

Abstract
A homogeneous, sensitive, cellular bioluminescent high throughput screen was developed for inhibitors of gyrase and other DNA damaging agents in Pseudomonas aeruginosa. The screen is based on a Photorhabdus luminescens luciferase operon transcriptional fusion to a promoter that responds to DNA damage caused by reduced gyrase levels and fluoroquinolone inhibition. This reporter strain is sensitive to levels of ciprofloxacin as low as ¼-MIC with Z’ scores above 0.5, indicating the assay is suitable for high-throughput screening. This screen combines the benefits of a whole cell assay with a sensitivity and target specificity superior to those of traditional cell-based screens for inhibitors of viability or growth. In duplicate pilot screens of 2,000 known bioactive compounds, 13 compounds generated reproducible signals ≥50% of that of the control (ciprofloxacin at ¼-MIC) using bioluminescence readings after 7h of incubation. Ten are fluoroquinolones known to cause accumulation of cleaved DNA-enzyme complexes in bacterial cells; the other three are known to create DNA adducts. Therefore, all 13 hits inhibit DNA synthesis, but by a variety of different DNA damaging mechanisms. This convenient, inexpensive screen will be useful for rapidly identifying DNA gyrase inhibitors and other DNA damaging agents, which may lead to potent new anti-bacterials.
Keywords: P. aeruginosa, gyrase, high throughput screen, luciferase
INTRODUCTION
Pseudomonas aeruginosa is an opportunistic pathogen in animals and humans. It is a common and extremely virulent cause of serious infections in immune-compromised/suppressed patients (e.g., HIV and cancer), cystic fibrosis patients, and those on mechanical ventilation or with burn wounds.1 Frequent antibiotic resistance and the highly virulent nature of P. aeruginosa make it deadlier than many other bacterial pathogens. P. aeruginosa exhibits intrinsic drug resistance due to the combined effects of a poorly permeable outer membrane, several multi-drug efflux pumps, and a chromosomally encoded cephalosporinase.1 New chemical classes of antibiotics are critical for continued effective therapy against P. aeruginosa, because such drugs are less likely to be subject to existing resistance mechanisms. In order to overcome these permeability and efflux obstacles and permit screening of a broad array of targets, we have developed P. aeruginosa cell-based assays for identifying inhibitors by using transcriptional reporters.
Transcriptional fusion of reporters to gene promoters, which are up-regulated in response to target depletion, whether by antibiotic inhibition or conditional under-expression, serves to transduce that depletion into a reporter signal. Several reporter assays of this type have been described in B. subtilis based on the response of cells to antibiotic treatments and using firefly luciferase as a reporter.2 We have extended this approach to a less permeable species, P. aeruginosa, and eliminated the need for luciferase substrate addition while achieving excellent sensitivity and highly positive Z’-scores. The resulting screen is simple, inexpensive, and homogeneous. In vivo reporter screens of this type offer substantial benefits, including (a) selection of permeable compounds, (b) ability to monitor multiple metabolic steps simultaneously (e.g., pathway screens), (c) sensitivity (e.g., superior to assays that simply detect growth inhibition), and (d) applicability to biochemically intractable targets (e.g., those with no known function or functions that are difficult to assay).
As a proof of concept, we focused on the known fluoroquinolone antibacterial target, DNA gyrase. This well-validated, druggable target is the essential A2B2 protein product of the gyrA and gyrB genes, which maintains the negative supercoiling of DNA during replication by removing positive supercoils in advance of the replication fork.3 The fluoroquinolone family of antibiotics has been developed through optimization of nalidixic acid, a gyrase inhibitor that was discovered serendipitously.3 A high throughput screen for inhibitors of gyrase has not been feasible because of the difficulty in measuring the substrates and products of the reaction, supercoiled and relaxed DNA, respectively. Previous attempts to screen for inhibitors have relied upon detecting inhibitors of the ATPase activity of the N-terminal fragment of GyrB. This approach has limited utility and runs the risk of identifying toxic compounds with broad anti-ATPase activity. In order to develop a reporter screen for gyrase inhibitors, we identified transcriptional units in P. aeruginosa that respond by up-regulation to both reduced expression of the gyrA gene and to inhibition of gyrase by the fluoroquinolone ciprofloxacin. Next, we fused responsive promoter regions to the Photorhabdus luminescens luxCDABE operon and integrated them into the P. aeruginosa chromosome. The most responsive promoter element is known to be up-regulated through the recA pathway by a variety of DNA damaging agents, including fluoroquinolones and compounds which create DNA adducts in vivo. We used this promoter to build and characterize a simple, sensitive, inexpensive, high throughput screen for compounds with anti-P. aeruginosa activity based on DNA damage.
MATERIALS AND METHODS
Strains, plasmids, and growth media
P. aeruginosa PAO-LAC, a version of strain PAO1 in which lacIq has been integrated into the ΦCTX site in the chromosome,4 E. coli TOP10 (Invitrogen®), E. coli DB3.1 (Gateway® host, Invitrogen®), and E. coli S17-1 (ATCC 47055), were used as hosts for molecular cloning. Luria-Bertani (LB) medium (liquid and agar) was purchased from Difco. LBIG is LB containing 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and 10 μg/ml gentamicin. Opaque, white, flat-bottom, 96-well microplates (Nunc Cat No. 236108; VWR International) were covered with gas permeable sealant (AeraSeal BS-25; Phenix Research Products, Candler, NC) for reporter screens.
PCR and Primers
Synthetic oligonucleotide primers were designed using web-based PRIMER3 (Whitehead Institute) and purchased from Operon, Inc. (see Table 1). Primers were used at 10 μM in PCR amplifications with Failsafe polymerase (Epicentre®), Buffer G (Epicentre®), and 4% DMSO for P. aeruginosa chromosomal DNA templates.
TABLE 1.
