

Summary
Members of the uncultured bacterial genus Candidatus Accumulibacter are capable of intracellular accumulation of inorganic phosphate (Pi) in activated sludge wastewater treatment plants (WWTPs) performing enhanced biological phosphorus removal (EBPR), but were also recently shown to inhabit freshwater and estuarine sediments. Additionally, metagenomic sequencing of two bioreactor cultures enriched in Candidatus Accumulibacter, but housed on separate continents, revealed the potential for global dispersal of particular Candidatus Accumulibacter strains, that we hypothesize is facilitated by the ability of Candidatus Accumulibacter to persist in environmental habitats. In the current study, we used sequencing of a phylogenetic marker, the ppk1 gene, to characterize Candidatus Accumulibacter populations in diverse environments, at varying distances from WWTPs. We discovered several new lineages of Candidatus Accumulibacter which had not previously been detected in WWTPs, and also uncovered new diversity and structure within previously detected lineages. Habitat characteristics were found to be a key determinant of Candidatus Accumulibacter lineage distribution, while, as predicted, geographic distance played little role in limiting dispersal on a regional scale. However, on a local scale, enrichment of particular Candidatus Accumulibacter lineages in WWTP appeared to impact local environmental populations. These results provide evidence of ecological differences among Candidatus Accumulibacter lineages.
Introduction
Removal of inorganic phosphate (Pi) from wastewater is a key step in wastewater treatment to prevent eutrophication of downstream water bodies impacted by treated effluent. One increasingly popular mechanism employed for Pi removal in wastewater treatment plants (WWTPs), known as enhanced biological phosphorus removal (EBPR), relies on the ability of some microorganisms in activated sludge to accumulate polyphosphate and thereby remove excess phosphorus from the water via biomass settling and removal. In most lab-scale reactors mimicking the EBPR processes used in full-scale WWTPs, the organism primarily responsible for Pi accumulation is a β-proteobacterium affiliated with the Rhodocyclus group, named Candidatus Accumulibacter phosphatis, (Hesselmann et al., 1999; Crocetti et al., 2000). This organism and related species have since been detected in many full-scale WWTPs, where they have also been shown to accumulate polyphosphate (Zilles et al., 2002a; Zilles et al., 2002b; Kong et al., 2004; He et al., 2008). Therefore it is likely that polyphosphate accumulation is widespread in the genus Candidatus Accumulibacter (henceforth referred to as Accumulibacter), although evidence suggests that other organisms also play a role in WWTPs treating industrial wastewaters (Kong et al., 2005). Although much has been learned about the biochemical steps required for EBPR, unpredictable perturbations to the process continue to plague treatment plant operations (Neethling et al., 2005; Oehmen et al., 2007). One impediment that has hampered EBPR research is the fact that while Accumulibacter can be enriched in bioreactors to levels over 90% of total cells (Lu et al., 2006), the organisms remain recalcitrant to isolation in pure culture, hence the taxonomic status of Candidatus (Murray and Stackebrandt, 1995).
Recent metagenomic sequencing of two bioreactors enriched in Accumulibacter, and housed on separate continents (in Wisconsin, USA and Queensland, Australia), confirmed many hypotheses regarding the biochemical functions required to accumulate Pi, but surprisingly, the genomes of the dominant strains enriched in the two reactors shared >95% nucleotide sequence identity over 79% of the assembled US genome (Garcia Martin et al., 2006). One explanation for the high degree of similarity between the two geographically remote genotypes is that Accumulibacter can be dispersed via environmental reservoirs. In support of this hypothesis, the Accumulibacter genome encodes numerous functions not required for growth in the nutrient replete habitat of WWTPs that would be useful in an oligotropic environment, including carbon and nitrogen fixation, high-affinity Pi transport, and motility via flagella (Kunin et al., 2008). Additionally, a PCR survey of freshwater, terrestrial and marine samples found Accumulibacter to be common in freshwater sediments, and occasionally present in freshwater, soil and estuarine sediment samples (Kunin et al., 2008). However, a survey of the Accumulibacter spp. present in full-scale WWTPs revealed that the lineages enriched in bioreactors represented only a subset of total Accumulibacter diversity; three major lineages were detected in WWTPs in addition to the two initially found in bioreactors, and four of the five total lineages were found to be dominant in at least one treatment plant (He et al., 2007). This result suggests that global abundance of particular Accumulibacter lineages is not the only determinant of dominance of a given lineage in any particular environment, but rather, ecological differences between lineages, or ecotypes, lead to differences in their relative competitiveness under different environmental conditions.
Although the initial investigations into Accumulibacter population structure described above (Kunin et al., 2008) suggest that geographic barriers do not limit global lineage distribution, studies of other bacterial populations indicate that geographic isolation and environmental parameters can affect both distribution, to varying degrees depending on characteristics of the particular organisms (reviewed in (Martiny et al., 2006)) and the genetic loci used to infer phylogeny. Several studies using loci other than the 16S rRNA gene suggest that geographic distance plays an important role in structuring populations of some extremophiles with limited habitat ranges such as hyperthermophilic Archaea (Whitaker et al., 2003), hot spring cyanobacteria (Papke et al., 2003) and symbionts of mussels living in deep sea hydrothermal vents (DeChaine et al., 2006). Patterns for organisms with wider habitat ranges appear to vary. Some studies have found evidence for endemism, or restriction of genotypes to particular locations, such as for fluorescent pseudomonads (Cho & Tiedje, 2000), while others have found evidence for widespread distribution of genotypes, such as freshwater actinobacteria (Newton et al., 2007).
Accumulibacter spp. distribution differs from the examples described above in that the organisms are present both at high densities in specialized, discrete habitats (WWTPs), and at low densities in more widespread, continuous environments (freshwater sediment; (Kunin et al., 2008)). Thus, the lineage is a compelling model with which to explore how the opposing forces of habitat filtering and competition act in the face of rapid dispersal to structure bacterial populations. Furthermore, a more complete understanding of the parameters affecting Accumulibacter population structure in WWTPs and bioreactors requires a broader characterization of Accumulibacter ecology in non-wastewater environments. For example, it is not clear whether the five lineages detected to date in WWTPs and lab-scale bioreactor environments (He et al., 2007) represent the complete diversity of Accumulibacter, or whether environmental reservoirs harbor additional lineages as yet undetected. It is also unclear to what extent enrichment of particular lineages in WWTPs impacts Accumulibacter population structure in surrounding environments. In the present study, we used sequencing of a high resolution single copy marker gene (ppk1), encoding polyphosphate kinase (He et al., 2007; Kunin et al., 2008)) to characterize population structure of Accumulibacter in a variety of freshwater and marine habitats, at varying distances from WWTPs performing EBPR, and in two geographic regions: Wisconsin, USA, and California, USA.
