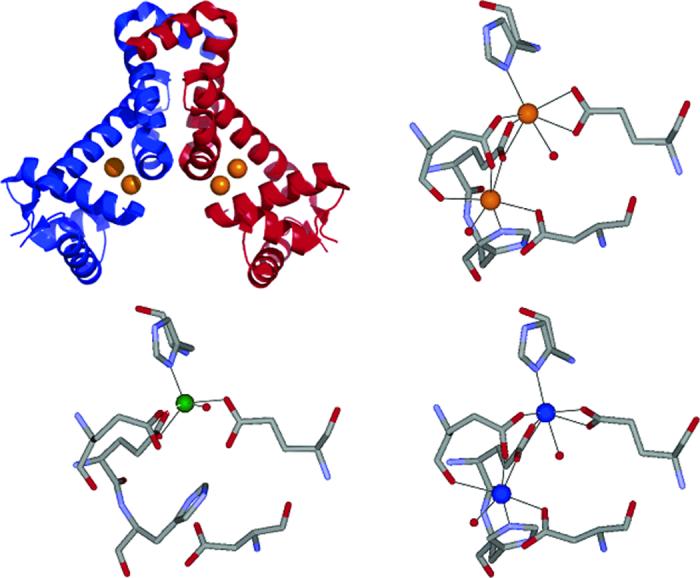
Ribbon diagram (upper left) of MntR.Cd2+ (orange spheres, PDB ID: 2EV0), with the monomers colored separately in red and blue. Diagrams of the active site coordination environments in MntR·Cd2+ (AC form, upper right, Cd2+ shown as orange spheres, PDB ID: 2EV0), MntR·Zn2+ (A form, lower left, Zn2+ shown as green sphere, PDB ID: 2EV6), and MntR·Mn2+ (AC form, lower right, Mn2+ shown as blue spheres, PDB ID: 2F5F).
MntR is involved in manganese homeostasis through regulation of two manganese transporters, MntABCD and MntH (11). The proposed scheme for metal ion regulation by MntR suggests that when the manganese requirements of the cell are met, MntR binds Mn2+, resulting in DNA binding and repression of genes responsible for encoding the aforementioned manganese transporter proteins. Presumably, when manganese levels drop below those required for B. subtilis, the metal ions dissociate from MntR, and MntR is released from the mnt operons allowing for transcription of the transporter proteins.
Prior studies on MntR have focused on the metal activation profile, which showed that in vitro an excess of Mn2+ or Cd2+ ions (1.0 mM, >100-fold concentration relative to protein present) results in tight DNA binding (12). Other metal ions, such as Fe2+ and Co2+, were found to elicit moderate operator binding as well (17). Previous studies suggest that binding of these activating metal ions to MntR result in a change in protein tertiary structure, while the secondary structure remains largely unaltered (12, 17). Despite the insight gained from these investigations, the studies were typically performed in the presence of excess metal ions (>100-fold excess), complicating the interpretation and physiological relevance of these findings. To better understand these results, detailed knowledge of the metal binding of MntR is required. Two recent reports have examined metal binding in MntR and related homologs. In the first study, several new crystal structures, with Ca2+, Mn2+, Zn2+, and Cd2+, along with calorimetry data on metal binding was reported for MntR (15). In the second study, a close homologue of MntR, AntR from B. anthracis, was probed by electron paramagnetic resonance (EPR) spectroscopy to examine metal binding and oligomerization behavior (18). Herein, we present the metal-binding affinities of MntR for several transition metal ions in an attempt to complement earlier biophysical studies and to compare our findings to the recent reports on MntR and AntR. By measuring the metal-binding affinities of MntR and relating them to the reported DNA-binding activation profile, we can refine our interpretation of these prior findings, as well as determine whether a high metal-binding affinity is essential for DNA binding activation of MntR. The results presented here also point to a possible explanation for the Mn2+/Cd2+ dual responsiveness of MntR.
MATERIALS AND METHODS
General
All buffers were prepared using water purified by a Labconoco Water Pro Plus purification system. All biochemical reagents were obtained from commercial suppliers and were used as provided unless otherwise specified. 8-Anilino-1-naphthalenelsulfonic acid (ANS) was acquired from Sigma-Aldrich and stock solutions were prepared as previously described (17). MnCl2.4H2O, CoCl2.H2O, CdCl2.H2O, NiCl2.6H2O, and ZnCl2 (99.99+%) were obtained from Aldrich. Protein purification was carried out as previously described (12, 17). The metal content/purity of all ultrapure water, buffers, protein preparations, and metal titrant solutions were determined as previously described (12) using a Perkin-Elmer Optima 3000 DV inductively coupled plasma optical emission spectrometer (ICP-OES) located at the Analytical Facility at the Scripps Institute of Oceanography. All fluorescence intensity and anisotropy experiments were performed on a Perkin-Elmer LS-55 luminescence spectrometer using a thermally jacketed cell holder that was maintained at 25 °C. All absorption spectra were collected on a Perkin-Elmer Lambda 25 spectrophotometer at ambient temperature.
Fura-2 and Mag-fura-2 were purchased from Invitrogen; stock solutions were prepared by resuspending the lyophilized powders in 1 mL of degassed ultrapure water. Dye concentrations were determined by absorption spectroscopy by using the reported extinction coefficients (28 000 M-1cm-1 at 363 nm for Fura-2 and 22 000 M-1 cm-1 at 369 nm for Mag-fura-2). Experiments with these fluorimetric dyes were carried out in either metal-binding buffer (10 mM HEPES, pH 7.2, 100 mM KCl) or protein storage buffer (20 mM HEPES, pH 7.2@4 °C, 200 mM NaCl, 5% glycerol).
ANS Experiments
In an 800 μL microcuvette, a 400 μL solution containing 200 μM ANS dye and 2 μM MntR in protein storage buffer was prepared. Metal stock solutions were titrated (from 0.001 to 1000 μM) into the ANS/MntR mixture (17). The fluorescence emission of ANS was monitored, and the intensity at 507 nm was plotted as a function of metal ion concentration. The data were fit with a 1:1 binding isotherm by using least-squares regression analysis software (KaleidaGraph, Synergy Software) to determine the relative affinities for Mn2+, Co2+, and Cd2+.
Protein Competition with Fura-2
In an 800 μL microcuvette, a 400 μL solution containing 5 μM Fura-2 and 50 μM MntR in protein storage buffer was prepared; the solution was incubated for 20 min at ambient temperature. To the equilibrated solution was added 5 μM metal ion (Mn2+, Co2+, or Cd2+), and the resulting absorption spectra were recorded for ∼16 h in 20 min intervals. The resulting spectra were compared to the spectrum just prior to addition of protein. A control experiment where 200-700 μM EDTA was added to the microcuvette (in lieu of MntR) was used to obtain the apo Fura-2 spectrum.
Metal-Binding Affinity/Stoichiometry of Mag-fura-2
For all fluorescence experiments employing the dye Mag-fura-2 the fluorescence excitation was scanned from 250 to 450 nm while monitoring emission at 505 nm. Excitation and emission slits were set at 10 and 5 nm, respectively. To determine the metal-binding stoichiometry of Mag-fura-2 with Mn2+, Co2+, Ni2+, Zn2+, and Cd2+ electronic absorption spectroscopy was used in combination with the method of continuous variation (Job’s method) (19). In an 800 μL microcuvette, a 400 μL solution containing Mag-fura-2 and the divalent metal ion of interest was prepared for a combined total concentration of 42 or 66 μM in metal-binding buffer. Nine solutions were prepared for each metal ion, with the mole fraction of Mag-fura-2 ranging from 0 to 1. Each solution was allowed to equilibrate at ambient temperature for 5 min before recording the absorbance spectrum from 200 to 500 nm. The absorbance of Mag-fura-2 alone was subtracted from each experimental spectrum, and the data were analyzed by plotting A369 vs mole fraction of Mag-fura-2 (XMf-2).
To determine the metal-binding affinity of Mag-fura-2 with Mn2+, Co2+, Ni2+, Zn2+, and Cd2+ ions two sets of experiments were carried out, one in metal-binding buffer and a second in protein storage buffer. In a 4 mL cuvette, a 2 mL solution containing 0.5 μM Mag-fura-2 and 1 or 10 mM metal ion was prepared; the solution was allowed to equilibrate at ambient temperature for 3 min. A competing metal ion was then titrated (0 to ∼5 mM, <10% total volume change) into the preincubated mixture, and the fluorescence excitation spectra were recorded. A 3 min equilibration time was allowed between additions of competing metal ion. In experiments designed to determine the affinity of Mag-fura-2 for either Co2+, Mn2+, or Ni2+ (binding of these metal ions quenches the Mag-fura-2 fluorescence, as previously observed for Fura-2) (20), Ca2+ was used as the preincubation metal (Kd = 25 μM, as determined by the manufacturer, Invitrogen) (21). In experiments designed to determine the affinity of Mag-fura-2 for Zn2+ or Cd2+ (binding of these metals generates a fluorescence excitation spectrum with a maximal λex ∼330 nm), the preincubation metal was Co2+, Mn2+, or Ni2+. The reported Mag-fura-2 Kd of 25 μM for Ca2+ was used as the reference for experiments performed in either metal binding or protein storage buffer. It has been reported that Mag-fura-2 metal-binding affinities do not change significantly in the pH range of 6.4-7.5 and within ionic strengths 0.1-0.2 M (22, 23); therefore, the Kd of 25 μM should be satisfactory under both buffer conditions used here. The fluorescence intensity at 505 nm when excited with an excitation wavelength of λex = 330 nm was fit using a custom DYNAFIT script that describes the competition between two metal ions for the dye (Supporting Information); reported Kd values are an average of at least three individual experiments. The program DYNAFIT does not utilize a single explicit equation (binding isotherm expression) to fit thermodynamic data, but fits the data employing simultaneous nonlinear algebraic equations (24, 25).