Oligonucleotide Primers Used in this Study
| No. | Primer Name | Primer Sequence |
|---|---|---|
| 1 | PaeGyrAupF+GWL | TACAAAAAAGCAGGCTtctggagcgaatgaaagagg |
| 2 | gyrA UpR+Tc | TCCTGCGTTATCCCCTGATTCTGTGGATAActtcgatattgaccgggaga |
| 3 | gyrA DwnF+Tc | GCTAACGGATTCACCACTCCAAGAATTGGAcggaaggcaacgaagagtaa |
| 4 | PaeGyrAdwnR+GWR | TACAAGAAAGCTGGGTctggcatcgatggtcttgta |
| 5 | PaeGyrA-outF | aggaggtctggctcgaacac |
| 6 | PaeGyrA-outR | ggatcgggttggtgtagaag |
| 7 | PaeGyrA-F+SD+GWL | TACAAAAAAGCAGGCTaggaaacagctatgggcgaactggccaaagaaat |
| 8 | PaeGyrA-R+GWR | TACAAGAAAGCTGGGTccgagccttactcttcgttg |
| 9 | pPS880Tet-FRT-F | ttatccacagaatcaggggataacgcagga |
| 10 | pPS880Tet-FRT-R | tccaattcttggagtggtgaatccgttagc |
| 11 | GW-Universal-attB1 | ggggacaagtttgtacaaaaaagcaggct |
| 12 | GW-Universal-attB2 | ggggaccactttgtacaagaaagctgggt |
| 13 | pUCP24-UP | TCCCCCGGGGTGTGAAATTGTTATCCGCTCACAATTCCACAC |
| 14 | pUCP24-DN | TCCCCCGGGAACGTCGTGACTGGGAAAACC |
| 15 | PlacO-F+GWL | TACAAAAAAGCAGGCTTTAATTAAtcgaGGCGCGCCatcgCACtatGTGattaatgcagctggcacgac |
| 16 | PlacO-R+Dra4CTX | tatttgccatccatttaatgCACcgcGTGtgtgaaattgttatccgctca |
| 17 | Lux-F5 | cattaaatggatggcaaatATGa |
| 18 | Lux-R2+GWR | TACAAGAAAGCTGGGTcgcaagcattccacttacaa |
| 19 | PA0612-F+Dra | catcgctccCACtatGTGggcaatctacagaccgatgg |
| 20 | PA0612-R+Dra | catcgctccCACcgcGTGgaaagcctccctggcgt |
| 21 | PA0614-F+Dra | catcgctccCACtatGTGcTGAgttcctggaccggata |
| 22 | PA0614-R+Dra | catcgctccCACcgcGTGcctggggacgcaccttta |
| 23 | PA0617-F+Dra | catcgctccCACtatGTGccacacccatTGAacatacg |
| 24 | PA0617-R+Dra | catcgctccCACcgcGTGgctccggcagagacagg |
Complemented deletion of gyrA
A deletion of codons 15-918 of gyrA marked with a tetracycline resistance (TcR) marker was constructed in PAO-LAC by allelic exchange.5-7 Briefly, N-terminal (1,020 bp) and C-terminal (951 bp) regions flanking gyrA were amplified using the primer pairs 1+2 and 3+4, respectively (Table 1, Fig. 1A). The amplified regions were joined to the TcR element gene from plasmid pALTER-1 (Promega) (amplified with primers 9+10, Table 1) in a three-fragment splicing by overlap extension (SOE) PCR,8 using the outside primers 1+4 (Table 1). This was followed by several amplification cycles with GW-Universal-attB1/GW-Universal-attB2 to complete the Gateway® sites for cloning. Products were gel-purified and cloned using the Gateway system into pEX18GWAp, a Gateway®-adapted pEX18Ap (GenBank AF004910), provided by Drs. X. Liang and S. Lory (Harvard Medical School). The resulting construct was conjugated from E. coli S17-1 into P. aeruginosa PAO-LAC (Fig. 1B). A merodiploid resulting from a single cross-over integration was transformed to GmR by electroporation with mini-Tn7-GW-lux carrying the promoter region from P. aeruginosa gene PA0614 (see below for construction) and helper plasmid pTNS29 (Fig.1C). The GmR element was removed by electroporation to SpR with pFLP2-SpR (AF048702, modifed by replacement of the XbaI GmR fragment with SpR) followed by screening for GmS colonies (Fig. 1D). Cells lost pFLP2-SpR rapidly upon growth in the absence of spectinomycin selection. Finally, a wild-type copy of the P. aeruginosa gyrA gene was amplified with PCR primers 7+8 (Table 1) and inserted into pUCP24GW, a Gateway® (Invitrogen) adapted version of pUCP24 (GenBank U07167) which was constructed as follows. The multiple cloning site and a portion of the lacZ-alpha gene were deleted using an outward PCR reaction with primers pUCP24-UP and pUCP24-DN. A Gateway® Vector Conversion System (Cat. No. 11828-029, Invitrogen, Inc.) cassette was ligated with blunt ended PCR product and used to transform E. coli DB3.1 cells to GmR. Complemented gyrA deletions were isolated by selection for growth on sucrose in the presence of 1 mM IPTG and screened for a TcR ApS GmR phenotype (Fig. 1E). Deletions were confirmed by PCR with primers outside the region carried on pEX18GWAp (PaeGyrA-outF/PaeGyrA-outR). Deletions could only be isolated in the presence of the complementing gyrA copy on pUCP24GW-gyrA, and deletions failed to grow in the absence of IPTG, consistent with the essentiality of the gyrA gene.
Figure 1.