Results
Detection of Accumulibacter in environmental samples
Accumulibacter were detected by PCR in diverse aquatic habitats from previously described samples collected in Contra Costa County, California (Kunin et al., 2008), and in samples described in the current study taken from sites in Dane and Green Counties, Wisconsin. We sought to characterize Accumulibacter population structure in samples representative of the diversity of aquatic habitats located in both regions, and collected at varying distance from WWTP practicing EBPR. The sites from which we recovered verified Accumulibacter ppk1 gene sequences (based on phylogenetic analysis, sites shown in Figure 1) include sediment from two of three water storage reservoirs sampled in CA, sediment from the estuary of a small CA stream, sediment from four points along the small Sugar River or its tributaries in WI, and sediment from two eutrophic WI lakes: shallow (4 m maximum depth), polymictic Lake Wingra and from the deepest point (25 m) of dimictic Lake Mendota. Sediment from three CA streams located 13.7, 12.1 and 8.4 km from an EBPR-practicing WWTP tested positive for Accumulibacter using a highly sensitive, nested PCR approach, but the only CA stream from which we recovered Accumulibacter ppk1 sequences was located 0.3 km from a WWTP using EBPR (Kunin et al, 2008). Positive stream and river samples in WI were collected from within 100 m of an EBPR-practicing WWTP discharge point, and 20.9 km upstream or 33.4 km downstream of the WWTP (Figure 1). However, we note that the frequent need for two rounds of PCR to detect amplicons suggested that Accumulibacter populations were generally in low abundance in these habitats.
Figure 1.
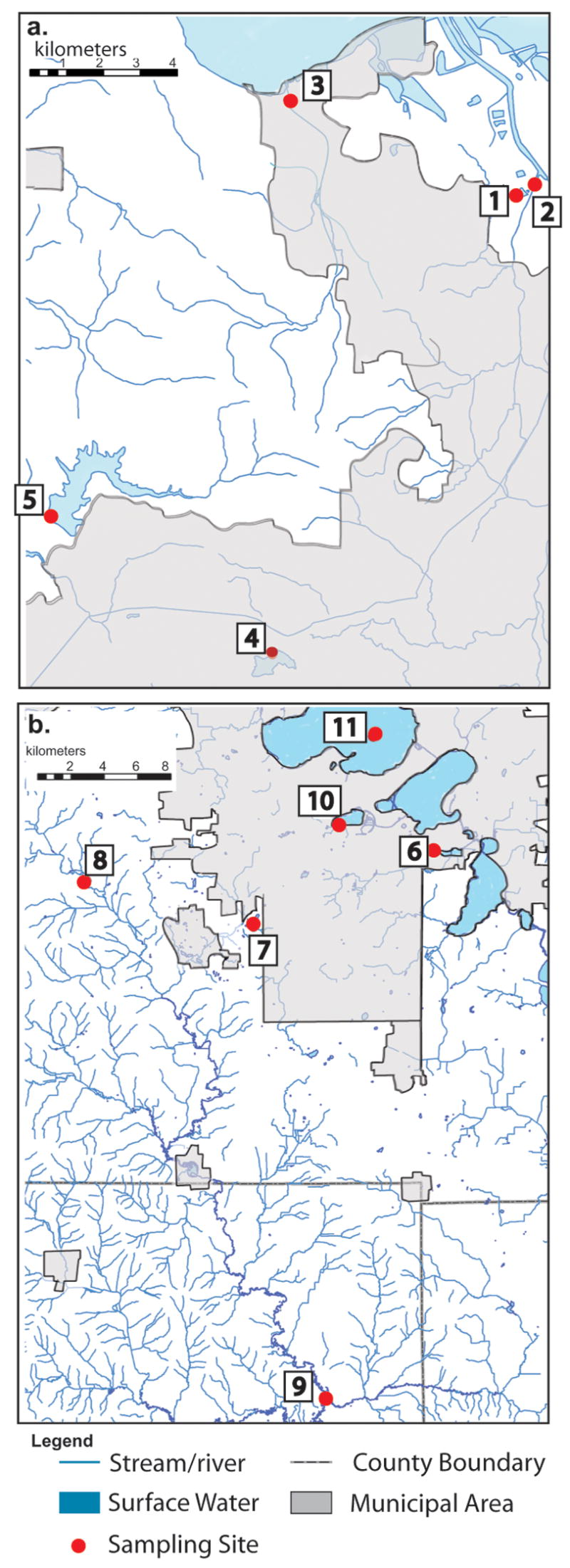
Sources of samples from which ppk1 clone libraries were built and sequenced. a) Contra Costa County, CA sampling sites: 1. Contra Costa WWTP; 2. Walnut Creek; 3. Alhambra Creek Estuary; 4. Lafayette Reservoir; 5. Briones Reservoir. b) Dane County and Green County, WI sampling sites: 6. Nine Springs WWTP; 7. Badger Mill Creek (both 100m upstream and 100m downstream of Nine Springs discharge point); 8. Sugar River tributary; 9. Sugar River, downstream; 10. Lake Wingra; 11. Lake Mendota.
Phylogenetic diversity of environmental Accumulibacter
We prepared and sequenced Accumulibacter ppk1 gene clone libraries from PCR amplicons obtained from four California environmental samples, six Wisconsin environmental samples (two of which were taken from sites separated by only 200 m), and samples from the Contra Costa WWTP (CCWWTP) EBPR sludge and Nine Springs WWTP effluent (Figure 1). Most retrieved sequences were affiliated with previously recognized Accumulibacter clades, but from a few samples we also recovered sequences more closely related to Dechloromonas, indicating a degree of non-specificity of the primers with complex DNA templates while using a lower annealing temperature than originally described (McMahon et al., 2007) to facilitate capture of a broader diversity of sequences (data not shown). Also, a relatively high number of chimeric sequences (10% of total verified Accumulibacter sequences) were identified in the libraries, likely as a result of the high number of PCR cycles required to obtain products from most samples. These were removed from subsequent analysis.
All of the Accumulibacter ppk1 sequences fell into two major lineages (Figure 2), previously designated Type I and Type II based on analysis of Accumulibacter ppk1 sequences from WWTPs (He et al., 2007). Within each major grouping, environmental sequences contributed several new clades. Among the Type I sequences, new clades IB, ID and IE consisted solely of sequences obtained from the Alhambra Creek estuary sample, the only non-freshwater sample included. Many of the environmental sequences from Type II clustered with clades previously detected in WWTPs (IIA–IIE). However, sequences forming one new group, designated IIF, were detected in the CCWTP and nearby Walnut Creek. Sequences recovered from several lake, stream sediment, and WWTP effluent samples from both regions formed a second new group, designated IIG. A few rare sequences appear to represent additional new clades, although in some cases, all sequences placed in one clade came from a single clone library, which raises the concern that these sequences may represent PCR artifacts. The environmental sequences also considerably increased the diversity and structure within clade IID, which had previously been only rarely detected in WWTPs.
Figure 2.

