

SUMMARY
Synapses undergo slow homeostatic adaptation when neuronal activity is chronically perturbed. Such adaptation is well-studied in cultured neurons, but not in more physiological networks where distinct synaptic circuits are preserved. We characterized adaptive modifications at three sets of excitatory synapses in organotypic hippocampal slices whose neuronal firing had been abolished by three to four days of exposure to tetrodotoxin. Adaptation to inactivity was strikingly synapse-specific. Hippocampal throughput synapses, dentate-to-CA3 and CA3-to-CA1, were upregulated, conforming to prevailing notions about homeostatic gain control to avoid extreme limits of neuronal firing. However, chronic inactivity decreased mini frequency at CA3-to-CA3 synapses, which were isolated either pharmacologically or surgically. This downregulation of recurrent synapses was opposite to that expected for conventional homeostasis, thus seeming to antagonize gain control. However, the changes at recurrent synapses contributed to an inactivity-induced shortening of reverberatory bursts generated by feedback excitation among CA3 pyramids, safeguarding the network from possible runaway excitation. Thus, synapse-specific adaptations of synaptic weight not only contributed to homeostatic gain control but also dampened reverberant, epileptogenic network activity.
INTRODUCTION
Neuronal circuits in the brain contain different kinds of synapses, arranged in a hierarchy of stages. In contrast to the extensive description of Hebbian plasticity at multiple levels, little is known about how various stages adapt to chronic changes in activity. The prevailing picture of such adaptation focuses on homeostasis of a single type of neuron, not diverse members of a multi-stage network. Adjustments in synaptic input strength and intrinsic excitability are seen as keeping neuronal firing away from upper and lower limits, within a range useful for information flow (Burrone and Murthy, 2003; Davis, 2006; Desai et al., 1999; Liu and Tsien, 1995; Turrigiano and Nelson, 2004). For example, excitatory synaptic weights are generally enhanced in response to chronic inactivity. Synaptic homeostasis has been framed as a process of uniform scaling that preserves information stored as relative synaptic weights by multiplying all weights to the same degree (Turrigiano et al., 1998).
A simple extrapolation of concepts derived from single neurons to neuronal circuits of realistic complexity faces potential difficulties. Experimental findings at odds with uniform scaling have been obtained even among purely excitatory synapses onto individual neurons (Moulder et al., 2006; Thiagarajan et al., 2005): regulation of synaptic weight can be nonuniform, affecting some synapses far more than others. Another concern arises from consideration of feedback connections made by recurrent collaterals (e.g., synapses between hippocampal CA3 pyramids): their homeostatic upregulation might have different outcomes from that of feedforward synapses (e.g., dentate gyrus to CA3 in hippocampus). In contrast to information processing in purely feedforward circuits, neuronal activity in feedback circuits is more prone to reverberating transmission and runaway excitation (Furshpan and Potter, 1989; Rutecki et al., 1985; Schwartzkroin and Prince, 1978; Wong et al., 1986). In this setting, homeostatic boosting of synaptic gain may run the risk of runaway excitation (Beggs and Plenz, 2003; Houweling et al., 2005). Indeed, chronically lowered neuronal activity can induce epileptiform activity in recurrent circuits such as hippocampus and neocortex (Echlin and Battista, 1963; McKinney et al., 1997), possibly because of “homeostatic” strengthening of synaptic excitation (Houweling et al., 2005; Trasande and Ramirez, 2007).
In sum, previous work suggests that inactivity-induced synaptic adaptation might lead to sharply divergent outcomes: an advantageous preservation of feedforward signaling but an inappropriate promotion of seizures. This led us to probe the adaptive responses of particular types of synapses and their consequences for the neuronal circuit as a whole. We used organotypic hippocampal cultures because they retain realistic circuits (albeit with more exuberant connectivity than in vivo) (Zimmer and Gahwiler, 1984), and are amenable to activity deprivation over days. Several studies have already capitalized on some of these advantages for studies of inactivity-induced homeostatic plasticity (Aptowicz et al., 2004; Bausch et al., 2006; Buckby et al., 2006; Karmarkar and Buonomano, 2006; Trasande and Ramirez, 2007; Tyler and Pozzo-Miller, 2003). However, it remains unclear how different kinds of synapses adapt to chronic inactivity.