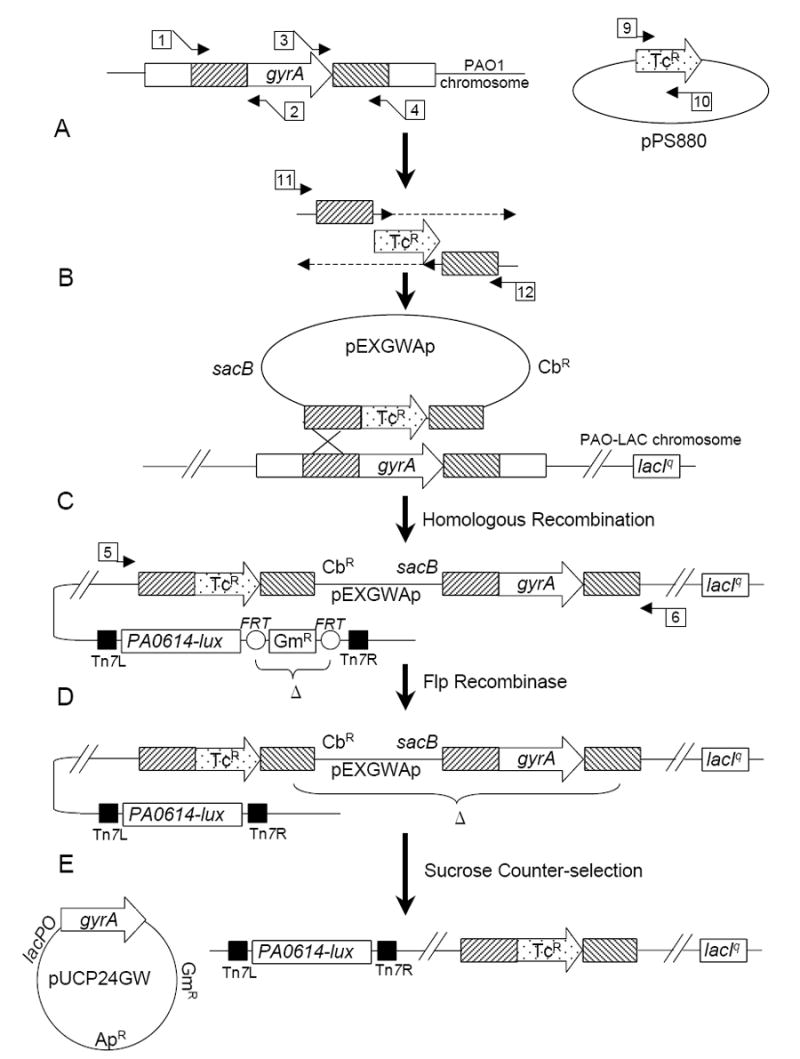
Construction of reporter strain consisting of a P. aeruginosa gyrA deletion complemented by a lac-regulated copy of gyrA on plasmid pUCP24GW and carrying a chromosomally inserted mini-Tn7 element expressing P. luminescens luxCDABE (lux) from the PA0614 gene promoter. A. SOE PCR was used to replace gyrA with a TcR element between ~1 kb of flanking homology (primers numbered as in Table 1); B. the PCR product was cloned into pEXGWAp using Gateway® technology and conjugated into P. aeruginosa PAO-LAC, resulting in integration of the pEXGWAp deletion construct into the P. aeruginosa chromosome (confirmed by PCR with primers outside of the region cloned); C. A mini-Tn7-GW-Gm construct containing the PA0614 promoter fused to luxCDABE was co-electroporated into the merodiploid together with the helper transposase plasmid pTNS2 resulting in integration of the mini-Tn7 element at the attTn7 locus; D. Plasmid pFLP2-SpR was introduced by electroporation to eliminate the GmR marker, then pFLP2-SpR was lost by growth without selection; E. A pUCP24GW construct carrying a lac-regulated complementing copy of gyrA was introduced by electroporation, and a deletion of the chromosomal copy of gyrA was selected on sucrose-containing medium and confirmed by PCR.
Construction and use of mini-Tn7-GW-luxCDABE
We modified the P. aeruginosa site-specific integrating vector, mini-Tn7-GW-Gm (GenBank AY737004)9 to create a vector for directionally cloning luciferase transcriptional fusions to promoter fragments generated by PCR. We generated a 2-way SOE PCR product8 consisting of the lac promoter and operator region bounded on both sides by DraIII restriction endonuclease sites and fused to the P. luminescens luxCDABE operon. Primers 15+16 and 17+18 (Table 1) were used to amplify and join fragments from the plasmid pUC-lux.10, 11 The 3-nt ambiguity in the DraIII recognition site was used for directional cloning of promoter fragments in place of the lacOP stuffer fragment. The SOE-PCR product was cloned into mini-Tn7 -GW-Gm by Gateway®, and a suitably luminescent clone was selected by its strong emission of light on medium containing 1 mM IPTG. Promoter regions to be inserted into the vector were amplified by PCR from genomic DNA by using primers containing two different DraIII sites as tails (e.g., PA0612-F+Dra/PA0612-R+Dra; PA0614-F+Dra/PA0614-R+Dra; and PA0617-F+Dra/ PA0617-R+Dra; see Table 1). DraIII digestion of the gel-purified PCR products and ligation into DraIII-cut mini-Tn7-GW-GM-LUX resulted in directionally cloned promoter regions upstream of luxCDABE. Resulting constructs were integrated into PAO1 or into the complemented gyrA deletion strain by co-electroporation with helper plasmid pTNS2.9 The GmR marker could be eliminated readily by electroporation with pFLP2-SpR, which was lost after a few generations of growth without spectinomycin selection. This maneuver allowed the re-use of the GmR marker (see above).