Protein Competition with Mag-fura-2
In an 800 μL microcuvette, a 400 μL solution containing 0.5 μM Mag-fura-2 and MntR (10-100 μM) was prepared in protein storage buffer. The mixture was incubated at ambient temperature for 3 min. The metal ion of interest was titrated into the preincubated solution with a 3 min equilibration time between additions. Fluorescence excitation spectra were recorded from 250 to 450 nm at each point in the titration. The fluorescence intensity at 505 nm with an excitation wavelength of λex = 330 nm (Zn2+, Cd2+) or λex = 380 nm (Co2+, Mn2+, Ni2+) was fit using a custom DYNAFIT scripts that describe the competition between the dye and the protein for the metal ion (Supporting Information). Alternatively, the data can be fit by adapting a series of binding isotherm equations used for competition absorption spectrophotometry (19, 26, 27). Fitting of the data to a single metal-binding site by the latter procedure gives values consistent with those obtained from the more explicit DYNAFIT procedure (Supporting Information, Figure S10). Reported Kd values are an average of at least three individual experiments.
Using absorption spectroscopy an analogous competition experiment was also carried out. In an 800 μL microcuvette, a 400 μL solution containing 6.5 μM Mag-fura-2 and 50 μM MntR was prepared in protein storage buffer. The mixture was incubated at ambient temperature for 3 min. A Co2+ solution was titrated from 0.03 to 200 μM into the preincubated solution with a 3 min equilibration time between additions. Absorbance spectra were recorded from 250 to 500 nm, and binding of Co2+ to the dye was monitored by disappearance of the apo Mag-fura-2 absorbance at ∼362 nm. Data were modeled using a custom DYNAFIT (24) script that fit competition between the dye and the protein for the metal ion (Supporting Information); reported Kd values are an average of at least three individual experiments.
Fluorescence Anisotropy
Fluorescence anisotropy experiments were performed as previously described (12). In a 4 mL cuvette were placed 10 nM dsmntH26 (fluorescein-labeled strand of dsmntH26 = 5′-6F-GAATAATTTGCCTTAAGGAAACTCTC-3′) (12) and 10, 100, or 1000 μM (final concentration) of divalent metal ion (Mn2+, Co2+, or Cd2+). To this solution was added increasing amounts of MntR (0-1.5 μM). Measurements were collected with an excitation wavelength of 492 nm (slit width 15 nm), an emission wavelength of 520 nm (slit width 20 nm), and a 1.0 s integration time. The g-factor for all of the experiments was 1.15 ± 0.08. The robs versus [MntR]total data were fit to a 1:1 binding isotherm model (non-dissociable dimer) (12) by using least-squares regression analysis software (KaleidaGraph, Synergy Software).
EPR Spectroscopy
X-band (9 GHz) EPR spectra were recorded on a Bruker ESP 300 spectrometer equipped with an Oxford ESR 910 cryostat for low-temperature measurements. The microwave frequency was calibrated with a frequency counter, and the magnetic field was calibrated with a NMR gaussmeter. The temperature was calibrated with resistors (CGR-1-1000) from LakeShore. A modulation frequency of 100 kHz was used for all EPR spectra. All experimental data were collected under nonsaturating microwave conditions. Room temperature measurements used a flat quartz cell positioned in an E-field null plane of the microwave cavity.
EPR spectra were analyzed by diagonalization of the spin Hamiltonian H = -2JS1.S2 + Hdipolar + H1 + H2, where J is the isotropic exchange coupling between metal sites, Hdipolar is the through space Mn-Mn magnetic dipolar interaction, and H1 and H2 contain the Zeeman and zero-field terms for the individual Mn2+ ions (28). The simulations were generated with consideration of all intensity factors relative to a spin standard (CuEDTA), which allowed computation of simulated spectra for a specific sample concentration. The simulations therefore allow a quantitative determination of protein signal intensities. The Windows software package SpinCount was used and is available for general application to any mono- or dinuclear metal complex by contacting the corresponding authors (M.P.H.).
For single value decomposition (SVD) of data (29), the spectra were arranged into an N rows by M columns matrix, A, with each column representing a spectrum with N data points. This matrix can be expressed in terms of three matrices U, S, and V such that A = USVT. In this representation, S is an N × N diagonal matrix of non-negative elements containing the singular values of matrix A. The diagonal elements of the matrix S2 are the eigenvalues, and the columns of V are the corresponding eigenvectors of ATA. Within the context of a physical model, the entries in V can be related to the amplitudes of the base spectra for each of the titration points. The physical model in our particular application is the sequential binding two metal sites to the protein,
| (1) |
where Mn, E, EMn, and EMn2 represent the concentrations of Mn(H2O)62+, and apo-, mono-, and dimetal bound protein species, respectively. The total concentrations of protein and metal are ET and MnT, and Ki are the equilibrium constants. From eq 1, the concentrations of Mn, EMn, and EMn2 can be determined in terms of the binding constants K1 and K2 (30). In the limit K1 ⪡ K2, the low concentration of the EMn species will render it undetectable. Thus, the mononuclear binding step is not measurable, and the fit of the data will be primarily sensitive to the product K1K2. The experimental matrix V is calculated from A using a standard SVD routine (31). A computer program was constructed that then leastsquare fits the value of the product K1K2 to give the best match of the theoretical matrix CP constructed from eq 1 to the experimental matrix V. C is an n × r matrix containing the species concentrations as a function of total added metal concentration (for our application, r = 2 species, n = 7 titration points), and P is a set of linear parameters (29).
EPR samples were prepared by titrating Mn2+ into a solution of apoMntR. Stock solutions of MnCl2 (Aldrich) were prepared daily in protein buffer with doubly distilled water. Quantitation of Mn2+ stock solutions were determined through double integration of the EPR spectra and compared against atomic absorption standards for Mn2+ (Aldrich). For each metal addition, MntR was titrated with the appropriate amount of stock solution in successive additions. For room temperature measurements, after each addition the sample was incubated for 10 min at 4 °C prior to recording the EPR spectrum. For low temperature measurements, each sample was incubated for 10 min at 4 °C and then frozen in liquid N2, followed by recording of the spectrum. Unless noted, the buffer for all samples was 20 mM HEPES (Sigma), pH 7.2, 300 mM NaCl, 5% glycerol.
RESULTS
ANS Experiments
The fluorescence of the ANS dye increases upon binding to hydrophobic surfaces; prior studies with ANS and MntR were used to monitor changes in protein tertiary/quaternary structure at several metal ion concentrations (17). The fluorescence of ANS in buffered solution increases upon addition of MntR and then successively decreases upon addition of increasing amounts of activating metal ions (e.g., Mn2+), indicating a change in protein structure and a burying of hydrophobic surfaces within MntR. Using this decrease in fluorescence as a probe, much like the quenching of tryptophan fluorescence, ANS can be used as an exogenous reporter (7, 9). Titration curves of fluorescence emission intensity versus metal ion concentration were obtained for MntR with Mn2+, Co2+, and Cd2+. Under these conditions, MntR binds Mn2+, Co2+, and Cd2+ with Kd values of 92, 13, and 0.5 μM, respectively (Figure 2). This initial study revealed that the cognate metal ion, Mn2+, is bound with the weakest affinity relative to other metal ions that activate MntR for DNA binding. Because of several limitations of using ANS as a probe for metal ion binding (vide infra), a more quantitative method for determining the metal-binding affinities of MntR was sought. The fluorimetric dyes Fura-2 and Mag-fura-2 were used in competition experiments with MntR to obtain quantitative data.
FIGURE 2.

Titration of Mn2+ (○), Co2+ (■), or Cd2+ (×) into a solution of MntR (2 μM) and ANS (200 μM) in protein storage buffer. The ANS fluorescence at 507 nm is quenched as MntR binds to the metal ions. Fits of the data using a 1:1 binding isotherm are shown as solid lines. T = 25 °C.