Here we show that synaptic homeostasis is heterogeneous, varying in both a target- and afferent-specific manner. While feedforward connections were upregulated by chronic inactivity as predicted by conventional homeostasis, the feedback recurrent synapses in CA3 showed a striking downregulation overall. Further experiments revealed that this unusual synaptic adaptation promoted network stability by curtailing reverberatory firing, whereas conventional homeostasis maintained the gain of throughput signaling.
RESULTS
Differences in synaptic homeostasis between hippocampal CA1 and CA3
To study adaptation of hippocampal neurons to inactivity, we chronically blocked neuronal firing by treating rat organotypic slice cultures with 1 µM tetrodotoxin (TTX) for 3–4 days. We then recorded miniature excitatory postsynaptic currents (mEPSCs or minis) to assess changes in synaptic events in activity-deprived neurons. Among the many types of synapses (Figure 1A), we focused first on synapses onto CA1 pyramidal cells, predominantly Schaffer collateral inputs. Activity deprivation increased the mean amplitude of mEPSC from 14.2±0.85 pA (n=7) in untreated slices to 17.4±0.77 pA (n=7) in TTX-treated slices [p<10−9, Kolmogorov-Smirnov (K–S) test; Figures 1B and 1C]. The increase in mEPSC amplitude was a conventionally expected outcome of activity deprivation, consistent with previous findings in CA1 neurons in slice cultures (Tyler and Pozzo-Miller, 2003) and earlier data in neuronal cultures (Ju et al., 2004; O'Brien et al., 1998; Sutton et al., 2006; Turrigiano et al., 1998). Chronic inactivity left the distribution of inter-mEPSC intervals unchanged (p>0.2, K–S test; Figure 1D). The mean frequency of mEPSCs was 0.65±0.11 Hz (n=7) in untreated slices and 0.62±0.15 Hz (n=7) in TTX-treated slices.
Figure 1. Adaptation of mEPSCs onto CA1 pyramidal cells following chronic exposure to TTX.

(A) Hippocampal excitatory networks. CA3 pyramidal cells receive synaptic inputs from dentate gyrus (DG) via mossy fibers (M.F.) and from other CA3 cells via recurrent collaterals (R.C.), and make synapses onto CA1 pyramids via Schaffer collaterals (S.C.). DCG-IV, an mGluR2 agonist, selectively blocks the mossy fiber transmission. Dotted line, st. pyramidale or granulosum; triangles, pyramidal or dentate granule cells.
(B) Voltage clamp recordings of mEPSCs from CA1 pyramidal cells in hippocampal slice cultures that were either untreated or treated with 1 µM TTX for 3–4 days. Holding potential, −70 mV. Right, ensemble-averaged traces of mEPSCs.
(C) Cumulative histogram of mEPSC amplitude. n=7 each for untreated or TTX-treated group. *, p<10−9, K–S test. Dashed line, TTX-treated cells.
(D) Cumulative histogram of inter-mEPSC interval. p>0.1, K–S test.
We asked if other types of synapses in the same preparation express a similar homeostasis, turning next to CA3 pyramidal cells. These neurons receive two different synaptic inputs (Figure 1A): mossy fiber inputs from dentate granule cells onto proximal dendrite [stratum (st.) lucidum], and recurrent collateral inputs from other CA3 pyramidals at distal dendrites (st. radiatum). Minis recorded from a CA3 pyramidal cell could result from either of these inputs. In TTX-treated CA3 pyramids, neither mean amplitude nor mean frequency of mEPSCs was altered (Figures 2A–2C). The mean mEPSC amplitude was 17.0±1.5 pA in untreated cells (n=8) and 16.1±0.94 pA in TTX-treated cells (n=8)(p>0.1, K–S test; Figure 2B). The mean frequencies for untreated and TTX-treated cells were 1.43±0.41 Hz (n=8) and 1.47±0.44 Hz (n=8) respectively (p>0.1, K–S test on inter-mEPSC intervals; Figure 2C). At first glance, it seemed that CA3 pyramidal cells simply lacked the ability to undergo homeostatic adaptation. However, a close examination of the mEPSC waveform revealed a significant change in their decay kinetics. Chronic inactivity made mEPSCs decay slightly faster: mean 90-10% decay time was 11.64±0.30 ms (n=8) in untreated cells and 10.06±0.40 ms (n=8) in TTX-treated cell (p<0.01, t-test). An acceleration of mini decay kinetics is one of the effects of chronic inactivity in hippocampal CA3-CA1 cultures (Thiagarajan et al., 2005). Thus, our data suggested that CA3 neurons had responded somehow to TTX treatment, despite the apparent lack of change in mini amplitude and frequency.