Measurement of bioluminescence of reporters
Strains were grown overnight from frozen stocks at 37°C on LB agar containing 1 mM IPTG and 10 μg/ml gentamicin. Cells from the agar plate were used to inoculate liquid LB medium containing IPTG and gentamicin at an OD600 ~0.05. Cultures were grown for about two hours to an OD600 = 0.4, and then 200 μl of culture was added to each well of a 96-well microtiter dish containing compound (2 μl in DMSO) to initiate an assay or screen. Luminescence was measured at various times in kinetic mode or at a single time in endpoint mode in a Wallac Microbeta Trilux Luminometer or a Wallac Victor2V 1420 Multilabel HTS counter. In some cases, relative luminescence units (RLU) were normalized to cell number by using OD600. Inhibition was measured as luminescence relative to that of the positive control containing 0.5-fold MIC of ciprofloxacin (0.03 μg/ml) in the pilot screen and was calculated as follows: % of positive control = [RLU of sample – Average RLU of negative control) / (Average RLU of positive control – Average RLU of negative control)] × 100, with the negative control consisting of incubation with 1% DMSO but without compound addition. Z and Z’ scores were calculated as previously described12 based on positive and negative controls as described above. The MIC of ciprofloxacin for P. aeruginosa strain PAO1 was determined according to NCCLS recommendations13 and is consistent with that of previous reports.14, 15
RESULTS
Construction of the reporter strain
The principle of the type of whole-cell reporter assay described here is the coupling of the transcriptional regulatory response produced by the depletion of an antibacterial target to a suitable reporter. A detectable signal, in this case bioluminescence, is produced when the quantity of active target is reduced by inhibition. First, it is necessary to identify promoters that respond to depletion of the potential target or to treatment with a known antibacterial agent which affects that target. We chose gyrase as the target for establishing the proof of principle for this type of reporter screen because it is a well-validated anti-bacterial target with several known inhibitors, many of which are marketed as antibiotics.3 Recently, Brazas and Hancock compared the transcription profile of P. aeruginosa cells grown in the presence and absence of the fluoroquinolone ciprofloxacin.16 They demonstrated that several genes, including a cluster of genes encoding the R and F type pyocins17 and spanning a contiguous region of over 28 kb in the genome (genes PA0612 – PA0648), were up-regulated significantly in cells treated with 0.3× and 1.0× MIC of the antibiotic. We have confirmed this result by expression profiling with microarrays and with luciferase fusions, and we have also demonstrated that the pyocin-encoding region is among the loci up-regulated when gyrA gene expression is reduced in a deletion strain complemented by a regulated copy of gyrA. We tested the entire upstream region from each of three of the earliest genes in the cluster for the ability to drive luciferase production in a ciprofloxacin-regulated manner as follows. We fused 451nt, 353 nt, and 150 nt of predicted non-coding sequence upstream from PA0612, PA0614, and PA0617,18,19 respectively, to the P. luminescens luxCDABE operon in the site-specific integrating vector mini-Tn7-Gm-GW-LUX and integrated them into the chromosome of P. aeruginosa PAO-LAC (Fig. 1C). The resulting strains were grown without ciprofloxacin and with a range of three different concentrations of ciprofloxacin, and the ratio of luminescence +/- ciprofloxacin was measured. In this experiment an initial inoculum of 0.2 ml of cell suspension grown in LB to an OD600 of 0.4 was incubated at 37°C +/- ciprofloxacin for 330 min. As shown in Figure 2, the response of the PA0614 promoter region was at least 10-fold stronger than those of the other two regions. For example, at 0.03 μg/ml ciprofloxacin (0.5 × MIC) the increase in bioluminescence as compared to untreated cells was 1.9-fold, 11.2-fold, and 0.8-fold for PA0612, PA0614, and PA0617 promoter regions, respectively.
Figure 2.
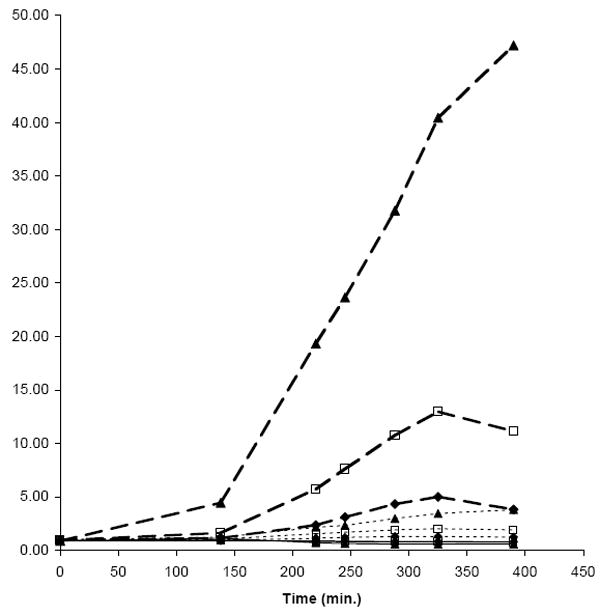
Ratio of luminescence during growth in the presence and absence of ciprofloxacin by P. aeruginosa strains carrying three different pyocin promoter regions fused to luxCDABE. Cells with appropriate mini-Tn7 luciferase fusions integrated into the chromosome were grown to OD600 = 0.2 in LB, and 0.2 ml were added to each well of a 96-well opaque white microplate. The ratios of relative luminescence units (RLU) from cells grown in the presence of ciprofloxacin at the following concentrations to that of cells grown in the absence of ciprofloxacin (24 wells each) is plotted, ♦, 0.015 μg/ml, □, 0.03 μg/ml; and ▲, 0.06 μg/ml (1× MIC). Promoter regions were the entire predicted untranslated sequence upstream from PA0612 (451 nt), dotted line; PA0614 (353 nt), dashed line; PA0617 (150 nt), solid line.
To measure the effect of GyrA depletion on bioluminescence, we inserted mini-Tn7-Gm-GW-LUX carrying the PA0614 promoter region into PAO-LAC and generated a deletion of the gyrA gene complemented by a lac repressor-regulated copy on the replicating extrachromosomal vector pUCP24GW (Fig. 1). The conditionally-complemented ΔgyrA strain failed to grow in LB + 0.001 mM IPTG, and growth was impaired at 0.0125 mM IPTG, confirming dependence of growth on expression of the complementing gene copy. Growth of cells in low IPTG (0.0125 mM) vs. high IPTG (1 mM) concentrations resulted in a decrease in gyrA mRNA levels by about 50% as judged by microarray analysis (data not shown) and an increase in bioluminescence of almost 2-fold (average of 10,764 +/-916 vs. 5,797 +/-713 RLU/OD600 in low vs. high IPTG-grown cells in two independent experiments).
In summary, as predicted by transcriptional profiling, the PA0614 upstream region behaved as a promoter that is up-regulated by ciprofloxacin and decreased GyrA levels. Since the ciprofloxacin-induced up-regulation of the PA0612 and PA0617 upstream regions was less significant than that of the PA0614 region, we used the PA0614 construct as the basis of a reporter screen. The resulting reporter assay strain, MBX-623, consists of the promoter region from PA0614 fused to P. luminescens luxCDABE operon and integrated at the Tn7 attachment site in the PAO-LAC chromosome. The strain carries a TcR-marked deletion of gyrA in the chromosome and is complemented by a lac repressor-regulated copy of gyrA on pUCP24GW carrying a GmR marker.