Protein Competition with Fura-2
Originally designed as a molecular sensor for Ca2+, the binding affinity of Fura-2 for several transition metal ions has been determined (20, 32-34), and this fluorophore has been employed as a tool to study metal binding with the metalloregulatory protein NikR (6). Dyes such as Fura-2 are useful in this capacity as both their absorption and fluorescence spectra change upon binding to metal ions, providing a direct spectroscopic handle for evaluating metal-binding affinities when in competition with metalloproteins. Competition experiments between Fura-2 and MntR were carried out by preincubating 5 μM dye and 50 μM MntR in protein storage buffer, after which 5 μM of the metal ion of interest (Mn2+, Co2+, Cd2+) was added to the solution, and absorption spectra were recorded overnight at 20 min intervals. In all experiments, the spectrum of dye-metal complex remained unperturbed (data not shown), implying that MntR was incapable of competing with the dye for metal binding. Because MntR was present in a 10-fold excess over Fura-2, the affinity of MntR for these metal ions must be at least an order of magnitude weaker than that of Fura-2. This places the lower limit on the Kd values of MntR at 30 nM for Mn2+, 90 nM for Co2+, and 10 pM for Cd2+. It was concluded that MntR was unable to compete with Fura-2 for these metal ions; therefore, a fluorescent dye with weaker metal ion affinities was sought.
Metal-Binding Affinity/Stoichiometry of Mag-fura-2
Mag-fura-2 (21), a dye structurally similar to Fura-2 (Figure S1, Supporting Information) but possessing a weaker Ca2+ affinity, was investigated as an alternative to Fura-2. It was assumed that the weaker Ca2+ affinity of Mag-fura-2 relative to Fura-2 would also reflect a decrease in transition metal ion affinities for Mag-fura-2. Unlike Fura-2, for which a number of metal-binding affinities have been reported (20, 32-34), the affinity of Mag-fura-2 has only been reported for the transition metal ion Zn2+ (35). Therefore, we determined both the affinity and stoichiometry of Mag-fura-2 for Mn2+, Co2+, Ni2+, Zn2+, and Cd2+ to perform our studies with MntR.
Similar to Fura-2, Mag-fura-2 metal binding is accompanied by changes in its absorbance and fluorescence spectra. In particular, complexation of Mag-fura-2 with the closed-shell ions Ca2+, Zn2+, or Cd2+ results in loss of fluorescent excitation at ∼369 nm (indicative of apo-Mag-fura-2, λem) 505 nm) with subsequent generation of a excitation band centered at ∼330 nm (indicative of the dye-metal complex). Complexation of Mag-fura-2 with the paramagnetic ions Mn2+, Co2+, and Ni2+ results in quenching of the excitation band at ∼369 nm. Using these spectral features, the dissociation constants for Mag-fura-2 were determined in both metal binding and protein storage buffer. A representative example of the spectra and model of the data for each type of competition titration (Co2+ and Cd2+) are shown in Figures S2 and S3 (Supporting Information). The Kd values range from 0.04 to 1 μM (Table 1), with Zn2+ < Ni2+ ∼2+Co∼ Cd2+ < Mn2+ ∼ Co2+ With exception the of Zn2 35 ∼ + (), the affinities of Mag-fura-2 ∼ for transition metal ions decrease by several orders of magnitude relative to Fura-2, consistent with that expected based on the difference in affinity of these two dyes for Ca2+ (21, 32). The experiments performed in metal-binding buffer are typical of the conditions often used for determining binding constants of fluorescent dyes and other small molecule ligands (32, 36). The Kd values for Mag-fura-2 with these metal ions were also measured in protein storage buffer (Table 1), to ensure that the effects of pH and ionic strength (relative to metal-binding buffer) were minimal and would not adversely impact the competition experiments with MntR. Finally, by using absorption spectroscopy and Job’s method (Figure S4, Supporting Information) a 1:1 metal/Mag-fura-2 stoichiometry was confirmed for all of the metal ions examined.
Table 1.
Dissociation Constants (Kd, with Standard Deviations) for Mag-fura-2 with Several Metal Ions as Determined in Metal Binding and Protein Storage Buffer.
| metal binding buffer (μM) | protein storage buffer (μM) | |
|---|---|---|
| Ca2+ | 25a | n/d |
| Mn2+ b | 0.97 ± 0.17 | 0.89 ± 0.03 |
| Co2+ b | 0.93 ± 0.04 | 0.92 ± 0.06 |
| Ni2+ b | 0.13 ± 0.01 | 0.13 ± 0.01 |
| Zn2+ c | 0.036 ± 0.001d | 0.037 ± 0.001 |
| Cd2+ c | 0.10 ± 0.01 | 0.16 ± 0.01 |
Provided by the manufacturer, Invitrogen.
Determined by competition against Ca2+.
Determined by competition against Mn2+, Co2+, or Ni2+.
20 nM, I = 0.15, T = 37 °C, see ref 35.
Protein Competition with Mag-fura-2
Mag-fura-2 was used in competition experiments against MntR. These competition experiments were performed in a manner consistent with others described in the literature (37), with the metal ions of interest titrated into a solution containing both Mag-fura-2 and MntR. The competition between MntR and Mag-fura-2 for the titrated metal ion was monitored by fluorescence spectroscopy and in one case also confirmed by absorption spectroscopy (Co2+, Figure S5, Supporting Information). Fluorimetric titration of Mn2+, Co2+, and Ni2+ into a solution of Mag-fura-2 with an excess MntR results in the quenching of the Mag-fura-2 fluorescence excitation band at ∼369 nm (λem = 505). Figure 3 shows a representative example of a titration performed with Co2+. Titration of Zn2+ and Cd2+ results in the disappearance of a band at ∼369 nm and formation of a new band at ∼330 nm in the fluorescence excitation spectrum indicative of the metaldye complex. Figure 4 shows a representative competition titration in the presence of Cd2+. The data from all competition titrations were analyzed using DYNAFIT (24, 25) with a model that incorporated the relevant equilibria (Supporting Information); for Mn2+, Co2+, and Ni2+ experiments data were fit using the fluorescence excitation at 380 nm, while for Zn2+ and Cd2+ titrations the data were fit using the fluorescence excitation at 330 nm. The dissociation constants measured from these experiments are summarized in Table 2.
FIGURE 3.

Fluorescence excitation spectra (top) of a competition experiment between MntR (15 μM) and Mag-fura-2 (0.5 μM) titrated with Co2+ in protein storage buffer. The initial spectrum is shown as a heavy solid line, and the final spectrum is shown as a heavy dashed line (binding of Co2+ quenches Mag-fura-2 fluorescence). The spectra represent the emission intensity at 505 nm (ordinate) as a function of excitation wavelength (abscissa). Fit (solid line) of the fluoresence excitation intensity at 380 nm (bottom) as a function of metal ion concentration. T = 25 °C.
FIGURE 4.

Fluorescence excitation spectra (top) of a competition experiment between MntR (15 μM) and Mag-fura-2 (0.5 μM) titrated with Cd2+ in protein storage buffer. The initial spectrum is shown as a heavy solid line, and the final spectrum is shown as a heavy dashed line (binding of Cd2+ changes the maxima of Mag-fura-2 fluorescence). The spectra represent the emission intensity at 505 nm (ordinate) as a function of excitation wavelength (abscissa). Fit (two-site model, solid line) of the fluoresence excitation intensity at 330 nm (bottom) as a function of metal ion concentration. T = 25 °C.
Table 2.
Dissociation Constants, Based on a Single Site Model, for MntR with Several Metal Ions as Determined by Competition Titrations against Mag-fura-2 or with ANS Dye in Protein Storage Buffer (Kd, with Standard Deviations)
| Mag-fura-2 - Kd (μM) | ANS - Kd (μM) | |
|---|---|---|
| Mn2+ | ≥50a, 160b | 92 ± 14 |
| Co2+ c | 4.9 ± 1.3 | 13 ± 1 |
| Ni2+ | 2.1 ± 0.1 | n/d |
| Zn2+ | 0.013 ± 0.003 | n/d |
| Cd2+ | 0.100 ± 0.006d | 0.5 ± 0.1 |
Value provided is a lower limit of the affinity estimated by modeling the data.
∼160 μM as measured by EPR spectroscopy T = 25 °C (Kd1 = 900 μM and Kd2 = 30 μM).
8.87 ± 1.48 as measured by absorption spectroscopy (Figure S5, Supporting Information).
Best fit with a two-site model (Kd1 = 0.10 μM and Kd2 = 3.9 μM).
Overall, the affinities of MntR appear to loosely follow the Irving-Williams series, with Kd values ranging from 0.01 to ≥50 μM, with Zn2+ < Cd2+ < Ni2+ ∼ Co2+, Mn2+. The results of the competition titrations between Mag-fura-2 and MntR are in agreement with the findings of the ANS experiments (vide supra). Importantly, the Mn2+ affinity could not be measured under the presented conditions; even a > 200-fold excess of MntR (110 μM) could not compete with Mag-fura-2 for Mn2+ (Figure S9, Supporting Information). On the basis of simulations of the data using DYNAFIT (24, 25), these experiments show that MntR has a surprisingly weak affinity for its cognate metal ion, with a lower limit for the Kd value of Mn2+ binding to MntR at ≥50 μM. Indeed, all of the other metal ions tested, including Ni2+ and Zn2+, which do not effectively activate MntR for DNA binding, have a higher binding affinity for MntR than Mn2+. These findings unambiguously demonstrate that the selective DNA-binding response of MntR is not due to a thermodynamic preference for binding Mn2+.