Maximum likelihood tree of Accumulibacter based on comparative analysis of aligned ppk1 gene nucleotide sequences. Branch points supported by bootstrap resampling are indicated by bootstrap proportions >70% on interior nodes. Rhodocyclus tenuis was used as the most closely related outgroup sequence, although more extensive sets of outgroups were used to determine Accumulibacter monophyly (data not shown). Two primary lines of descent in Accumulibacter, Type I and II, are indicated by brackets to the right of the figure. Monophyletic lineages within these Types are shown mostly as compressed wedges. The number of sequences within each wedge is indicated inside the wedge. Fully expanded trees for these lineages are shown in Figs. S1–S7. Bar, 0.10 changes per sequence position
Distribution of Accumulibacter lineages across environments
Accumulibacter clades were unevenly distributed across clone libraries from different samples (Figure 3). Some clades were only detected in a single sample, such as IB, ID and IE, all of which were only detected in the Alhambra Creek estuary. With the exception of a single sequence from clade IIB from the Sugar River tributary sample, clades IIB and IIA were detected only in the two WWTPs and nearby stream samples, and not in any of the environmental samples collected from further away from the WWTPs. Clade IIE was only detected in the CCWWTP and nearby stream. In contrast, sequences from clades IID and IIG were broadly distributed among environments; five of the twelve clone libraries contained sequences from the IIG clade and all but the libraries from the Alhambra Creek estuary and the Nine Springs WWTP contained sequences from clade IID.
Figure 3.
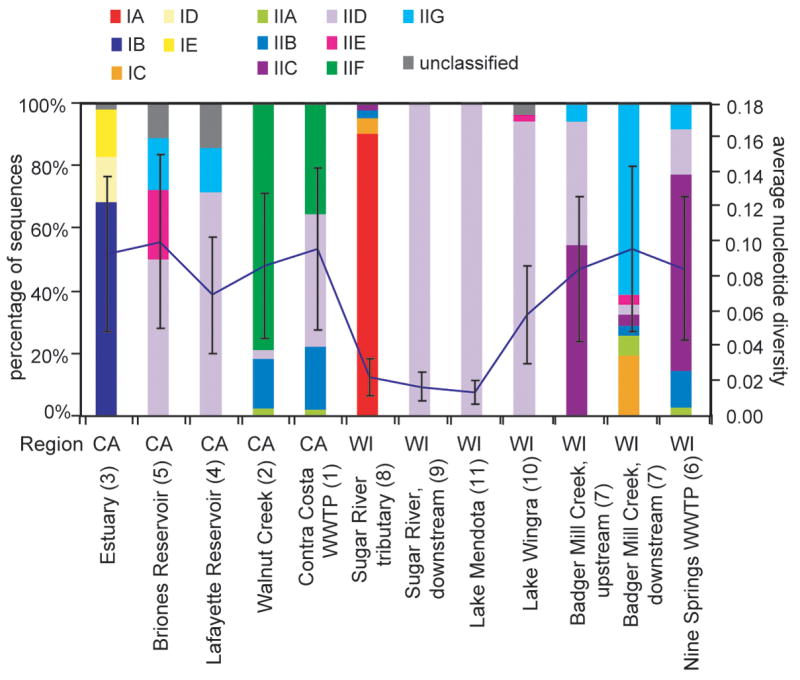
Distribution of Accumulibacter clades among samples, and average nucleotide diversity values for each ppk1 clone library. Sample location numbers corresponding to Figure 1 are shown in parentheses. Percentages represent the fraction of Accumulibacter ppk1 clones from a given library that cluster in the indicated clade, without removing duplicate or highly similar sequences. Average nucleotide diversity of ppk1 sequences in each library was calculated using Arlequin, and means +/− one standard error are shown.
The uneven distribution of clades across environments suggests that environmental parameters, geographic isolation, or both, restrict distribution of Accumulibacter clades. We assessed the relative importance of geographic distance and environmental parameters for determining clade distribution using the Unifrac environment clustering function. The Unifrac metric measures the degree of shared history in phylogenies constructed from sequences obtained from different samples, optionally weighted by the frequency of sequence (or operational taxonomic unit (OTU)) observation (Lozupone et al., 2006; Lozupone et al., 2007). Using the qualitative, unweighted clustering function, few clusters containing multiple environments received significant bootstrap support (Figure 4a), likely because many clades were detected in multiple environments (Figure 3). However, the quantitative clustering, in which OTUs (defined in this study as sequences belonging to the same, smallest clade with significant (>60%) bootstrap support in maximum likelihood phylogenetic analysis, which generally included sequences with a difference in genetic distance of less than 0.02 based on nt comparisons) were weighted by frequency of observation in a given clone library, revealed many significant clusters between environments (Figure 4b).
Figure 4.
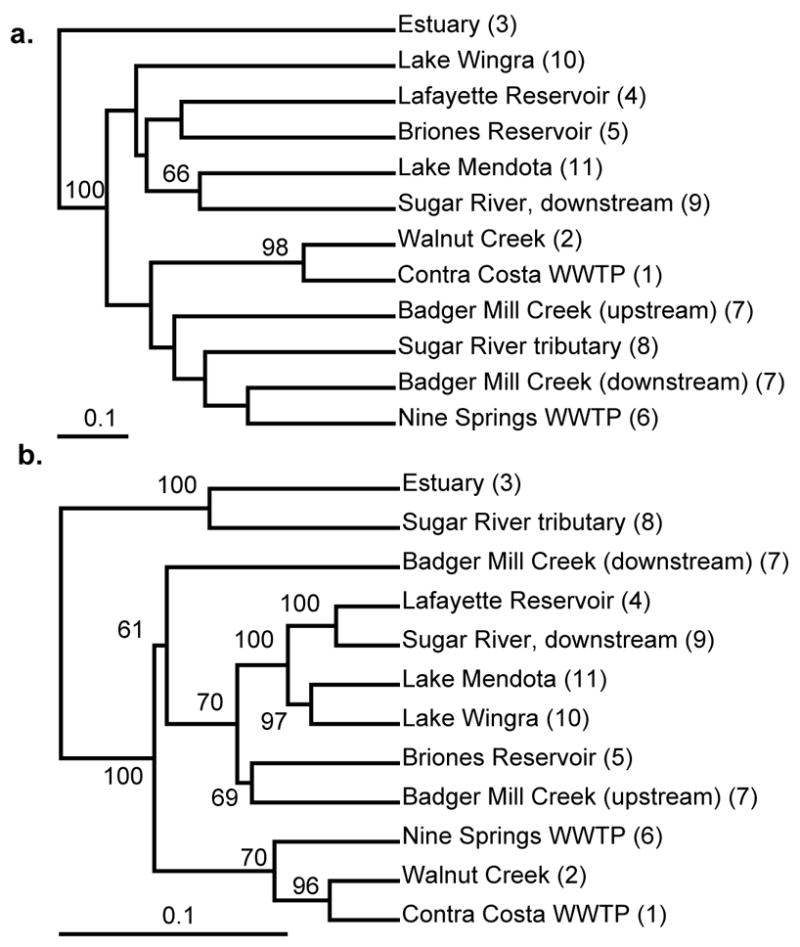
Unifrac environment cluster analysis of Accumulibacter ppk1 gene sequences, both without (a) and with (b) weighting for sequence frequency. Sample location numbers corresponding to Figure 1 are shown in parentheses. Jackknife values greater than 60 (for 100 iterations) are shown.