Optimization of the assay
We examined the sensitivity of the assay by testing two-fold dilutions of ciprofloxacin over a 128-fold range from 0.24 μg/ml (equivalent to 4-fold above the MIC) to 0.002 μg/ml (equivalent to 32-fold below the MIC). Strain MBX-623 produced sufficient luminescence to yield Z’ scores at certain time points above 0.5 for all ciprofloxacin concentrations from 0.24 ug/ml through 0.015 μg/ml (4-fold below the MIC), but not for ciprofloxacin concentrations below that. We have not examined ciprofloxacin concentrations greater than 4-fold above the MIC, but it is interesting that the luminescence signal increased for 7 h even when cells were incubated with ciprofloxacin up to 4-fold MIC (data not shown). The kinetics of luminescence from reporter strain MBX-623 incubated with three concentrations of ciprofloxacin (0.015, 0.03, or 0.06 μg/ml, equivalent to 0.25, 0.5, and 1-fold MIC, respectively), and in the absence of ciprofloxacin are shown in Figure 3A. Cells were grown in LBIG to a cell density of OD600 ~0.4, added to microtiter wells containing ciprofloxacin, and incubated for the time indicated. Increased luminescence in response to ciprofloxacin treatment was apparent after about 3 h and peaked after about 7 h. Luminescence was directly proportional to the concentration of ciprofloxacin added and was significantly stimulated by as little as 0.25-fold MIC. Luminescence declined after 7 h, but remained elevated above the zero ciprofloxacin control after 23 h. We calculated the Z’ score, which is a measure of the variability and signal to background ratio, at several time points (Fig. 3B). A Z’ score > 0.5 is indicative of the suitability of a screen for HTS.12 Cells treated with 0.03 μg/ml and 0.06 μg/ml ciprofloxacin exhibited Z’ scores >0.5 from 350 min through the last measurement at 1,385 min (23 hr), indicative of an adequate window for detection of inhibitors. Cells treated with the lowest ciprofloxacin concentration (0.015 μg/ml) exhibited positive Z’ scores from 350 min, but the Z’ score failed to reach 0.5 until the 23 hr timepoint. Concentrations of DMSO up to 2% had no significant effect on luminescence signal intensity (data not shown). The assay was tested with initial cell densities ranging from OD600 = 0.25 to 0.50. All produced robust signals at 6-7 h, but lower initial cell densities yielded slightly higher luminescence readings.
Figure 3.
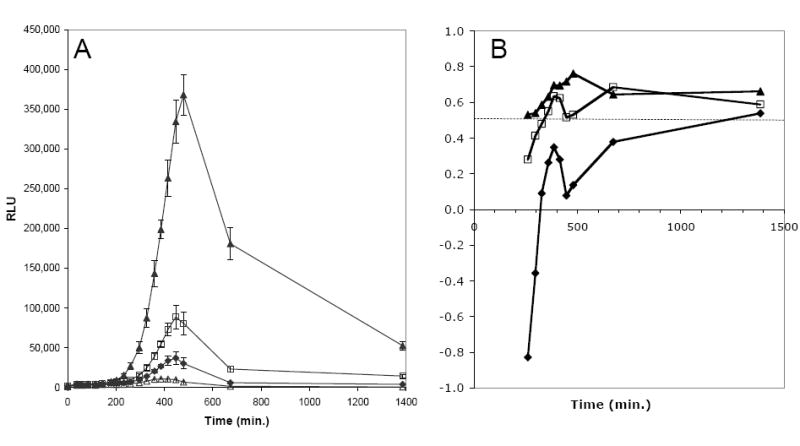
Luminescence and Z’ scores of strain MBX-623 vs. time and ciprofloxacin concentrations. Cells of MBX-623 were grown to OD600 = 0.4 in LBIG, and 0.2 ml were added to each well of a 96-well opaque white microplate. Ciprofloxacin was present at the following concentrations (24 wells each): Δ, 0 μg/ml; ♦, 0.015 μg/ml, □, 0.03 μg/ml; and ▲, 0.06 μg/ml (1× MIC). Relative luminescence units (RLU) were measured in each well in a Wallac Victor2V 1420 Multilabel HTS Counter at the times indicated. Error bars represent the standard deviation over 24 wells per time point. A. Kinetics of luminescence of reporter assay strain MBX-623. B. Z’ scores throughout the time course of the MBX-623 reporter assay of 3A. Z’ scores were calculated for the luminescence at each ciprofloxacin concentration vs. the no ciprofloxacin
In principle, the assay strain should be genetically stable in un-supplemented growth medium (LB) since all modules are integrated stably into the chromosome (promoter-luxCDABE at the Tn7 attachment site; stable deletion of P. aeruginosa gyrA gene) or on pUCP24GW, which is maintained by expression of the essential gyrA gene. IPTG is the only necessary supplement; however, gentamicin was also added in the pilot screen to ensure that the pUCP24GW-gyrA plasmid levels were maintained at a high level. Steady-glow bioluminescence from the P. luminescens luxCDABE operon products requires no reagent addition. Growth medium and minimal aeration (plates were covered with a gas permeable sealant but not shaken) are required for cells to produce ATP and light.
Application of the assay to known compounds -- a pilot screen
We screened 2,000 compounds from a biologically active and structurally diverse set of known drugs, experimental bioactives, and pure natural products (“Spectrum” library; Microsource Discovery, Inc.). To initiate the screen, 200 μl of a fresh culture of strain MBX-623 (OD600 ~ 0.40) in LBIG was added directly to opaque white flat bottom 96-well microtiter dishes containing 2 μl of library compounds (25 μM final concentration in 1% DMSO). Positive controls consisted of ciprofloxacin at 0.06 μg/ml (~1× MIC) and at 0.03 μg/ml (~0.5× MIC, the concentration used to calculate % of positive control values) final, and negative controls consisted of wells with DMSO but no compound addition. Plates were incubated at 37°C, and bioluminescence was read at 7 h in a Wallac Microbeta Trilux Luminometer plate reader. Percent bioluminescence of the positive control was calculated as described in Materials and Methods.