EPR Spectroscopy
Samples of 0.25 mM apoMntR were titrated with MnCl2 in successive additions up to approximately 6 equiv. The room-temperature EPR spectra of the titration showed a six-line hyperfine pattern typical of 55Mn2+ (inset of Figure 5). This spectrum is typical of Mn-(H2O)62+ species. All titration points showed the same spectra but with varying intensity of the signal. The concentration of Mn(H2O)62+ in the titration series was determined by normalizing the intensity to a sample of a known amount of MnCl2 added to the same buffer and recorded under the same conditions. The amount of Mn2+ detected in these spectra was a fraction of the total amount added to the protein sample, indicating binding of Mn2+ to the protein is an equilibrium process. Mn2+ complexed to protein reduces the coordination of weak-field water, increasing spin-orbit interactions and zero-field energies relative to that of Mn(H2O)62+. Consequently, the signals from the protein bound Mn2+ species are too broad to be detectable at room temperature (38). A plot of the Mn(H2O)62+ equivalents observed versus the total equivalents of Mn2+ added to the samples is shown in Figure 5. The results were repeated for two independent titration experiments.
FIGURE 5.

Equivalents (open circles) of the Mn(H2O)62+ species versus equivalents of added Mn2+, as measured from the intensity of the room-temperature EPR spectra. The mathematical curves show the equivalents of the Mn(H2O)62+ species (solid line) and the protein bound dinuclear Mn2+ species (dashed line) generated from the sequential binding model of eq 1 with Kd1 = 900 μM, Kd2 = 30 μM. Inset: room-temperature EPR spectrum of 0.19 mM MntR with 1.15 mM MnCl2 added. Spectral conditions: microwaves, 9.78 GHz, 20 mW; modulation, 0.1 mTpp.
The equivalence of Mn(H2O)62+ data can be fit to the sequential metal-binding model of eq 1. The theoretical curve for equivalents of 2+ dimers (28) as a function of temperature, microwave frequency, and microwave orientation. The complexes of this previous study have sufficiently large spin exchange (J ≫ D, gβB), to give well-isolated excited spin manifolds and corresponding subspectra from each manifold. For MntR, the spin exchange energy is small, thus the splitting between the spin manifolds is comparable to the zero-field and Zeeman energies. Consequently, there are no isolated spin manifolds or subspectra, the spin levels all mix, and we expect that the many overlapping transitions will produce a broad featureless spectrum. The spectrum should have greatest intensity near g) 2, since the transitions with the highest probability occur here. In addition, at temperatures as low as 14 K, each spin level has nearly equal population, and we expect the spectra should show Curie law behavior.
Figure 7C shows simulations of the MntR dinuclear Mn2+ site at temperatures of 2 and 14 K. The simulated spectra were calculated for equivalent Mn2+ sites (parameters given in Figure 7). As we have demonstrated in our previous work, the software allows quantitative comparisons of experimental spectra with simulations. From the SVD results, the concentration of the dinuclear species in Figure 7B is [Mn2-MntR]) 0.13 mM. The simulations of Figure 7C are calculated for the same concentration of the dinuclear Mn2+ species. While the simulation does not match all of the features of the spectrum, the intensity is in approximate agreement with that of the experimental spectrum. The experimental spectra do not display sufficient resolution to allow determination of any of these parameters with certainty, except for the exchange interaction. The assignment to a dinuclear species is unambiguous. For an antiferromagnetic exchange coupling of J)-0.20(5) cm-1 (H = -2JS1.S2) for the dinuclear Mn2+ site, the temperature dependence of the simulations matches that of the experimental data shown in Figure 6A. The temperature dependence of the data cannot be reproduced with only a magnetic dipolar interaction between the Mn2+ ions, since this represents an energy contribution to the system of less than 0.05 cm-1.
FIGURE 7.

EPR basis spectra derived from the SVD decomposition of the spectra of the titration of MntR with MnCl2: (A) Mn-(H2O)62+, (B) Mn2-MntR. The basis spectra are derived using the consecutive binding model (eq 1) for the 14 K spectra, and K1K2 = 5 × 108 M-2. A 1:1 addition of (A) and (B) gives the 1.2 eq EPR spectrum at 14 K of Figure 6A. (C) Simulations of the Mn2-MntR species at temperatures of 14 K (solid line) and 2.3 K (dashed line). The intensity scale of the 14 K simulation is calculated for 0.13 mM Mn2-MntR (fully load protein). Spectrum B is plotted for this same scale. The intensity of 2.3 K simulation is scaled for 1/T relative to the 14 K simulation. Simulation parameters are for two identical Mn2+ ions: J = -0.2 cm-1 (-2JS1·S2), S = 5/2, I = 5/2, A = 250 MHz, D = 0.04 cm-1, E/D = 0.21, rMnMn = 4.4 Å, rΘ = 45°.
FIGURE 6.

EPR spectra of 0.22 mM MntR with additions of (A) 0.29 mM MnCl2 and (B) 1.14 mM MnCl2 at temperatures of 14 K (solid line) and 2.3 K (dashed line). The intensity of the spectra is plotted as signal × temperature after normalization for instrumental parameters. The scale of A has been increased by 10× for clarity. Spectral conditions: microwaves, 9.65 GHz, 0.2 mW(14 K) and 0.002 mW (2 K); modulation, 1.0 mTpp.
Fluorescence Anisotropy Experiments
To get a better idea of how the metal binding of MntR affects DNA binding, we revisited our prior fluorescence anisotropy experiments, which were previously performed in the presence of 1.0 mM metal ion (12, 17), which was in excess of >100-fold over the concentration of MntR. To probe the effect of metal ion affinity, the binding of MntR to the consensus sequence oligonucleotide dsmntH26 was measured under lowered metal ion concentrations. The binding of MntR to dsmntH26 was unchanged when the concentration of Mn2+, Co2+, or Cd2+ was dropped from 1.0 mM to 100 μM. In contrast, at 10 μM metal ion (Figure 8), the binding of MntR to dsmntH26 was preserved with Cd2+ but was virtually abolished with Mn2+. In the presence 10 μM Co2+, MntR binding to dsmntH26 is diminished but not totally eradicated. These observations are wholly consistent with the measured metal-binding affinities; at 10 μM M2+, the protein can still bind Cd2+ and to a lesser degree Co2+ because the amount of available metal exceeds the measured Kd values (Table 2). In contrast, 10 μM Mn2+ is below the Kd value for this metal ion leading to loss of DNA binding ability. The observed effects of lowered metal ion concentrations on the DNA binding ability of MntR are in good qualitative agreement with the measured metal-binding affinities of MntR.
FIGURE 8.

DNA binding assays with 10 nM dsmntH26 titrated with MntR in presence of 1000 μM (○), 100 μM (■), or 10 μM (×) of metal ion. Results are shown for Cd2+ (top), Co2+ (middle), and Mn2+ (bottom). Buffer = 20 mM HEPES pH 7.2@4 °C, 500 mM NaCl, 5% (v/v) glycerol. T = 25 °C.
DISCUSSION
Our prior work with MntR has focused on elucidation of the metal-mediated DNA binding properties of this protein (12, 17) and studies on how metal binding affects protein structure (17). Two new reports describe some metal-binding affinities of MntR and a homolog AntR (15, 18); our results are compared with these recent findings below. Measuring the metal-binding affinities is an essential component for understanding how MntR selectively responds to its cognate metal ion and elucidating its mechanism of action.
In preliminary experiments to gauge the metal-binding affinities of MntR, the hydrophobic dye ANS was utilized as an exogenous fluorescent reporter. Changes in ANS fluorescence were used as an indicator of metal-induced structural organization; Kd values could be estimated based on changes in ANS fluorescence intensity as the dye was excluded from hydrophobic surfaces on MntR. The Kd values determined by this method were 92 μM for Mn2+, 13 μM for Co2+, and 0.5 μM for Cd2+ (Table 2). These results were the first to indicate that MntR does not show tight binding for its cognate metal ion Mn2+. ANS is an environmental probe, thereby its fluorescence depends on the interaction of the dye with hydrophobic portions of MntR. As the location of binding, binding affinity, and number of binding sites for ANS on MntR are not known, the values determined by this approach are subject to several sources of error/bias (e.g., ANS might compete for the metal-binding sites), and the Kd values obtained are best interpreted as relative affinities. With these limitations in mind, an alternative method to corroborate the values obtained from the ANS experiments was sought.
Competition titrations between the protein and a spectroscopically active competitor ligand were selected as a means to quantitatively measure the metal-binding affinities of MntR. Fura-2 and Mag-fura-2 are fluorescent dyes that have been previously employed for studying other metalloregulatory proteins (6, 37) and thus were selected as a starting point for our investigation. Both dyes are frequently employed as ratiometric Ca2+/Mg2+ chelators but can also bind transition metal ions with related spectral changes. The affinities of Fura-2 for several transition metal ions has been determined (20, 32-34), while for Mag-fura-2 only the binding constant with Zn2+ has been reported (35).