Generally, cluster analysis indicated little impact of geographic limits to distribution of Accumulibacter clades. Many clusters with significant bootstrap support contained samples collected in both Wisconsin and California (Figure 4b). A second line of evidence to support the lack of geographic restriction to distribution of Accumulibacter lineages on a regional scale was the presence of ppk1 sequences with zero or only one to two nucleotide differences from clone libraries built from California and Wisconsin samples (Figures S1–S7). However, despite the lack of regional geographic patterns of clade distribution, the Accumulibacter population in Walnut Creek water closely resembled that of the Contra Costa WWTP, located within 0.5 km, suggesting that enrichment for particular clades in the WWTP may impact nearby freshwater Accumulibacter populations, or less likely vice versa. Equivalent correspondence between the Nine Springs WWTP and nearby Badger Mill Creek was not observed, but the Badger Mill Creek sampling site, while within 100 m of a WWTP effluent discharge point, was nearly 12 km from the WWTP itself, rather than 0.5 km. Our interpretation of this result is that Accumulibacter is dispersed via aerosols from the open activated sludge aeration basins enriched in this organism, and not via effluent.
Significant clustering of phylogenetic structure of samples obtained from geographically distant sites suggests that environmental parameters play a larger role overall than geographic isolation, in determining Accumulibacter clade distribution. When clone frequency was weighted in the cluster analysis, three clusters representing distinct environment types emerged: freshwater habitats, WWTP samples (and the Walnut Creek sample taken from within 0.5 km of a WWTP, see above), and the sole estuarine sample. One freshwater sample, taken from a Sugar River tributary in Wisconsin, clustered with the estuary sample, but closer examination of the sequences recovered from the two samples indicated that while both were the only two samples dominated by Type I ppk1 sequences (Figures 2, S1 and S2), all but 4 of the 94 Sugar River sequences fell into clade IA, while the estuary sequences affiliated with clades IB, ID and IE. Among the freshwater samples, several other clusters were apparent, but the small number of samples obtained from any given specific freshwater environment limits the conclusions that can be drawn regarding distribution of clades among these habitats.
Accumulibacter population structure across environments
One measure of population structure, the average nucleotide diversity of sequences in a clone library, varied between samples (Figure 3). For bacterial communities, this diversity index has the advantage over traditional diversity indices such as the Shannon and Simpson indices of not requiring an arbitrary sequence distance cutoff to designate OTUs (Martin, 2002). Most clone libraries contained similar (within one standard deviation) average nucleotide diversity. However, the average nucleotide diversity of the two Sugar River clone libraries and the Lake Mendota clone library were significantly lower than that of the other libraries (Figure 3). These three clone libraries with reduced nucleotide diversity were each dominated by sequences from a single clade; clade IA dominated the Sugar River tributary library and clade IID dominated the Lake Mendota and downstream Sugar River libraries (Figure 3). It is unlikely that dominance of clades IA or IID in these samples resulted solely from preferential amplification of these clades by the ppk1 primers, because clade IA was previously detected as a minority member of other clone libraries (He et al., 2007), and IID was a minority member of the clone libraries constructed from several of the samples in this study (Figure 3, Figure S6).
As a second means to compare population structure across communities, we examined the phylogenetic species variability (PSV) and evenness (PSE) for each sample, and across samples (Helmus et al., 2007a). PSV provides a measure of the average degree of relatedness among species within a community. PSE also measures phylogenetic relatedness within communities, but weighted by species abundance. Two permutation tests can be performed to compare the average PSV and PSE values observed across communities to null hypotheses regarding the relationship between phylogeny and community composition to generate hypotheses about which forces may be most strongly shaping species distribution.
The first null hypothesis assumes that all species (in this case, the lineages designated in Figure 2) are equally prevalent. Permutation under this hypothesis maintains the observed number of species in each community, but randomly selects species from the global pool (i.e. the entire dataset) and then calculates the average PSV (or PSE) value. Null hypothesis 1 is rejected if there is a phylogenetic pattern in the prevalence of species in the global pool (e.g., there is a group of related species that is very prevalent in all communities regardless of environmental conditions). Permutation under the second null hypothesis maintains the observed species abundance, but assumes equal species richness across communities. Null hypothesis 2 tests for phylogenetic structure within local communities independent of any structure caused by species prevalence.
Using our full dataset, we found the observed average PSV and PSE values to be significantly (p<0.001) lower than those expected under either null 1 or null 2 (Figure 5a) hypotheses. Thus, clades found in a given environment were more closely related, regardless of their global abundance, than expected by chance, a pattern known as phylogenetic underdispersion. Underdispersion is expected to result if habitat filtering selects for related species that share common adaptations to local environmental conditions (Webb et al., 2002; Helmus et al., 2007a). Because the composition of the sole estuary sample was considerably different from the other samples (Figure 3), we also calculated observed and expected PSV and PSE values under both null hypotheses with the estuary community and clades only found in the estuary (IB, ID and IE) removed. Removing the estuary data did not change the result of significant underdispersion (data not shown).
Figure 5.