The pilot screen was performed in duplicate. The first screen was accomplished over a period of three days, testing 8-10 plates from the 25-plate library each day; the duplicate screen was done in a single day. Z’ scores, signal-to-background (S/B), and coefficient of variation (CV; standard deviation of positive control as a percentage of positive control) values were calculated from the positive (0.5× MIC ciprofloxacin, 0.03 μg/ml) and negative controls (no ciprofloxacin) for each day of the screen. These values, Z’ (S/B, %CV) for the three-day screen were 0.48 (6.8, 12%), 0.45 (5.2, 13%), and 0.30 (5.8, 16%), and for the single day screen were 0.45 (6.8, 12%). The luminescence for one of the replicates was read after overnight incubation for 23 h, yielding a Z’ score of 0.56 (8.7, 11%). Bulk reagent dispensing was done by hand with multi-channel pipets, suggesting that better Z’ scores could be obtained with more automated dispensing of cells.
Thirteen of the 2,000 tested compounds produced luminescence ≥50% of that of the positive control in both replicates at 7 h, and were defined as hits in the screen (Table 2). Results of the two duplicate screens were in agreement with regard to the compounds identified as hits. Variation between the two values averaged 18% for the hits, but was higher (57%) for those compounds that caused luminescence below the level defined as a hit. Ten of the hits were identified as fluoroquinolones and are thus “true” positives in the same drug class as ciprofloxacin. Four early generation quinolones, cinoxacin, pipemidic acid, piromidic acid, and oxolinic acid, and a 1st generation fluoroquinolone, flumequine, are present in the library but are not very potent vs. P. aeruginosa, exhibiting MICs ~25 μg/ml (~80 μM).20-22 These were not detected as hits, but all except cinoxacin and piromidic acid (the least potent with MIC’s >64 and 200 μg/ml, respectively) produced modest luminescence increases of 12-47% of the positive control in the cell-based reporter screen, consistent with potencies at the limit of detection of the screen. These compounds represent all of the quinolones and fluoroquinolones in the screening library. The three non-quinolone hits are known to affect DNA synthesis by forming DNA adducts. These include a nitrofuran, furazolidone,23 a bifunctional alkylating agent, mechlorethamine,24 and an anti-neoplastic agent trichlormethine.25 Clearly, all thirteen hits share a common mechanism of damaging DNA by forming adducts, either directly or catalyzed by gyrase/topoisomerase action. Thus, the false positive rate in this pilot screen was <0.05% (<1/2,000).
TABLE 2.
Compounds Producing Reproducible Significant Inhibition in the Pilot Screen
| Compound | Therapeutic Use | Class/Activity | % of Control, 7h (1st replicate) | % of Control, 7h (2nd replicate) | % of Control, 23h |
|---|---|---|---|---|---|
| Enoxacin | antibacterial | Fluoroquinolone | 361 | 215 | 429 |
| Pefloxacine mesylate | antibacterial | Fluoroquinolone | 314 | 336 | 429 |
| Lomefloxacin HCl | antibacterial | Fluoroquinolone | 331 | 246 | 153 |
| Gatifloxacin | antibacterial | Fluoroquinolone | 277 | 259 | 153 |
| Moxifloxacin HCl | antibacterial | Fluoroquinolone | 252 | 164 | 138 |
| Ofloxacin | antibacterial | Fluoroquinolone | 332 | 246 | 133 |
| Ciprofloxacin | antibacterial | Fluoroquinolone | 182 | 101 | 24 |
| Levofloxacin | antibacterial | Fluoroquinolone | 167 | 223 | 145 |
| Norfloxacin | antibacterial | Fluoroquinolone | 132 | 72 | 107 |
| Sarafloxacin HCl | antibacterial | Fluoroquinolone | 127 | 299 | 127 |
| Trichlormethine | antineoplastic | Alkylating agent | 141 | 214 | 6 |
| Furazolidone | antibacterial | Forms DNA adducts | 125 | 85 | 15 |
| Mechlorethamine | antineoplastic | Alkylating agent | 50 | 135 | 7 |
| Flumequine | antibacterial | Fluoroquinolone | 30 | 20 | -2 |
| Cinoxacin | antibacterial | Quinolone | 1 | -8 | 0 |
| Pipemidic acid | antibacterial | Quinolone | 15 | 47 | -1 |
| Oxolinic acid | antibacterial | Quinolone | 12 | 21 | -2 |
| Piromidic acid | antibacterial | Quinolone | 2 | -12 | 2 |
| Sildenafil | impotency therapy | Phosphodiesterase inhibitor | 22 | 3 | 60 |
| Carboplatin | antineoplastic | Alkylating agent | 2 | 19 | 97 |
| Polymyxin B sulfate | antibacterial | Membrane active cyclic lipopeptide | -19 | -33 | 80 |
The “Spectrum” library of known bioactives (2,000 compounds) was screened at 25 μM in duplicate. The whole cell reporter screen was incubated at 37°C, and readings were taken in a Wallac Microbeta Trilux Luminometer. The first duplicate was read after both 7 h and at 23 h; the second duplicate was only read after 7 h. A screening hit is defined as a compound causing ≥50% of the luminescence observed from the 0.5× MIC ciprofloxacin control, calculated as follows: % of control = [(LUXcpd – LUXNegCtl)/(LUXPosCtl – LUXNegCtl)] × 100, where PosCtl = 0.03 μg/ml ciprofloxacin, and NegCtl = no addition
Several of the 132 compounds in the library annotated as having antibacterial activity are non-quinolone, anti-pseudomonal compounds, including carbenicillin, piperacillin, cefotaxime, ceftriaxone, tobramycin, amikacin, gentamicin, spectinomycin, and minocycline.1 The screening strain carries a gentamicin-resistance marker on pUCP24GW, and no effect was observed on RLU values by gentamicin. However, tobramycin, piperacillin, cefotaxime, and minocycline reduced luminescence slightly (-14%, -3%, -13%, and -9%, respectively, of the positive control at 7 h) consistent with their inhibition of the growth or viability of the screening strain without affecting DNA or DNA synthesis. Carbenicillin, ceftriaxone, amikacin, and minocycline had no detectable effects on the luminescence values at 7 h. Spectinomycin produced a slight increase in luminescence, 7% of the positive control at 7 h. None of these anti-pseudomonal compounds had a significant effect on the screen.