Fura-2 was selected for initial experiments because its affinities for Mn2+, Fe2+, Co2+, and Cd2+ ions are known and are in a range (picomolar to nanomolar) observed for many metalloregulatory proteins (40). Surprisingly, in our competition experiments between Fura-2 and MntR, where MntR was present in a 10-fold excess over Fura-2, MntR proved unable to compete for the metal ions Mn2+, Co2+, and Cd2+. On the basis of the inability of MntR to compete with Fura-2 for binding these metal ions, an approximate lower limit on the Kd values for MntR could be inferred: Mn2+ > 30 nM, Co2+ > 90 nM, and Cd2+ > 0.01 nM. Compared to Fura-2, Mag-fura-2 lacks several ligating atoms (Figure S1, Supporting Information) and has Ca2+/Mg2+ affinities 2 orders of magnitude weaker than that of Fura-2 (21, 32). Because the binding affinities of Fura-2 for transition metal ions are several orders of magnitude stronger than they are for alkali earth metals, it was anticipated that Mag-fura-2 would show a similar binding constant trend (e.g., Kd values for transition metal ions in the nanomolar to micromolar range). Previously, Zn2+ was the only transition metal ion for which the binding constant with Mag-fura-2 had been reported (35). Therefore, the stoichiometry and metal-binding affinity of Mag-fura-2 for Mn2+, Co2+, Ni2+, Zn2+ (as a control), and Cd2+ were measured (at I = 0.11 M and 25 °C) using the approach originally employed for determining Fura-2 affinities for Mn2+ and Fe2+ (32). Specifically, Fura-2 fluorescence quenches upon binding to these paramagnetic ions, and the binding affinities can be determined by monitoring this quenching as the transition metal ions compete with Ca2+ for the dye. Mag-fura-2 shows similar fluorescence behavior, with Mn2+, Co2+, and Ni2+ quenching the fluorescence of the dye and Zn2+ and Cd2+ exhibiting fluorescence spectra similar to that generated upon binding Ca2+. On the basis of this observation, the affinities of Mag-fura-2 for paramagnetic ions were measured by competition against Ca2+, while affinities for Zn2+ and Cd2+ were measured by competition against the paramagnetic ions (after the Kd values were determined). As anticipated, the measured Kd values (Table 1) fall in the low micromolar to nanomolar range, which should prove useful in studies involving relatively weak metal-ligand or metal-biomolecule interactions.
Metal Binding Affinities of MntR
After determining Mag-fura-2 affinities for several transition metal ions, dye-protein competition titrations were performed to determine metal-binding affinities of MntR. Overall, the binding behavior loosely follows the Irving-Williams series (41), with Kd values (Table 2) following the order Zn2+ < Cd2+ < Ni2+ ∼ Co2+, Mn2+. The model used to evaluate the metal binding of MntR utilizes only a single binding constant; however, crystallographic analysis of MntR loaded with Mn2+, Co2+, Zn2+, and Cd2+ show that only Mn2+ and Cd2+ form dinuclear sites (13, 15), while MntR binds only one Co2+ or Zn2+ ion per monomer (15) (A. Glasfeld personal communication). Attempts to analyze Cd2+ binding to MntR with a two-site model were found to improve the fit to the data (Kd1 = 0.10 μM and Kd2 = 3.9 μM), and the Kd values are provided as a footnote in Table 2. Under our present experimental conditions, this two binding site analysis for Cd2+ is the most robust model of the data we can provide.
The weak binding affinity of MntR for Mn2+ determined by competition with Mag-fura-2 qualitatively agrees with the measured dissociation constants determined by ANS dye experiments and EPR data presented here (vide infra), as well as those determined by EPR for the homolog AntR (18). In contrast, the dissociation constant determined in a recent report by calorimetry (∼3 μM for two independent sites, vide infra) (15) is significantly stronger and does not agree with either of the aforementioned studies. Furthermore, the dissociation constants measured by ITC between MntR and Zn2+ (2-6 μM) and Cd2+ (7-17 μM) are 300- and 180-fold larger than those determined here by competition ∼ with Mag-fura-2 (15). The slightly higher affinity of MntR for Mn2+ versus Cd2+ reported by ITC is inconsistent with the DNA-binding data reported here, where in the presence of low concentrations of Cd2+ MntR is competent to bind DNA, while at the same concentration of Mn2+ DNA binding is completely abolished (Figure 8). Differences in the buffer conditions for the ITC versus the experiments reported here (500 mM NaCl, pH 8, 10% for ITC vs 200 or 300 mM NaCl, pH 7.2, 5% glycerol) seem inadequate to explain the disparate findings from these two studies. The ITC experiments were reported in large part to confirm the metal/protein monomer binding stoichiometry (2:1 for Mn2+ and Cd2+, 1:1 for Zn2+) observed crystallographically (15). On the basis of the aforementioned discrepancies, we propose that the ITC values are a useful gauge of binding stoichiometry but that the Mag-fura-2 and EPR studies presented here are a more reliable determination of the binding constants.
The EPR study of AntR reported a binding affinity of this protein for Zn2+ of 152 (47 μM based on a 2:1 binding stoichiometry per AntR monomer with infinite cooperativity between the binding sites (18). This value is substantially different than the Kd value of 13 ±3 nM determined here for MntR and Zn2+ based on a single binding site. The disparity between these findings is surprising, considering the good agreement for the Mn2+ Kd values and the extremely high homology between these two proteins. Both AntR and MntR are 142 aa in length, and an alignment analysis (ClustalW v1.83, data not shown) shows that the proteins share 82% sequence homology and all of the metal-binding residues in MntR are conserved in AntR. Again, the solution conditions of these experiments are insufficient to explain the large difference in the Zn2+ Kd values. At the moment, the difference in the studies is puzzling; however, the activation profile for AntR may provide an explanation. For AntR, it is reported that the Zn2+ form is a good analogue of the Mn2+ bound form, with the former being activated for binding the cognate DNA sequence (18). This finding is in sharp contrast to MntR, for which Zn2+ is an extremely poor activator of DNA binding (12). This comparative evidence suggests that despite the very high homology of these two proteins, they may well have different activation profiles and hence different metal-binding sites. This explanation remains highly speculative, and further biophysical and structural characterization of AntR is required to resolve this apparent inconsistency in the AntR/MntR findings.
EPR Spectroscopy
The crystal structure of MntR identifies four binding sites for Mn2+ per protein dimer, which forms two dinuclear Mn2+ centers. To date, no other direct spectroscopic probe of the electronic environment of the metal has been available. Here we report the first EPR spectra to show direct evidence for the quantitative formation of a dinuclear Mn2+ center from solution samples. The temperature-dependent data and simulations indicate a weak antiferromagnetic exchange coupling of J = -0.2 cm-1. The exchange interaction for dinuclear Mn2+ model complexes with H2O/dicarboxylato bridges are in the range -1 to -3 cm-1 (42, 43), and hydroxo-/dicarboxylato bridges are -9 cm-1 (44). Dinuclear Mn2+ model complexes with only dicarboxylate bridges have exchange couplings near -1cm-1 (45-47). The exchange coupling for MntR is significantly smaller than that of dinuclear Mn2+ centers in these model complexes. A dinuclear Mn2+ model complex with a single carboxylato bridge has been characterized with J = -0.19 cm-1 (48), a value close to that for MntR.
The crystal structures of MntR identify two protein conformations that bind Mn2+ with differing protein and solvent coordination. The structure with an Mn···Mn distance of 3.3 Å with a bridging hydroxyl species would give characteristically different EPR spectra and a larger exchange value than observed for MntR in these studies. The other structure has a significantly longer Mn-Mn distance of 4.4 Å. For this structure, the manganese ions are bridged by μ1,3-carboxylato-(Glu99) and μ(O)-carboxylato-(Glu102) in two different configurations. In both configurations, both carboxylato groups have a long Mn-O bond (2.5 Å). These long bonds will weaken the exchange pathway, ∼ which is consistent with the lower exchange value observed here for MntR. We have made many attempts to facilitate the formation of the structure with the shorter Mn···Mn distance. We found no change in the dinuclear Mn2+ complex at pH 7.2 and 8.5. In addition, higher salt levels, a cryo-protectant (glycerol), and flash-freezing of protein all had no effect on the signals. These results suggest a role for crystal packing forces in the structure with the shorter Mn···Mn distance and that the structure with the longer Mn···Mn distance is relevant in solution studies.
The ratio of the binding constants indicates that the binding of the first Mn2+ to MntR is significantly weaker than that of the second Mn2+. This ratio indicates a cooperative binding process which is perhaps better described by the overall equilibrium for the binding of two Mn2+ ions (2Mn2+ + apoMntR ↔Mn2-MntR) with an equilibrium constant K = K1K2. Our dissociation constants (Kd1 = 900 μM, Kd2 = 30 μM,) are comparable to those determined previously from a AntR (Kd1 = 210 μM and Kd2 = 17 μM, ) (18) but significantly greater than those obtained from ITC measurements (Kd1 = 10 μM and Kd2 = 1 μM, ) (15). The value of is more certain than the individual values [radical1] and are also consistent with the results of the ANS and Mag-fura-2 experiments reported here.