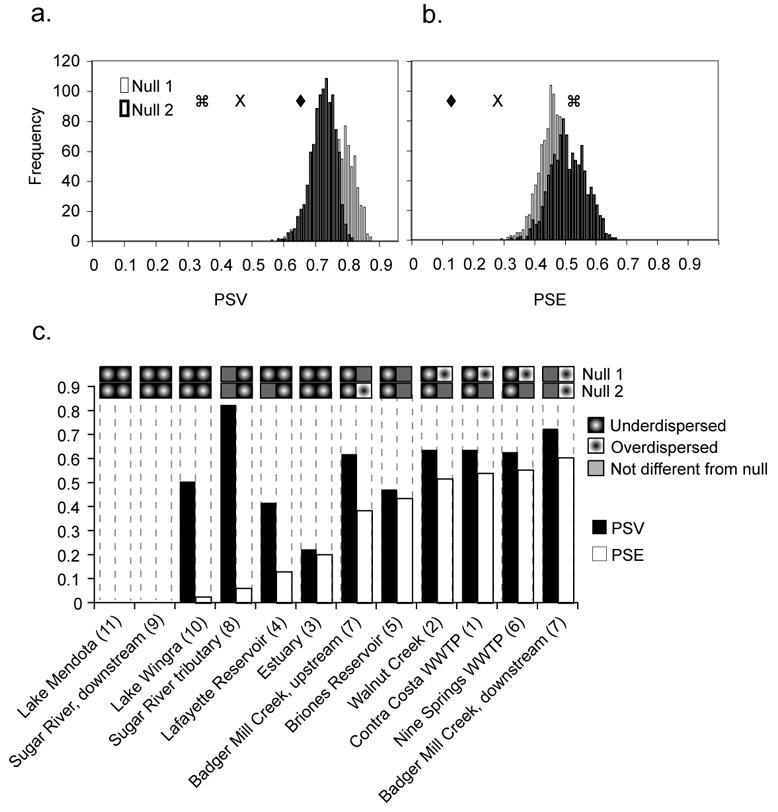
Measurements of phylogenetic structure in Accumulibacter communities. (a) Distribution of expected mean (a) PSV and (b) PSE values obtained from iteration under null hypotheses 1 and 2. The observed mean PSV and PSE across all communities (X), for WWTPs and nearby stream samples (♦), and for freshwater and estuarine samples collected more distantly from WWTPs (
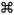 ). (c) PSV and PSE values calculated for Accumulibacter communities in each sample. Sample numbers corresponding to Figure 1 are shown in parentheses. PSV and PSE values for the Sugar River downstream sample and Lake Mendota were zero, because these clone libraries were each dominated by a single clade. If measured PSV or PSE values were significantly different (α=0.05) than the null distribution, the coded boxes across the top of panel (c) indicate if the sampled communities were under- or overdispersed.
). (c) PSV and PSE values calculated for Accumulibacter communities in each sample. Sample numbers corresponding to Figure 1 are shown in parentheses. PSV and PSE values for the Sugar River downstream sample and Lake Mendota were zero, because these clone libraries were each dominated by a single clade. If measured PSV or PSE values were significantly different (α=0.05) than the null distribution, the coded boxes across the top of panel (c) indicate if the sampled communities were under- or overdispersed.
The values for PSV or PSE can also be compared between communities, to determine if the overall pattern of phylogenetic distribution across communities is maintained consistently in different environments. With the exception of the Sugar River sample discussed below, PSV values were higher for all samples taken from WWTPs or from streams nearby than for more distant freshwater samples, with the estuary sample having an intermediate PSV (Figure 5). PSE values generally followed the same trend, although Briones Reservoir had a slightly higher PSE than the Badger Mill Creek sample taken from 100 m upstream of the Nine Springs WWTP outlet (Figure 5c). However, the average PSV values for each of the two categories of samples were still significantly lower than the expected average under both null hypotheses. Together, these observations indicate that while all habitats showed indications of phylogenetic underdispersion based on PSV, the trend was more pronounced for freshwater samples taken far from WWTPs. Based on measures of PSE, the aggregate Accumulibacter community across all samples was also underdispersed. However, samples collected from habitats far from WWTPs did not have measurable phylogenetic structure while communities from habitats nearby WWTPs were more strongly underdispersed.
Discussion
Compared to the wealth of accumulated research into macroorganismal population biology, the study of microbial populations remains in its infancy. Given the small size of bacteria, their large population sizes, and their potential for widespread distribution and lateral gene transfer between distantly related individuals, it is unknown to what extent bacterial populations will conform to principles governing populations of macro-scale organisms (Prosser et al., 2007). Development of a theoretical framework for understanding bacterial population biology requires a strong underpinning of empirical observations of bacterial populations. In this study, we characterized the diversity and population structure in twelve different samples for one industrially important bacterial genus, Accumulibacter. Our observations expand the known diversity of Accumulibacter and document the relationship of Accumulibacter population structure with habitat type and geographic location.
For a growing number of bacterial lineages, geographic restriction to dispersion appears to play a role in determining population structure (Cho & Tiedje, 2000; Papke et al., 2003; Whitaker et al., 2003; Glaeser & Overmann, 2004; Foti et al., 2006). In most of these studies, detection of geographic restraints on bacterial dispersal relied on high-resolution methods to describe bacterial genetic diversity, such as multilocus sequencing typing (Whitaker et al., 2003), repetitive extragenic palindromic PCR-based genomic footprinting (Cho & Tiedje, 2000; Foti et al., 2006) or 16S–23S intergenic spacer region sequencing (Cho and Tiedje, 2000; Papke et al., 2003). Using the sequence of the Accumulibacter ppk1 gene, which was previously shown to afford greater phylogenetic resolution than the 16S rRNA gene sequence (He et al., 2007), we found habitats with similar characteristics were more likely to harbor similar Accumulibacter lineages than habitats that were geographically clustered. The importance of habitat filtering in determining local Accumulibacter population compositions was also revealed through phylogenetic species variability analysis, which found strong evidence for phylogenetic underdispersion. Underdispersion is predicted to result when adaptations to particular environmental parameters are shared between related lineages, and are more important in determining the distribution of species than competition between closely related lineages (Webb et al., 2002; Helmus et al., 2007a; Newton et al., 2007). Bacterial communities were also found to be underdispersed using the 16S rRNA locus in freshwater mesocosms and soil, as were ammonia oxidizing populations based on analysis of ammonia monooxygenase genes in estuary sediments (Horner-Devine & Bohannan, 2006). Recently described microcosm experiments with freshwater sediments provided experimental evidence to support a similar importance for environmental selection in determining clade distribution for sulfur-oxidizing Achromatium sp. (Gray et al., 2007). Although environmental parameters appear to be the primary determinant of the distribution of major clades within Accumulibacter, a finer-scale analysis than that afforded by ppk1 gene sequencing will be required to ascertain the extent to which geography plays a role in limiting dispersal of strains within particular Accumulibacter lineages. Such studies might also reveal any ecologically relevant differentiation among strains or lineages that could be significant for phosphorus cycling activity either within WWTPs or in natural environments.
Given that we previously retrieved nearly identical Accumulibacter ppk1-gene sequences from bioreactors operated in Wisconsin, USA and Queensland, Australia, the widespread dispersal of Accumulibacter clades in the environment was not surprising. For example, identical ppk1 sequences belonging to clade IID were identified in freshwater samples from California (Lafayette Reservoir) and Wisconsin (Sugar River, downstream). The mechanism by which Accumulibacter achieves such widespread distribution remains unclear, but the relatively high abundance of these organisms in WWTPs may play a role. Surveys of full-scale WWTP demonstrate that Accumulibacter can represent up to 20% of the total bacterial population (Zilles et al., 2002b; Kong et al., 2005; He et al., 2007). In contrast, the need for high numbers of PCR cycles or nested PCR to detect Accumulibacter in freshwater sediment and the absence of Accumulibacter-related sequences in previous surveys of freshwater bacterial diversity (based on BLAST analysis of Accumulibacter 16S rRNA gene sequences against sequences deposited in Genbank) suggest that abundance of Accumulibacter populations is much lower in natural habitats. Despite the lack of regional limits to dispersal of Accumulibacter clades, we did observe that populations in samples taken from a stream (sampling site 2 in Fig. 1) near one WWTP (sampling site 1 in Fig. 1) clustered with the respective WWTP samples, suggesting that organisms are dispersing from the WWTP, likely via aerosols, into nearby waterways. Whether such populations persist in the environment is unknown.