Interestingly, nine of the ten fluoroquinolones, but none of the three DNA adduct forming compounds identified as hits at 7h exhibited luminescence ≥50% of the positive control at 23 h. Thus, while the 23 h data exhibit adequate Z’ scores (see Fig. 3), they fail to identify one fluoroquinolone and three known DNA damaging agents, trichlormethine, furazolidone, and mechorethamine, suggesting a false negative rate of 0.2% (4/2,000). Furthermore, the 23 h data identify as inhibitors two compounds that do not act by damaging DNA, polymyxin B and sildenafil, suggesting a false positive rate of ≥0.1% (2/2,000). Finally, the DNA alkylating agent carboplatin26 only qualifies as a hit at 23 h, and thus, could be considered a false negative at 7 h.
DISCUSSION
The whole cell reporter screen described here is a successful transduction of the effects of depletion or inhibition of GyrA in P. aeruginosa to a bioluminescent readout. The approach of determining the cellular transcriptional response to depletion of a target and fusing appropriate promoter regions to a luciferase operon is a general one, which may be applied to many antibacterial targets. A similar approach has been used to build B. subtilis reporter screening strains, but use of those strains for HTS required an additional pipeting step for the addition of luciferase substrate prior to measuring luminescence.2 We used the entire luciferase operon from P. luminescens to build strain MBX-623. The resulting reporter strain requires no reagent addition for production of bioluminescence, is genetically stable, and produces high levels of luminescence in 4-7 hours of incubation. Despite the inherent variability of cell-based screens, strain MBX-623 produced acceptable Z’ scores and no known false positives in the pilot screen reported here. The number of false negatives is more difficult to assess since many compounds fail to gain entry into P. aeruginosa, or are effluxed rapidly, and thus would not be expected to generate luminescence even if their mechanism involved DNA damage. For example, five of the fifteen quinolones in the library, cinoxacin, pipemidic acid, piromidic acid, oxolinic acid, and flumequine, are weakly potent against P. aeruginosa and were not detected as hits, but this result is not surprising considering the screening concentration was significantly less than the MIC values for these compounds. If these compounds are considered true negatives, then the false negative rate for this pilot screen is < 0.05% (1/2000).
While this reporter screen was developed using ciprofloxacin and gyrase as the model inhibitor-target pair, the promoter chosen to drive the luciferase reporter is known to respond to DNA damage in general. All three genes, PA0612, PA0614, and PA0617, for which promoters were examined in this study, are part of a bactericin locus (pyocin) that responds to DNA damage through the RecA pathway.27 This property of regulation by DNA damage is shared with the B. subtilis genes used by Hutter et al.2 to generate B. subtilis reporter strains responding to fluoroquinolones, but the P. aeruginosa and B. subtilis genes and their predicted proteins are unrelated by sequence homology. The three P. aeruginosa genes encode products (Zn++ finger protein, holin, and assembly protein W, respectively) related to the tails of contractile (myoviridae) bacteriophages, but produce an R-type pyocin with the capability of killing certain strains of P. aeruginosa.17 The pronounced response of the promoter region upstream of PA0614 to DNA damage is fortuitous and extremely useful for reporter strain construction. This regulation by DNA damage is consistent with the results of the pilot screen, since the three non-quinolones among the 13 replicated hits are all known to damage DNA. It may be possible to construct a more gyrase-specific reporter screen by using other promoter regions to drive luciferase. In fact, several additional genes and apparent operons are up-regulated in response to ciprofloxacin treatment.16,28 However, this general DNA damage pathway reporter screen is useful for both identifying potential antibacterial candidates and also for eliminating DNA-damaging agents from further consideration.
The sensitivity of the screen might be improved by modifiying the assay strain or screening protocol. First, an efflux deficient assay strain could increase sensitivity of the assay. The efflux pump encoded by the mexAB-oprM locus is known to play a major role in fluoroquinolone resistance in P. aeruginosa.29 Screening with a strain carrying markerless deletions of mexAB-oprM and other efflux pumps would likely permit identification of a wider range of hits, some of which might be optimised to avoid efflux from wild-type P. aeruginosa cells. In addition, we reasoned that screening at lower IPTG levels might sensitize the assay to inhibitors by reducing the amount of gyrase in the cell, thereby lowering the inhibitor concentration threshold required for luciferase induction. However, such experiments failed to increase the sensitivity to ciprofloxacin, possibly because energy and light production as well as growth were limited due to insufficient gyrA expression. Therefore, the assay strain could be simplified to include the PA0614 promoter region driving luxCDABE without the complemented gyrA deletion. In fact, we have examined such a strain and found it to be as responsive to DNA damaging agents as MBX-623. However, the complemented deletion assay strain configuration offers a significant advantage because it allows the substitution of heterologous gyrA genes for inhibitor screening and profiling of hits. For example, we have substituted the Burkholderia pseudomallei gyrA gene for the P. aeruginosa gyrA gene to permit screening for inhibitors of this Select Agent bacterium in a surrogate BSL2 strain (DTM and MD, unpublished).
In summary, the results of the pilot screen support the utility of this whole cell reporter screen for DNA damaging agents. Analysis of the results indicate the following: (a) a reasonable inhibition cut-off value (e.g., 50% of the RLU produced by 0.5xMIC ciprofloxacin) may be used to eliminate all false positives, at least in this set of 2,000 compounds at a 7 h endpoint; (b) hits in the assay share a common mechanism of damaging DNA by forming adducts, either directly or catalyzed by gyrase/topoisomerase action; and (c) the screen may used as a simple rapid method for identifying DNA damaging agents in compound libraries. Note that some hits may provide sufficient selectivity to be suitable for optimization studies as potential anti-bacterials.
Acknowledgments
We gratefully acknowledge helpful discussions with Dr. Stephen Lory (Harvard Medical School) and Dr. Donald E. Woods (U. Calgary) on the development of this reporter assay. This research was funded by SBIR grant 1-R43-AI056644 awarded by NIAID.