The EPR measurements detect a small change (factor of 4) toward tighter binding upon freezing MntR. The crystallographic data finds two different conformations of the Mn2 site that depend on crystallization conditions having a short (3.3 Å) and long (4.4 Å) Mn···Mn distance (15). We do not attribute the change in the binding constant to a switch between these conformations, for the following two reasons. First, the increase in the stability of the complex upon freezing is in the range expected for the enthalpy change due to metal-ligand binding. For example, the enthalpy change for binding of Mn2+ to two histidines is -5.2 kcal/mol (22). A change in temperature from 25 °C to 0 °C will increase the stability of the complex by a factor of 2. Second, the EPR spectra of samples that were frozen over 1 min were identical to spectra of flash frozen samples. If the interconversion between the two conformations (e.g., short Mn···Mn at room temperature, long Mn···Mn when frozen) is due to a relatively slow protein dependent event, then the flash frozen samples should have shown a significant change in the EPR spectrum. Thus, we conclude that the change in the binding constant is an enthalpic effect rather than a change in Mn···Mn distance.
Metal Affinities of MntR Versus Other Metalloregulatory Proteins
Recent calorimetry studies on DtxR were able to dissect a high (2 × 10-7 M) and low (6.3 10-4 M) affinity site for the binding of two Ni2+ ions per protein monomer (10); however, in DtxR the metal-binding sites are well separated and possess substantially different ligating residues, a factor likely contributing to the measurable difference in affinities. In contrast, Mn2+ binding to MntR is best fit with a cooperative binding model. The coordination environments of the two metal ions in the dinuclear site of MntR are very similar: both are essentially hexacoordinate with one nitrogen and five oxygen based ligands (13, 15). The proximity of the two sites also supports the observation that the binding of Mn2+ is cooperative. Similarly, metal binding was also found to be highly cooperative in AntR (18), further supporting the cooperative binding model found for Mn2+ binding to MntR.
The observation that metal binding in MntR generally follows the Irving-Williams series (41) is not unique to this metalloregulatory protein. Similar findings have been obtained with Fur, NikR, and NmtR, which have completely different metal response profiles from MntR (6, 7, 49). The observation that several classes of metalloregulatory proteins show similar metal binding trends clearly indicates that selective allosteric regulation is not merely a reflection of metal-binding affinity. This is particularly noteworthy for MntR as the Irving-Williams series dictates (and our data support) that the cognate metal ion Mn2+ will be bound with lower affinity than most other biologically relevant, divalent transition metal ions. This finding shows that the selectivity of MntR must be of a different origin, which likely involves a critical active site geometry imposed by Mn2+ (15) or perhaps specific Mn2+ delivery via a metallochaperone (50), consistent with the arguements of Robinson and Giedroc (49-51).
With the metal-binding affinities of MntR determined, we can now re-evaluate the selective activation of MntR for DNA binding. As already detailed, the cognate metal ions, Mn2+ and Cd2+, are bound quite weakly and tightly, respectively. Co2+, an ion that leads to modest DNA binding at saturating levels, as well as Ni2+ and Zn2+, which do not effectively activate DNA binding even at saturation, are all bound by MntR more tightly than Mn2+. This indicates that although MntR is competent for binding Co2+, Ni2+, and Zn2+, the metals do not induce the appropriate allosteric change to induce tight DNA binding. In the case of Co2+ and Zn2+, incomplete activation is in part due to failure to form a dinuclear site (15) (A. Glasfeld, personal communication). Ni2+ may also fail to form a dinuclear site, or the geometry of such a site with this metal ion may be significantly different, which again fails to produce the requisite allosteric change in MntR. Overall, we would concur with the hypothesis of Glasfeld et al. (15) that the geometry and stoichiometry (dinuclear) of metal binding to MntR is more significant to the mechanism of activation than metal ion affinity.
It is interesting that Fur and DtxR family members exhibit metal affinities generally much weaker than members of MerR and ArsR/SmtB families, the latter of which bind cognate ions in the 10-9 to 10-21 M range (8, 37, 40, 52-57). One explanation for this apparent trend may come from considering the function of these metalloregulatory protein families. MerR and ArsR/SmtB metalloregulators generally regulate genes (as repressors in their apo form) required to export toxic metal ions or an overabundance of an otherwise essential metal ion (40, 52). The removal of such harmful metal ions may require a very sensitive response to manage the threat before damage is incurred on the cell. In contrast, Fur- and DtxR-family proteins are generally responsible for controlling genes involved in essential metal ion import (acting as repressors in their holo forms) (58). Because cellular acquisition of an essential metal ion may be more tolerant to a slight excess than a deficiency of the ion, the proteins that regulate metal ion influx may be responsive at much higher metal concentrations to ensure that adequate metal stores are obtained before suppressing import genes. In this context, one can rationalize why members of Fur/DtxR families have weaker cognate metal-binding affinities when compared to MerR/ArsR families.
The aforementioned hypothesis may also be an argument for contextualizing the difference in binding affinities of MntR for Mn2+ and Cd2+. It is known that several Mn2+ transporting proteins are capable of also transporting Cd2+ (59). At saturation concentrations, both Mn2+ and Cd2+ were found to be equally strong activators of MntR for DNA binding (12); however, the studies here show that MntR binds Mn2+ at least 1000-fold less tightly than Cd2+. Therefore, in presence of Mn2+, MntR functions, as expected, like a member of the DtxR family, allowing the accumulation of the necessary levels of this essential metal ion before shutting down the uptake machinery. In contrast, it may be that when B. subtilis is faced with Cd2+ in its surroundings, MntR functions more like a member of the MerR/ArsR family of proteins, responding with great sensitivity to the presence of the toxic metal ion and suppressing the corresponding uptake genes.
The question remains: under physiological conditions how does MntR selectively respond to Mn2+ when the protein has such a low binding affinity for this metal ion? At present, no information about the B. subtilis metallome is available; however, the Escherichia coli metallome has been determined (56, 60) and can be used to make an comparative analysis. In E. coli, the total metal concentrations in the cell is ∼100 μM for Zn2+ and Fe2+, ∼10 μM for Mn2+ and Cu2+, and submicromolar for Ni2+ and Co2+ (60). However, the concentrations of “free” metal ions (those not bound to proteins or other biological ligands) are likely less than the total ion concentrations listed. For example, in the case of Cu2+ and Zn2+ the free concentration of these ions is estimated to be less than one free metal ion per cell (∼10-9 M) (54, 56). If B. subtilis accumulates metal ions in a similar fashion, the amounts of total Co2+ and Ni2+ and free Cu2+ and Zn2+ would be well below the binding affinity of the MntR, rendering it non-responsive to these metals (although the binding constant for Cu2+ and MntR is not available, it would be quite surprising for MntR to demonstrate sufficiently tight binding to Cu2+ to overcome the extremely low concentrations of this metal ion). “Loosely bound” Fe2+ concentrations in E. coli have been estimated at ∼10 μM (61), and although we do not have quantitative binding data for MntR with Fe2+, these concentrations could potentially compete with Mn2+. Indeed, some mutants of MntR can be activated by Fe2+ in vivo (62). Although no direct measurement of free Mn2+ is available for E. coli or B. subtilis, evidence suggests that these microorganisms might be more tolerant to higher cellular concentrations of Mn2+ relative to other transition metal ions. Toxicity tests with E. coli grown on agar plates containing increasing concentrations of transition metal ions showed that the minimal inhibitory concentration for Mn2+ was 20 mM, approximate 20 times higher than that found for Co2+, Ni2+, Cu2+, and Zn2+ (1 mM) and 40 times higher than that of Cd2+ (0.5 mM) (63). Furthermore, in several Bacillus species, the minimal Mn2+ concentration required for normal vegetative growth is ∼10-7 M, 10-6-10-3 M for production of secondary metabolites, and 10-4-10-3 M for cultural longevity (64). On the basis of these studies, one could conclude that Mn2+ may indeed be the only metal ion for which MntR has sufficient affinity and is present in suitable free concentrations within the cell, which would bind to and regulate MntR during normal cellular homeostasis. Clearly, the full metallome and concentrations of free metal ions in B. subtilis must be determined to validate this hypothesis.
CONCLUSIONS
We have determined the affinity of Mag-fura-2 for several transition metal ions, which fall in the nanomolar to micromolar range, making this dye useful for competition experiments with the metalloregulatory protein MntR. Evaluation of metal binding by MntR shows this protein belongs to a class of metalloregulatory proteins that possess a weak affinity for its cognate metal ion, while still eliciting tight DNA binding upon complete metal loading. EPR studies have provided evidence for a weakly coupled, dinuclear metal center consistent with the revised crystallographic report of MntR (15). While additional structural studies will be necessary for elucidating the complete mechanisms of metal activation, the results presented here unambiguously show that the selective metal ion response of MntR does not correlate with metal-binding affinities. Furthermore, the high binding affinity of MntR for Cd2+ versus Mn2+ suggests a dual role for this repressor in vivo—to maintain essential levels of Mn2+, while excluding toxic Cd2+ with exquisite sensitivity.
ACKNOWLEDGMENT
We thank Dr. Emmanuel Guedon and Prof. John D. Helmann (Cornell University) for providing the protein vectors, Prof. Arthur Glasfeld (Reed College) for many helpful discussions and disclosure of unpublished data, and Dr. Annette Deyhle (Scripps Institute of Oceanography) for access and assistance with the ICP-OES. We thank Dr. Petr Kuzmic for extensive discussions on the use of DYNAFIT, suggested improvements to experimental design, and assistance with the data fitting analysis.
Footnotes
This work was supported by the University of California, San Diego, the Hellman Family fund, a Cottrell Scholar Award from the Research Corporation (S.M.C.), and the National Institutes of Health GM077387 (M.P.H.).