In addition to the differences in phylogenetic composition of Accumulibacter communities found in different environments, we also observed differences in population structure across samples. For example, the Sugar River and Lake Mendota samples exhibited significantly lower diversity of Accumulibacter clades than other environments sampled. Interestingly, although both Sugar River samples were dominated by a single clade, the specific clade dominating was different in each case; clade IA dominated the Sugar River tributary sample, while clade IID was dominant downstream (Figure 3). The relatively low diversity of Accumulibacter clades in these environments may result from increased competition from other organisms; diversity of bacteria other than Accumulibacter in freshwater sediment is expected to be higher than in WWTP (Curtis et al., 2002). The differences in Accumulibacter clade composition in these environments with low Accumulibacter diversity could result from correlations between traits of these clades and environmental parameters. Alternatively, the stochastic niche theory of community assembly proposed by Tilman predicts that in environments harboring high diversity, infrequency of invasions combined with stocasticity inherent in dispersal of organisms can lead to differences in species composition in otherwise similar habitats (Tilman, 2004).
A second difference in Accumulibacter population structure across environments was revealed through PSV and PSE analysis. While generally habitat filtering appeared to play a key role in determining community composition, the measured effect was stronger for freshwater habitats distant from WWTPs than for WWTP communities or those in streams near WWTPs when the PSV metric was used. This suggests that competition between related clades may play a more important role in determining community composition in EBPR systems than in freshwater habitats, which could result from the relatively large proportion of Accumulibacter observed in EBPR systems compared to freshwater habitats (discussed above). Alternatively, the stresses imposed in the freshwater environment may impose more stringent selection for particular adaptations than those encountered by Accumulibacter populations in WWTPs. The underdispersion signal disappeared in habitats distant from WWTPs when clade prevalence was incorporated into the analysis through the use of the PSE metric. Since PSE is less sensitive to the presence/absence of rare taxa (Helmus et al., 2007a), we speculate that within-clade microdiversity contributed by less abundant populations depresses PSV and makes the Accumulibacter communities appear more underdispersed when clade prevalence is neglected.
The ecological role of Accumulibacter in the environment and ecological differences between clades remain completely unknown. Storage and cycling of polyphosphate by microorganisms has recently been suggested to play a key role in the phosphorus cycle in aquatic sediments (Hupfer et al., 2007), where Accumulibacter were most frequently detected. Fluctuations in the boundary between oxic and anoxic zones in sediments may facilitate polyphosphate accumulation similar to that carried out by Accumulibacter in EBPR sequencing batch reactors and WWTPs. The detection of some clades in a fraction of the environments sampled, such as types IB, ID and IE in the estuary, as well as the detection of multiple coexisting but distinct clades in single environmental samples suggest that these clades represent distinct ecotypes occupying a particular niche (Cohan, 2006). Additionally, although the clades depicted in Figure 2 represented cohesive clusters of sequences, we also detected an accumulation of fine-scale diversity of sequences within each clade in many environments (Figures S1–S7). Similar high, fine-scale diversity has previously been observed for 16S rRNA gene sequences from a marine sample (Acinas et al., 2004) and from sulfate-reducers in a salt marsh sample (Klepac-Ceraj et al., 2004), and is suggested to result from selective sweeps followed by accumulation of differences for which selection is not strong enough to purge (Acinas et al., 2004; Klepac-Ceraj et al., 2004).
This work provides a snapshot of diversity and population structure for one bacterial genus across different environments and geographic scales. Future repeated sampling of a particular environment over time, and expansion of the survey into additional ecosystems would provide additional insight into the dynamics of Accumulibacter population structure and the relationship between particular environmental parameters and the detection of different Accumulibacter clades. Additionally, genomic analysis of lineages other than the previously sequenced strains from lab-scale bioreactors (Garcia Martin et al., 2006) would yield insight into ecological differences between lineages. Our results demonstrate that Accumulibacter is an excellent model group of bacteria with which to conduct such studies.
Experimental Procedures
Sample collection and processing
Samples were collected from the sites in California and Wisconsin depicted in Figure 1. The California sites were sampled as previously described (Kunin et al., 2008). At the Wisconsin stream and river sampling sites, we aseptically collected one liter of surface water, 50 ml of sediment from near the stream bank, and a 50 ml container of soil from above all obvious high water marks. From Lake Wingra and Lake Mendota, we collected one liter of surface water, one liter of water from near the lake bottom, at the deepest point (~3 m and ~20 m, respectively), and 50 ml sediment from the lake bottom, also near the deepest point. The Nine Springs WWTP effluent sample (500 ml) was collected from the discharge location. The WWTP was operating as a modified University of Cape Town process with ultra-violet light disinfection as the final unit process in treatment. Additional information about plant configuration and performance can be found elsewhere (He et al., 2007; McMahon et al., 2007). We note that the effluent discharge to Badger Mill creek is 12 km away from the Nine Springs WWTP. Badger Mill creek and Nine Springs WWTP were sampled on June 6, the Sugar River on July 9, Lake Mendota on July 13 and Lake Wingra on July 19, all in 2006.
Sample processing and DNA extraction proceeded essentially as described (Kunin et al., 2008). Briefly, cells were collected from water samples on 0.2 mm pore size membrane filters (200–500 ml filtered, depending on concentration of particulate matter), and DNA was extracted from membranes or from 0.25 g soil and sediment samples using the bead-beating based Power Soil kit (MolBio). All DNA samples were resuspended in TE buffer and stored at −20°C.
PCR screening, clone library construction and sequencing
We screened Wisconsin samples for the presence of Accumulibacter using lineage-specific 16S rRNA and ppk1 gene targeted primers as described previously (Kunin et al., 2008). From sediment and WWTP samples collected in both WI (the current study) and CA (Kunin et al., 2008) that tested positive for Accumulibacter (sites shown in Figure 1), we gel-purified the products of ppk1 gene PCR with the Accumulibacter-targeted primers Acc-ppk1-254f and Acc-ppk1-1376 (McMahon et al., 2007) (25–40 cycles, annealing temperature of 58°C, extension time of 1 min 30 sec) and cloned them using the pCR4-TOPO vector (Invitrogen). As described previously (McMahon et al., 2007), these Accumulibacter ppk1-targeted primers were designed to specifically amplify sequences from this genus based on a multiple alignment of diverse ppk1 gene sequences retrieved using degenerate ppk1-targeted primers of broad specificity. It is possible that these primers fail to capture previously undetected diversity of Accumulibacter ppk1 gene sequences. However, a quantitative PCR study comparing total abundance of Accumulibacter sequences detected by ppk1 and 16S rRNA gene-targeted primers found comparable estimates of total Accumulibacter abundance using both methods in sludge from numerous full-scale treatment plants and lab-scale reactors, indicating that primers Acc-ppk1-254f and Acc-ppk1-1376 likely capture most Accumulibacter ppk1 gene sequences (He et al., 2007).