Figure 2. Adaptation of mEPSCs in CA3 following chronic TTX.

(A) Voltage clamp recordings of mEPSCs from CA3 pyramidal cells. Holding potential, −70 mV.
(B–C) Cumulative histogram of mEPSC amplitude (B) or inter-mEPSC interval (C). n=8 each for untreated or TTX-treated group. p>0.1, K–S test for both (B) and (C).
(D) mEPSC recordings at −70 mV in the acute presence of 1 µM DCG-IV in the bath solution. Dotted line, TTX-treated.
(E–F) Cumulative histograms of mEPSC amplitude (E) or inter-mEPSC interval (F) obtained with DCG-IV in the bath. n=6 each for untreated and TTX-treated groups. *, p<10−4, K–S test.
Differential adaptations of recurrent and mossy fiber synapses dissected with DCG-IV
Because the CA3 mEPSCs represent the aggregate activity of both mossy fiber and recurrent collateral synapses, it seemed possible that the individual populations of synapses were capable of adaptation. The contributions of the different types of synapses can be dissected by stimulation of type 2 metabotropic glutamate receptors (mGluR2), which are specifically expressed on mossy fiber terminals and not on recurrent collateral terminals (Petralia et al., 1996; Shigemoto et al., 1997). Agonists of mGluR2 selectively block mossy fiber transmission while sparing synaptic input from recurrent axons (Kakegawa et al., 2004; Kamiya et al., 1996; Yoshino et al., 1996). mGluR2 agonists such as DCG-IV suppress evoked EPSCs (eEPSCs) and mEPSCs, not only by reducing Ca2+ entry, but also by inhibiting exocytotic machinery (Kamiya and Ozawa, 1999; Scanziani et al., 1995). Accordingly, we recorded mEPSCs from CA3 neurons in the presence of 1 µM DCG-IV to isolate the contribution of recurrent synapses (Figure 2D). Mean mEPSC amplitude was still no different between untreated and TTX-treated cells (Figure 2E): 16.4±1.4 pA (n=6) for untreated cells and 16.3±1.5 pA (n=6) for TTX-treated cells (p>0.1, K–S test). Likewise, mini decay time was not changed (p>0.3, t-test). In contrast, mEPSC frequency in DCG-IV was significantly reduced by chronic inactivity, from 1.09±0.39 Hz (n=6) in untreated cells to 0.84±0.30 Hz (n=6) in TTX-treated cells (p<10−4, K–S test; Figure 2F). The diminished mEPSC frequency of recurrent synapses was opposite in direction to conventional homeostasis. In principle, the lowered mini frequency could arise from an attenuation of presynaptic release probability (Pr) across all synapses or a decreased number of active synapses.
In light of the reduction of the mEPSC frequency of recurrent synapses, the lack of overall change in the aggregate mEPSC frequency implied that the mEPSC frequency of mossy fibers actually increased but was masked when studied together with the recurrent collaterals. Such an alteration in mossy fiber input would constitute a typical form of homeostasis whereby synaptic strength is augmented in response to inactivity.
Changes in short-term plasticity of evoked EPSCs
The changes in mEPSC suggested a closer look at presynaptic properties might have been altered by chronic inactivity. We measured short-term plasticity of eEPSC to test for changes in Ca2+-dependent release not necessarily apparent from mEPSCs. To minimize polysynaptic transmission, AMPA receptor function was attenuated with a low concentration of NBQX (0.3 µM). St. lucidum was stimulated five times at 20 Hz in the presence and absence of DCG-IV (2 µM), and the mossy fiber EPSC was estimated by subtracting the post-DCG-IV EPSC from the pre-DCG-IV EPSC (Figures 3A–3C). Compared to a mild synaptic depression in untreated controls, TTX-treated cells showed pronounced depression with repeated stimulation, consistent with the idea of presynaptic modification. The accentuated depression indicated an increase in Pr, in line with an elevated mEPSC frequency inferred at the same synapses, in accord with a conventional synaptic homeostasis.