- ANS
- 8-anilino-1-naphthalenesulfonic acid
- AntR
- anthracis repressor
- DtxR
- diphtheria toxin repressor
- EPR
- electron paramagnetic resonance
- ICP-OES
- inductively coupled plasma optical emission spectroscopy
- Mag-fura-2
- 2-[2-(5-carboxy) oxazole]-5-hydroxy-6-aminobenzofuran-N,N,O-triacetic acid
- MntR
- manganese transport regulator
SUPPORTING INFORMATION AVAILABLE
Figures S1-S8 and Dynafit scripts. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Moore CM, Helmann JD. Metal ion homeostasis in Bacillus subtilis. Curr. Opin. Microbiol. 2005;8:188–195. doi: 10.1016/j.mib.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Outten FW, Outten CE, O’Halloran TV. Metalloregulatory systems at the interface between bacterial metal homeostasis and resistance. In: Storz G, Hengge-Aronis R, editors. Bacterial Stress Responses. ASM Press; Washington, DC: 2000. pp. 145–157. [Google Scholar]
- 3.O’Halloran TV. Transition metals in control of gene expression. Science. 1993;261:715–725. doi: 10.1126/science.8342038. [DOI] [PubMed] [Google Scholar]
- 4.Rangachari V, Marin V, Bienkiewicz EA, Semavina M, Guerrero L, Love JF, Murphy JR, Logan TM. Sequence of ligand binding and structure change in the diphtheria toxin repressor upon activation by divalent transition metals. Biochemistry. 2005;44:5672–5682. doi: 10.1021/bi047825w. [DOI] [PubMed] [Google Scholar]
- 5.Bloom SL, Zamble DB. Metal-selective DNA-binding response of Escherichia coli NikR. Biochemistry. 2004;43:10029–10038. doi: 10.1021/bi049404k. [DOI] [PubMed] [Google Scholar]
- 6.Wang SC, Dias AV, Bloom SL, Zamble DB. Selectivity of metal binding and metal-induced stability of Escherichia coli NikR. Biochemistry. 2004;43:10018–10028. doi: 10.1021/bi049405c. [DOI] [PubMed] [Google Scholar]
- 7.Mills SA, Marletta MA. Metal binding characteristics and role of iron oxidation in the ferric uptake regulator from Escherichia coli. Biochemistry. 2005;44:13553–13559. doi: 10.1021/bi0507579. [DOI] [PubMed] [Google Scholar]
- 8.Liu T, Golden JW, Giedroc DP. A zinc(II)/lead-(II)/cadmium(II)-inducible operon from the cyanobacterium Anabaena is regulated by AztR, an R3N ArsR/SmtB metalloregulator. Biochemistry. 2005;44:8673–8683. doi: 10.1021/bi050450+. [DOI] [PubMed] [Google Scholar]
- 9.Chou CJ, Wisedchaisri G, Monfeli RR, Oram DM, Holmes RK, Hol WGJ, Beeson C. Functional studies of the Mycobacterium tuberculosis iron-dependent regulator. J. Biol. Chem. 2004;279:53554–53561. doi: 10.1074/jbc.M407385200. [DOI] [PubMed] [Google Scholar]
- 10.D’Aquino JA, Tetenbaum-Novatt J, White A, Berkovitch F, Ringe D. Mechanism of metal ion activation of the diphtheria toxin repressor DtxR. Proc. Natl. Acad. Sci. U.S.A. 2005;102:18408–18413. doi: 10.1073/pnas.0500908102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Que Q, Helmann JD. Manganese homeostasis in Bacillus subtilis is regulated by MntR, a bifunctional regulator related to the diphtheria toxin repressor family of proteins. Mol. Microbiol. 2000;35:1454–1468. doi: 10.1046/j.1365-2958.2000.01811.x. [DOI] [PubMed] [Google Scholar]
- 12.Lieser SA, Davis TC, Helmann JD, Cohen SM. DNA-binding and oligomerization studies of the manganese(II) metalloregulatory protein MntR from Bacillus subtilis. Biochemistry. 2003;42:12634–12642. doi: 10.1021/bi0350248. [DOI] [PubMed] [Google Scholar]
- 13.Glasfeld A, Guedon E, Helmann JD, Brennan RG. Structure of the manganese-bound manganese transport regulator of Bacillus subtilis. Nat. Struct. Biol. 2003;10:652–657. doi: 10.1038/nsb951. [DOI] [PubMed] [Google Scholar]
- 14.Pohl E, Holmes RK, Hol WGJ. Motion of the DNA-binding domain with respect to the core of the diphtheria toxin repressor (DtxR) revealed in the crystal structures of apo- and holo-DtxR. J. Biol. Chem. 1998;273:22420–22427. doi: 10.1074/jbc.273.35.22420. [DOI] [PubMed] [Google Scholar]
- 15.Kliegman JI, Griner SL, Helmann JD, Brennan RG, Glasfeld A. Structural basis for the metal-selective activation of the manganese transport regulator of Bacillus subtilis. Biochemistry. 2006;45:3493–3505. doi: 10.1021/bi0524215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyd J, Oza MN, Murphy JR. Molecular cloning and DNA sequence analysis of a diphtheria tox iron-dependent regulatory element (dtxR) from Corynebacterium diphtheriae. Proc. Natl. Acad. Sci. U.S.A. 1990;87:5968–5972. doi: 10.1073/pnas.87.15.5968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Golynskiy MV, Davis TC, Helmann JD, Cohen SM. Metal-induced structural organization and stabilization of the metalloregulatory protein MntR. Biochemistry. 2005;44:3380–3389. doi: 10.1021/bi0480741. [DOI] [PubMed] [Google Scholar]
- 18.Sen KI, Sienkiewicz A, Love JF, vanderSpek JC, Fajer PG, Logan TM. Mn(II) binding by the anthracis repressor from Bacillus anthracis. Biochemistry. 2006;45:4295–4303. doi: 10.1021/bi052288g. [DOI] [PubMed] [Google Scholar]
- 19.Connors KA. Binding Constants: The Measurement of Molecular Complex Stability. John Wiley & Sons, Inc.; New York: 1987. [Google Scholar]
- 20.Kwan C-Y, Putney JW., Jr. Uptake and intracellular sequestration of divalent cations in resting and methacholine-stimulated mouse lacrimal acinar cells. J. Biol. Chem. 1990;265:678–684. [PubMed] [Google Scholar]
- 21.Raju B, Murphy E, Levy LA, Hall RD, London RE. A fluorescent indicator for measuring cytosolic free magnesium. Am. J. Physiol. 1989;256:C540–C548. doi: 10.1152/ajpcell.1989.256.3.C540. [DOI] [PubMed] [Google Scholar]
- 22.Martell AE, Smith RM. Critical Stability Constants. Vol. 4. Plenum Press; New York: 1974. [Google Scholar]
- 23.Lattanzio FA, Jr., Bartschat DK. The effect of pH on rate constants, ion selectivity and thermodynamic properties of fluorescent calcium and magnesium indicators. Biochem. Biophys. Res. Comm. 1991;177:184–191. doi: 10.1016/0006-291x(91)91966-g. [DOI] [PubMed] [Google Scholar]
- 24.Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- 25.Kuzmic P. A generalized numerical approach to rapidequilibrium enzyme kinetics: Application to 17 beta-HSD. Mol. Cell. Endocrinol. 2006;248:172–181. doi: 10.1016/j.mce.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 26.Mely Y, Cornille F, Fournie-Zaluski M-C, Darlix JL, Roques BP, Gerard D. Investigation of zinc-binding affinities of Moloney murine leukemia-virus nucleocapsid protein and its related zinc finger and modified peptides. Biopolymers. 1991;31:899–906. doi: 10.1002/bip.360310709. [DOI] [PubMed] [Google Scholar]
- 27.Walkup GK, Imperiali B. Fluorescent chemosensors for divalent zinc based on zinc finger domains. Enhanced oxidative stability, metal binding affinity, and structural and functional characterization. J. Am. Chem. Soc. 1997;119:3443–3450. [Google Scholar]
- 28.Golombek AP, Hendrich MP. Quantitative analysis of dinuclear manganese(II) EPR spectra. J. Magn. Reson. 2003;165:33–48. doi: 10.1016/j.jmr.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Henry ER, Hofrichter J. Singular value decomposition: application to analysis of experimental-data. Methods Enzymol. 1992;210:129–192. [Google Scholar]
- 30.Pierce BS, Elgren TE, Hendrich MP. Mechanistic implications for the formation of the diiron cluster in ribonucleotide reductase provided by quantitative EPR spectroscopy. J. Am. Chem. Soc. 2003;125:8748–8759. doi: 10.1021/ja021290h. [DOI] [PubMed] [Google Scholar]
- 31.Press WH, Teukolsky SA, Vetterling WT, Flannery BP. Numerical Recipes in FORTRAN: The Art of Scientific Computing. 2nd ed. Cambridge University Press; Cambridge: 1992. [Google Scholar]