Ninety-six clones from each clone library (except the Sugar River tributary library, from which 192 clones were picked) were sequenced in both directions using vector primers, and paired reads were quality- and vector-trimmed, and assembled using genelib (E. Kirton, JGI, unpublished). Sequences were checked for chimeras using Bellerophon (Huber et al., 2004), Mallard (Ashelford et al., 2006), and manually using partial treeing analysis.
Phylogenetic reconstruction and analysis
Assembled partial ppk1 nucleic acid sequences were aligned in ARB along with previously described reference sequences from diverse WWTP (He et al., 2007). Maximum likelihood phylogenetic reconstruction on aligned sequences with positions of less than 50% maximum frequency masked (a total of 1052 positions were retained) was performed using RAxML (Stamatakis et al., 2005), specifying a GTR (general time reversible) model with 100 bootstraps calculated. To perform Unifrac clustering analysis (Lozupone & Knight, 2005), representative sequences from each environment were selected from each cluster in the maximum likelihood phylogeny with significant (greater than 60%) bootstrap support. Generally, sequences falling into one cluster diverged by less than 0.02 units of genetic distance. The number of sequences from each environment falling into each cluster was calculated, and this was used to create an environment file in which each representative sequence was listed, along with the number of additional sequences detected in each environment in the cluster being represented. The web-based Unifrac program was then used to calculate clustering by environments on the basis of shared branch length on the maximum likely phylogeny and the frequency of occurrence of sequences, as described previously (Lozupone & Knight, 2005; Lozupone et al., 2006; Lozupone et al., 2007). We used Arlequin (Schneider et al., 2000) to calculate average nucleotide diversity of aligned ppk1 sequences from each sample which were all trimmed to be of equal length (1057 bp). Phylogenetic species variability (PSV) and eveness (PSE) were calculated as described previously (Helmus et al., 2007a; Helmus et al., 2007b; Newton et al., 2007), except only 1000 iterations were calculated for each permutation in testing null hypotheses 1 and 2. Sequences not falling into one of the designated clusters of sequences shown in Figure 2 were not included in the PSV and PSE calculations. A total of 289 ppk1 gene sequences have been deposited to GenBank under the accession numbers EU432585-EU433291.
Supplementary Material
Acknowledgments
4SBP was the recipient of an NIH training grant which supported a summer internship to the Microbial Ecology Program at JGI. Special thanks to Jason Flowers, Shaomei He, and Victor Kunin for thoughtful discussions; Ryan Newton, Stuart Jones and Daniel Dalevi for help on phylogenetic analysis, Stephanie Schmidt for help with GPS plotting, and Jo Handelsman for support of this work. The assistance of personnel in the WWTPs included in this study, and at the East Bay Municipal Utility District is greatly appreciated. The work was conducted in part under the auspices of the US Department of Energy’s Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Livermore National Laboratory under Contract No. W-7405-Eng-48, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231 and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396. This project was also funded by the National Science Foundation (BES 0332136 to KDM).
References
- Acinas SG, Klepac-Ceraj V, Hunt DE, Pharino C, Ceraj I, Distel DL, Polz MF. Fine-scale phylogenetic architecture of a complex bacterial community. Nature. 2004;430:551–554. doi: 10.1038/nature02649. [DOI] [PubMed] [Google Scholar]
- Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl Environ Microbiol. 2006;72:5734–5741. doi: 10.1128/AEM.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JC, Tiedje JM. Biogeography and degree of endemicity of fluorescent Pseudomonas strains in soil. Appl Environ Microbiol. 2000;66:5448–5456. doi: 10.1128/aem.66.12.5448-5456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohan FM. Towards a conceptual and operational union of bacterial systematics, ecology, and evolution. Philosophical Transactions of the Royal Society B-Biological Sciences. 2006;361:1985–1996. doi: 10.1098/rstb.2006.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocetti GR, Hugenholtz P, Bond PL, Schuler A, Keller J, Jenkins D, Blackall LL. Identification of polyphosphate-accumulating organisms and design of 16S rRNA-directed probes for their detection and quantitation. Appl Environ Microbiol. 2000;66:1175–1182. doi: 10.1128/aem.66.3.1175-1182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis TP, Sloan WT, Scannell JW. Estimating prokaryotic diversity and its limits. Proc Natl Acad Sci U S A. 2002;99:10494–10499. doi: 10.1073/pnas.142680199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChaine EG, Bates AE, Shank TM, Cavanaugh CM. Off-axis symbiosis found: characterization and biogeography of bacterial symbionts of Bathymodiolus mussels from Lost City hydrothermal vents. Environ Microbiol. 2006;8:1902–1912. doi: 10.1111/j.1462-2920.2005.01113.x. [DOI] [PubMed] [Google Scholar]
- Foti M, Ma S, Sorokin DY, Rademaker JLW, Kuenen JG, Muyzer G. Genetic diversity and biogeography of haloalkaliphilic sulphur-oxidizing bacteria belonging to the genus Thioalkalivibrio. FEMS Microbiol Ecol. 2006;56:95–101. doi: 10.1111/j.1574-6941.2006.00068.x. [DOI] [PubMed] [Google Scholar]
- Garcia Martin H, Ivanova N, Kunin V, Warnecke F, Barry KW, McHardy AC, et al. Metagenomic analysis of two enhanced biological phosphorus removal (EBPR) sludge communities. Nat Biotech. 2006;24:1263–1269. doi: 10.1038/nbt1247. [DOI] [PubMed] [Google Scholar]
- Glaeser J, Overmann J. Biogeography, evolution, and diversity of epibionts in phototrophic consortia. Appl Environ Microbiol. 2004;70:4821–4830. doi: 10.1128/AEM.70.8.4821-4830.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray ND, Brown A, Nelson DR, Pickup RW, Rowan AK, Head IM. The biogeographical distribution of closely related freshwater sediment bacteria is determined by environmental selection. ISME J. 2007;1:596–605. doi: 10.1038/ismej.2007.74. [DOI] [PubMed] [Google Scholar]