Figure 3. Changes in short-term plasticity of eEPSC by chronic TTX.

(A) Schematic diagram of recording and stimulation for mossy fiber EPSC.
(B) Short-term plasticity of mossy fiber synapses. EPSCs were evoked by stimulating st. lucidum (five times at 20 Hz) in the presence of gabazine (10 µM), D-AP5 (50 µM) and low dose of NBQX (0.3 µM). Mossy fiber EPSCs were measured by subtracting post-DCG-IV (2 µM) current (dotted line) from pre-DCG-IV current (thin solid line). Subtracted traces are shown as thick solid lines (subtr.). Scale bars, 50 pA and 50 ms.
(C) Group data of short-term plasticity at mossy fiber synapse shows larger depression in TTX-treated cells (n=5) than in untreated cells (n=4). *, p<0.01, Bonferroni t-test after two-way ANOVA.
(D) Diagram of recording and stimulation for recurrent synapse EPSC.
(E) EPSCs from recurrent synapses were recorded in 2 µM DCG-IV. Scale bars, 30 pA and 50 ms.
(F) Averaged short-term plasticity at recurrent synapses. Depression over stimulus is larger in TTX-treated cells (n=8) than in control (n=6). *, p<0.05, Bonferroni t-test after two-way ANOVA. For a given condition in a cell, 19–80 traces were averaged.
EPSCs at recurrent synapses were isolated by stimulating the border of st. oriens and st. pyramidale with mossy fiber transmission blocked with DCG-IV (Figure 3D). In untreated slices, recurrent EPSCs did not depress with repeated stimulation (5× at 20 Hz), but rather facilitated (Figures 3E and 3F), whereas TTX-treated cells displayed depression with repeated stimulation. This result supported the notion that presynaptic function had also been altered at recurrent synapses. However, the heightened depression at this synapse implied an increase in Pr of the active nerve terminals. At a first glance, the apparent increase in Pr seemed at odds with the overall decrease in mini frequency. However, the disparate effects could be reconciled if mini frequency were influenced by the fraction of active synaptic inputs whereas short-term plasticity were not–chronic TTX treatment might have reduced the proportion of active recurrent synapses while also augmenting their Pr.
We performed additional experiments to confirm that TTX-treated recurrent synapses underwent an increase in Pr and a decrease in the fraction of active synapses (Supplemental Figure 1). First, we verified that paired-pulse modulation responded as expected to an agent known to lower Pr. Indeed, 2-chloroadenosine (2-CA, 5 µM), which inhibits presynaptic release (Thompson et al., 1992; Yoon and Rothman, 1991) clearly increased paired-pulse facilitation (PPF) to a degree (1.61±0.079, n=5) significantly higher than in the absence of 2-CA (1.22±0.11, n=6)(p<0.05, t-test). Having confirmed the inverse relationship between Pr and paired-pulse ratio (Thomson, 2000; Zucker, 1989), we proceeded to assay the effect of chronic TTX in the presence of 2-CA (Supplemental Figure 1C). In TTX-treated slices, the PPF ratio was 1.14±0.057 (n=5) in 2-CA, significantly less than in the untreated slices (p<0.05, Bonferroni t - test after two way ANOVA). This confirmed that chronic TTX blunted PPF, and thus increased Pr of active recurrent synapses.