- 32.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 33.Hinkle PM, Shanshala ED, II, Nelson EJ. Measurement of intracellular cadmium with fluorescent dyes. J. Biol. Chem. 1992;267:25553–25559. [PubMed] [Google Scholar]
- 34.Atar D, Backx PH, Appel MM, Gao WD, Marban E. Excitation-transcription coupling mediated by zinc influx through voltage-dependent calcium channels. J. Biol. Chem. 1995;270:2473–2477. doi: 10.1074/jbc.270.6.2473. [DOI] [PubMed] [Google Scholar]
- 35.Simons TJB. Measurement of free Zn2+ ion concentration with the fluorescent probe mag-fura-2 (furaptra) J. Biochem. Biophys. Meth. 1993;27:25–37. doi: 10.1016/0165-022x(93)90065-v. [DOI] [PubMed] [Google Scholar]
- 36.Fahrni CJ, O’Halloran TV. Aqueous coordination chemistry of quinoline-based fluorescence probes for the biological chemistry of zinc. J. Am. Chem. Soc. 1999;121:11448–11458. [Google Scholar]
- 37.VanZile ML, Cosper NJ, Scott RA, Giedroc DP. The zinc metalloregulatory protein Synechococcus PCC7942 SmtB binds a single zinc ion per monomer with high affinity in a tetrahedral coordination geometry. Biochemistry. 2000;39:11818–11829. doi: 10.1021/bi001140o. [DOI] [PubMed] [Google Scholar]
- 38.Reed GH, Cohn M. Electron paramagnetic resonance spectra of manganese(II)-protein complexes. J. Biol. Chem. 1970;245:662–667. [PubMed] [Google Scholar]
- 39.Reiter NJ, White DJ, Rusnak F. Inhibition of bacteriophage lambda protein phosphatase by organic and oxoanion inhibitors. Biochemistry. 2002;41:1051–1059. doi: 10.1021/bi011577b. [DOI] [PubMed] [Google Scholar]
- 40.Busenlehner LS, Pennella MA, Giedroc DP. The SmtB/ArsR family of metalloregulatory transcriptional repressors: structural insights into prokaryotic metal resistance. FEMS Microbiol. Rev. 2003;27:131–143. doi: 10.1016/S0168-6445(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 41.Sigel H, McCormick DB. On the discriminating behavior of metal ions and ligands with regard to their biological significance. Acc. Chem. Res. 1970;3:201–208. [Google Scholar]
- 42.Caneschi A, Ferraro F, Gatteschi D, Melandri MC, Rey P, Sessoli R. Synthesis, structure and magneticproperties of a dinuclear manganese(II) complex with one μ-Aqua and 2 μ-carboxylato bridges. Angew. Chem. Int. Ed. 1989;28:1365–1367. [Google Scholar]
- 43.Yu S-B, Lippard SJ, Shweky I, Bino A. Dinuclear manganese(II) complexes with water and carboxylate bridges. Inorg. Chem. 1992;31:3502–3504. [Google Scholar]
- 44.Wieghardt K, Bossek U, Nuber B, Weiss J, Bonvoisin J, Corbella M, Vitols SE, Girerd JJ. Synthesis, crystal structures, reactivity, and magnetochemistry of a series of binuclear complexes of manganese(II), -(III), and -(IV) of biological relevance. The crystal structure of [L′MnIV(u-O) MnIV 3 L′]-(PF6)2, H2O containing an unprecedented short Mn···Mn distance of 2.296 Å. J. Am. Chem. Soc. 1988;110:7398–7411. [Google Scholar]
- 45.Cano J, DeMunno G, Sanz J, Ruiz R, Lloret F, Faus J, Julve M. Significant antiferromagnetic coupling in a terephthalate (ta)-bridged manganese(II) compound: preparation, crystal structure and magnetic properties of the chain [Mn2bipy4(ta)]-[ClO4]2 (bipy)2,2′-bipyridine) J. Chem. Soc. Dalton. 1994;3465:3469. [Google Scholar]
- 46.Nepveu F, Gaultier N, Korber N, Jaud J, Castan P. New polynuclear manganese(II) complexes with orotic acid and some of its derivatives: crystal structures, spectroscopic and magnetic studies. J. Chem. Soc. Dalton. 1995;4005:4013. [Google Scholar]
- 47.Oshio H, Ino E, Mogi I, Ito T. A weak antiferromagnetic interaction between Mn2+ centers through a TCNQ column: Crystal structures and magnetic properties of [MnII(tpa)(TCNQ)(CH3OH)](TCNQ)2, CH3CN, [MnII(tpa)(u-O2-CCH3)]2(TCNQ)2, 2CH3CN, and [MnII(tpa)(NCS)2], CH3CN (tpa) tris(2-pyridylmethyl)amine) Inorg. Chem. 1993;32:5697–5703. [Google Scholar]
- 48.Chen X-M, Tong Y-X, Xu Z-T, Mak TCW. Synthesis, structure and magnetic properties of a singly carboxylate-bridged dinuclear manganese(II) complex. J. Chem. Soc. Dalton. 1995;4001:4004. [Google Scholar]
- 49.Cavet JS, Meng W, Pennella MA, Appelhoff RJ, Giedroc DP, Robinson NJ. A nickel-cobalt-sensing ArsR-SmtB family repressor. J. Biol. Chem. 2002;277:38441–38448. doi: 10.1074/jbc.M207677200. [DOI] [PubMed] [Google Scholar]
- 50.Tottey S, Harvie DR, Robinson NJ. Understanding how cells allocate metals using metal sensors and metallochaperones. Acc. Chem. Res. 2005;38:775–783. doi: 10.1021/ar0300118. [DOI] [PubMed] [Google Scholar]
- 51.Pennella MA, Giedroc DP. Structural determinants of metal selectivity in prokaryotic metal-responsive transcriptional regulators. BioMetals. 2005;18:413–428. doi: 10.1007/s10534-005-3716-8. [DOI] [PubMed] [Google Scholar]
- 52.Brown NL, Stoyanov JV, Kidd SP, Hobman JL. The MerR family of transcriptional regulators. FEMS Microbiol. Rev. 2003;27:145–163. doi: 10.1016/S0168-6445(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 53.Busenlehner LS, Weng T-C, Penner-Hahn JE, Giedroc DP. Elucidation of primary (R3N) and vestigial (R5) heavy metal-binding sites in Staphylococcus aureus pI258 CadC: evolutionary implications for metal ion selectivity of ArsR/SmtB metal sensor proteins. J. Mol. Biol. 2002;319:685–701. doi: 10.1016/S0022-2836(02)00299-1. [DOI] [PubMed] [Google Scholar]
- 54.Changela A, Chen K, Xue Y, Holschen J, Outten CE, O’Halloran TV, Mondragón A. Molecular basis of metal-ion selectivity and zeptomolar sensitivity by CueR. Science. 2003;301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- 55.Eicken C, Pennella MA, Chen X, Koshlap KM, VanZile ML, Sacchettini JC, Giedroc DP. A metal-ligandmediated intersubunit allosteric switch in related SmtB/ArsR zinc sensor proteins. J. Mol. Biol. 2003;333:683–695. doi: 10.1016/j.jmb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 56.Outten CE, O’Halloran TV. Femtomolar Sensitivity of Metalloregulatory Proteins Controlling Zinc Homeostasis. Science. 2001;292:2488–2492. doi: 10.1126/science.1060331. [DOI] [PubMed] [Google Scholar]
- 57.VanZile ML, Chen X, Giedroc DP. Allosteric negative regulation of smt O/P binding of the zinc sensor, SmtB, by metal ions: a coupled equilibrium analysis. Biochemistry. 2002;41:9776–9786. doi: 10.1021/bi020178t. [DOI] [PubMed] [Google Scholar]
- 58.Hantke K. Iron atnd metal regulation in bacteria. Curr. Opin. Microbiol. 2001;4:172–177. doi: 10.1016/s1369-5274(00)00184-3. [DOI] [PubMed] [Google Scholar]
- 59.Kehres DG, Maguire ME. Emerging themes in manganese transport, biochemistry and pathogenesis in bacteria. FEMS Microbiol. Rev. 2003;27:263–290. doi: 10.1016/S0168-6445(03)00052-4. [DOI] [PubMed] [Google Scholar]
- 60.Finney LA, O’Halloran TV. Transition metal speciation in the cell: insights from the chemistry of metal ion receptors. Science. 2003;300:931–936. doi: 10.1126/science.1085049. [DOI] [PubMed] [Google Scholar]
- 61.Keyer K, Imlay JA. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. U.S.A. 1996;93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guedon E, Helmann JD. Origins of metal ion selectivity in the DtxR/MntR family of metalloregulators. Mol. Microbiol. 2003;48:495–506. doi: 10.1046/j.1365-2958.2003.03445.x. [DOI] [PubMed] [Google Scholar]
- 63.Nies DH. Microbial heavy metal resistance. Appl. Microbiol. Biotechnol. 1999;51:730–750. doi: 10.1007/s002530051457. [DOI] [PubMed] [Google Scholar]
- 64.Weinberg ED. Manganese requirement for sporulation and other secondary biosynthetic processes of Bacillus. Appl. Microbiol. 1964;12:436–441. doi: 10.1128/am.12.5.436-441.1964. BI0607406. [DOI] [PMC free article] [PubMed] [Google Scholar]