- He S, Gall D, McMahon K. Candidatus Accumulibacter” population structure in enhanced biological phosphorus removal sludges as revealed by polyphosphate kinase genes. Appl Environ Microbiol. 2007;73:5865–5874. doi: 10.1128/AEM.01207-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Gu AZ, McMahon KD. Progress toward understanding the distribution of Accumulibacter among full-scale enhanced biological phosphorus removal systems. Microb Ecol. 2008;55:229–236. doi: 10.1007/s00248-007-9270-x. [DOI] [PubMed] [Google Scholar]
- Helmus MR, Bland TJ, Williams CK, Ives AR. Phylogenetic measures of biodiversity. Am Nat. 2007a;169:E68–E83. doi: 10.1086/511334. [DOI] [PubMed] [Google Scholar]
- Helmus MR, Savage K, Diebel MW, Maxted JT, Ives AR. Separating the determinants of phylogenetic community structure. Ecol Lett. 2007b;10:917–925. doi: 10.1111/j.1461-0248.2007.01083.x. [DOI] [PubMed] [Google Scholar]
- Hesselmann RP, Werlen C, Hahn D, van der Meer JR, Zehnder AJ. Enrichment, phylogenetic analysis and detection of a bacterium that performs enhanced biological phosphate removal in activated sludge. Syst Appl Microbiol. 1999;22:454–465. doi: 10.1016/S0723-2020(99)80055-1. [DOI] [PubMed] [Google Scholar]
- Horner-Devine MC, Bohannan BJM. Phylogenetic clustering and overdispersion in bacterial communities. Ecology. 2006;87:S100–S108. doi: 10.1890/0012-9658(2006)87[100:pcaoib]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Huber T, Faulkner G, Hugenholtz P. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics. 2004;20:2317–2319. doi: 10.1093/bioinformatics/bth226. [DOI] [PubMed] [Google Scholar]
- Hupfer M, Gloess S, Grossart HP. Polyphosphate-accumulating microorganisms in aquatic sediments. Aquat Microb Ecol. 2007;47:299–311. [Google Scholar]
- Klepac-Ceraj V, Bahr M, Crump BC, Teske AP, Hobbie JE, Polz MF. High overall diversity and dominance of microdiverse relationships in salt marsh sulphate-reducing bacteria. Environ Microbiol. 2004;6:686–698. doi: 10.1111/j.1462-2920.2004.00600.x. [DOI] [PubMed] [Google Scholar]
- Kong Y, Nielsen JL, Nielsen PH. Identity and ecophysiology of uncultured actinobacterial polyphosphate-accumulating organisms in full-scale enhanced biological phosphorus removal plants. Appl Environ Microbiol. 2005;71:4076–4085. doi: 10.1128/AEM.71.7.4076-4085.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y, Nielsen J, Nielsen P. Microautoradiographic study of Rhodocyclus-related polyphosphate-accumulating bacteria in full-scale enhanced biological phosphorus removal plants. Appl Environ Microbiol. 2004;70:5383–5390. doi: 10.1128/AEM.70.9.5383-5390.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V, He S, Warnecke F, Peterson SB, Garcia Martin H, Haynes M, et al. A bacterial metapopulation adapts locally to phage predation despite global dispersal. Genome Res. 2008;18:293–297. doi: 10.1101/gr.6835308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, Hamady M, Knight R. UniFrac--an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics. 2006;7:371. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Oehmen A, Virdis B, Keller J, Yuan Z. Obtaining highly enriched cultures of Candidatus Accumulibacter phosphates through alternating carbon sources. Water Res. 2006;40:3838–3848. doi: 10.1016/j.watres.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Martin AP. Phylogenetic approaches for describing and comparing the diversity of microbial communities. Appl Environ Microbiol. 2002;68:3673–3682. doi: 10.1128/AEM.68.8.3673-3682.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL, et al. Microbial biogeography: putting microorganisms on the map. Nature Reviews Microbiology. 2006;4:102–112. doi: 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- McMahon K, Yilmaz S, He S, Gall D, Jenkins D, Keasling J. Polyphosphate kinase genes from full-scale activated sludge plants. Appl Microbiol Biotechnol. 2007;77:167–173. doi: 10.1007/s00253-007-1122-6. [DOI] [PubMed] [Google Scholar]
- Murray RGE, Stackebrandt E. Taxonomic Note: implementation of the provisional status Candidatus for incompletely described procaryotes. Int J Syst Bacteriol. 1995;45:186–187. doi: 10.1099/00207713-45-1-186. [DOI] [PubMed] [Google Scholar]
- Neethling JB, Bakke B, Benisch M, Gu AZ, Stephens HM. Water Environment Research Foundation. 2005. Factors influencing the reliability of enhanced biological phosphorus removal. Report number 01CTS3. [Google Scholar]
- Newton RJ, Jones SE, Helmus MR, McMahon KD. Phylogenetic ecology of the freshwater Actinobacteria acI Lineage. Appl Environ Microbiol. 2007;73:7169–7176. doi: 10.1128/AEM.00794-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oehmen A, Lemos PC, Carvalho G, Yuan Z, Keller J, Blackall LL, Reis MA. Advances in enhanced biological phosphorus removal: from micro to macro scale. Water Res. 2007;41:2271–2300. doi: 10.1016/j.watres.2007.02.030. [DOI] [PubMed] [Google Scholar]
- Papke RT, Ramsing NB, Bateson MM, Ward DM. Geographical isolation in hot spring cyanobacteria. Environ Microbiol. 2003;5:650–659. doi: 10.1046/j.1462-2920.2003.00460.x. [DOI] [PubMed] [Google Scholar]
- Prosser JI, Bohannan BJ, Curtis TP, Ellis RJ, Firestone MK, Freckleton RP, et al. The role of ecological theory in microbial ecology. Nat Rev Microbiol. 2007;5:384–392. doi: 10.1038/nrmicro1643. [DOI] [PubMed] [Google Scholar]
- Schneider S, Roessli D, Excoffier L. Arlequin: A software for population genetics data analysis. Ver 2.000. Genetics and Biometry Lab, Dept. of Anthropology, University of Geneva; 2000. [Google Scholar]
- Stamatakis A, Ludwig T, Meier H. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 2005;21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- Tilman D. Niche tradeoffs, neutrality, and community structure: a stochastic theory of resource competition, invasion, and community assembly. Proc Natl Acad Sci U S A. 2004;101:10854–10861. doi: 10.1073/pnas.0403458101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb CO, Ackerly DD, McPeek MA, Donoghue MJ. Phylogenies and community ecology. Annu Rev Ecol Syst. 2002;33:475–505. [Google Scholar]
- Whitaker RJ, Grogan DW, Taylor JW. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science. 2003;301:976–978. doi: 10.1126/science.1086909. [DOI] [PubMed] [Google Scholar]
- Zilles JL, Hung CH, Noguera DR. Presence of Rhodocyclus in a full-scale wastewater treatment plant and their participation in enhanced biological phosphorus removal. Water Sci Technol. 2002a;46:123–128. [PubMed] [Google Scholar]
- Zilles JL, Peccia J, Kim MW, Hung CH, Noguera DR. Involvement of Rhodocyclus-related organisms in phosphorus removal in full-scale wastewater treatment plants. Appl Environ Microbiol. 2002b;68:2763–2769. doi: 10.1128/AEM.68.6.2763-2769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.