References
- 1.Kiska DL, Gilligan PH. Pseudomonas. In: Murray PR, Baron EJ, Jorgensen JH, Pfaller MA, Yolken RH, editors. Manual of Clinical Microbiology. 8. Vol. 1. Washington, DC: ASM Press; 2003. pp. 719–728. [Google Scholar]
- 2.Hutter B, Fischer C, Jacobi A, Schaab C, Loferer H. Panel of Bacillus subtilis reporter strains indicative of various modes of action. Antimicrob Agents Chemother. 2004;48:2588–2594. doi: 10.1128/AAC.48.7.2588-2594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hooper DC. Mechanisms of action of antimicrobials: focus on fluoroquinolones. Clin Infect Dis. 2001;32(Suppl 1):S9–S15. doi: 10.1086/319370. [DOI] [PubMed] [Google Scholar]
- 4.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid. 2000;43:59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 5.Schweizer HP, Hoang TT. An improved system for gene replacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene. 1995;158:15–22. doi: 10.1016/0378-1119(95)00055-b. [DOI] [PubMed] [Google Scholar]
- 6.Wolfgang MC, Lee VT, Gilmore ME, Lory S. Coordinate regulation of bacterial virulence genes by a novel adenylate cyclase-dependent signaling pathway. Dev Cell. 2003;4:253–263. doi: 10.1016/s1534-5807(03)00019-4. [DOI] [PubMed] [Google Scholar]
- 7.Choi KH, Schweizer HP. An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol. 2005;5:30. doi: 10.1186/1471-2180-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herring CD, Blattner FR. Conditional lethal amber mutations in essential Escherichia coli genes. J Bacteriol. 2004;186:2673–2681. doi: 10.1128/JB.186.9.2673-2681.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, et al. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods. 2005;2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 10.Voisey CR, Marincs F. Elimination of internal restriction enzyme sites from a bacterial luminescence (luxCDABE) operon. Biotechniques. 1998;24:56–58. doi: 10.2144/98241bm11. [DOI] [PubMed] [Google Scholar]
- 11.Becher A, Schweizer HP. Integration-proficient Pseudomonas aeruginosa vectors for isolation of single-copy chromosomal lacZ and lux gene fusions. Biotechniques. 2000;29:948–950. 952. doi: 10.2144/00295bm04. [DOI] [PubMed] [Google Scholar]
- 12.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 13.NCCLS. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard M7-A6. Wayne, PA: NCCLS; 2003. [Google Scholar]
- 14.Li XZ, Livermore DM, Nikaido H. Role of efflux pump(s) in intrinsic resistance of Pseudomonas aeruginosa: resistance to tetracycline, chloramphenicol, and norfloxacin. Antimicrob Agents Chemother. 1994;38:1732–1741. doi: 10.1128/aac.38.8.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oliver A, Levin BR, Juan C, Baquero F, Blazquez J. Hypermutation and the preexistence of antibiotic-resistant Pseudomonas aeruginosa mutants: implications for susceptibility testing and treatment of chronic infections. Antimicrob Agents Chemother. 2004;48:4226–4233. doi: 10.1128/AAC.48.11.4226-4233.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brazas MD, Hancock RE. Ciprofloxacin induction of a susceptibility determinant in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2005;49:3222–3227. doi: 10.1128/AAC.49.8.3222-3227.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakayama K, Takashima K, Ishihara H, Shinomiya T, Kageyama M, Kanaya S, et al. The R-type pyocin of Pseudomonas aeruginosa is related to P2 phage, and the F-type is related to lambda phage. Mol Microbiol. 2000;38:213–231. doi: 10.1046/j.1365-2958.2000.02135.x. [DOI] [PubMed] [Google Scholar]
- 18.Winsor GL, Lo R, Sui SJ, Ung KS, Huang S, Cheng D, et al. Pseudomonas aeruginosa Genome Database and PseudoCAP: facilitating community-based, continually updated, genome annotation. Nucleic Acids Res. 2005;33:D338–343. doi: 10.1093/nar/gki047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, et al. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 20.Lumish RM, Norden CW. Cinoxacin: in vitro antibacterial studies of a new synthetic organic acid. Antimicrob Agents Chemother. 1975;7:159–163. doi: 10.1128/aac.7.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohler T, Michea-Hamzehpour M, Plesiat P, Kahr AL, Pechere JC. Differential selection of multidrug efflux systems by quinolones in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1997;41:2540–2543. doi: 10.1128/aac.41.11.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shimizu M, Takdase Y, Nakamura S, Katae H, Minami A. Pipemidic acid: its activities against various experimental infections. Antimicrob Agents Chemother. 1976;9:569–574. doi: 10.1128/aac.9.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sisson G, Goodwin A, Raudonikiene A, Hughes NJ, Mukhopadhyay AK, Berg DE, et al. Enzymes associated with reductive activation and action of nitazoxanide, nitrofurans, and metronidazole in Helicobacter pylori. Antimicrob Agents Chemother. 2002;46:2116–2123. doi: 10.1128/AAC.46.7.2116-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Alencar TA, Leitao AC, Lage C. Nitrogen mustard- and half-mustard-induced damage in Escherichia coli requires different DNA repair pathways. Mutat Res. 2005;582:105–115. doi: 10.1016/j.mrgentox.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Sykora I, Gandalovicova D. Trichlormethine hydrochloride and correlation of its mutagenic and toxic effects on male germ cells in mice. Mutat Res. 1992;266:291–297. doi: 10.1016/0027-5107(92)90196-9. [DOI] [PubMed] [Google Scholar]
- 26.Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Perez JM. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem. 2007;7:3–18. doi: 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- 27.Matsui H, Sano Y, Ishihara H, Shinomiya T. Regulation of pyocin genes in Pseudomonas aeruginosa by positive (prtN) and negative (prtR) regulatory genes. J Bacteriol. 1993;175:1257–1263. doi: 10.1128/jb.175.5.1257-1263.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cirz RT, O’Neill BM, Hammond JA, Head SR, Romesberg FE. Defining the Pseudomonas aeruginosa SOS response and its role in the global response to the antibiotic ciprofloxacin. J Bacteriol. 2006;188:7101–7110. doi: 10.1128/JB.00807-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob Agents Chemother. 2001;45:105–116. doi: 10.1128/AAC.45.1.105-116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]