The hypothesis that mEPSC frequency fell because of presynaptic silencing led to a testable prediction: the mini frequency decrease should disappear if the mini recordings were specifically oriented towards active terminals only. To accomplish this, we evoked EPSCs in the presence of Sr2+ (Supplemental Figures 1D–1G), adjusting stimulus strength to keep the eEPSCs within a limited range of amplitudes; accordingly, average EPSC amplitude was no different with or without TTX treatment (Supplemental Figure 1E; p>0.8, t-test). We then focused our analysis on the delayed shower of mEPSCs characteristic of recordings in Sr2+ (Abdul-Ghani et al., 1996; Bekkers and Clements, 1999), which arise from residual elevation of [Sr2+]i in the active terminals that generated the eEPSC. Compared to control (n=5), those minis were not larger following TTX pretreatment (n=6)(p>0.02, K–S test), nor was the distribution of inter-mEPSC intervals any different (p>0.2, K–S test; Supplemental Figures 1F and 1G). Thus, the change in interevent interval registered across all the recurrent presynaptic terminals (Figure 2F) vanished when sampling was restricted to active recurrent synapses. This result implied that the lowered mini frequency in Figure 2F could be due to a decreased number of active synapses, in line with our prediction.
At first glance, one might even expect to see an increased frequency of asynchronous EPSCs in Sr2+, given that Pr appears enhanced after chronic TTX as probed by short-term plasticity. However, the frequency of asynchronous EPSCs was determined by two factors, the number of active synapses and Pr at those active synapses. Thus, a decrease in the number of active synapses that are stimulated (N) would have offset the increase in Pr, resulting in no detectable change in mini frequency. Our adjustment of stimulation strength to match average eEPSC sizes (<EPSC>), with no change in quantal size (q), was likely to have contributed to holding N×Pr (=<EPSC>/q) fixed.
Adaptations of recurrent synapses isolated by anatomical exclusion of mossy fibers
To complement the pharmacological approach to isolating recurrent synapses, we used anatomical isolation by removing dentate gyrus at 1 DIV (Figure 4A). In such “dentateless” slices, the CA3 area is devoid of mossy fibers, and contains only recurrent synapses (Dailey et al., 1994). To confirm the absence of mossy fibers, we tested the effect of DCG-IV on mEPSC frequency (Figure 4B). DCG-IV (1 µM) reduced mEPSC frequency in intact slices by 37.8±4.8% (n=5; p<0.05, paired t-test), demonstrating a substantial mossy fiber input, but had little effect on mini frequency in dentateless slices (−2.1±6.2%; n=6; p>0.5, paired t-test). Thus, microsurgery effectively eliminated the functional contribution of mossy fiber synapses.
Figure 4. Inactivity-induced adaptation of CA3 mEPSC in slices devoid of dentate gyrus.

(A) Diagram of a dentateless slice. Dentate gyrus was surgically removed at 1 DIV. mEPSCs were recorded from CA3 pyramidal cells.
(B) Suppression of mEPSC frequency by 1 µM DCG-IV in intact (Intact; n=5) and dentateless slices (No DG; n=6). *, p<0.05, paired t-test between pre- and post-DCG-IV.
(C) mEPSC recording at −70 mV from an untreated (solid line) and a TTX-treated cell (dotted line).
(D–E) Cumulative histogram of mEPSC amplitude (D) or inter-mEPSC interval (E). n=6 each for untreated or TTX-treated group. *, p<10−8, K–S test.
(F) Possible mechanism of changes in mEPSC frequency at recurrent synapses. The less frequent minis after chronic TTX might be due to silencing a part of synapses. Dotted lines, inactive synapses.
(G) Adaptation of eEPSC can be explained by an increase in Pr at each synapse, depicted schematically as larger synapses. Stimulating electrodes (right) stimulate four (untreated) or two (TTX-treated) synapses. In Sr2+ (see text), the higher Pr after TTX treatment would tend to increase asynchronous EPSC frequency, but is offset by the reduced density of active synapses, resulting in no detectable change in frequency.
The effects of chronic TTX were retested in dentateless slices. Minis were less frequent in TTX-treated CA3 neurons than in their untreated counterparts: 1.00±0.17 Hz (n=6) in untreated slices, 0.62±0.11 Hz (n=6) in TTX-treated slices (p<10−8, K–S test; Figures 4C and 4E). mEPSC amplitude, however, was unchanged: 18.6±1.4 pA in untreated slices, 18.7±0.7 pA in TTX-treated slices (p>0.02, K–S test; Figure 4D). This finding was in close accord with results obtained by blocking mossy fiber inputs with DCG-IV in anatomically intact slices. Likewise, the mean decay time was again not significantly altered (p>0.1, t-test).
The excellent agreement between anatomical and pharmacological approaches to isolating recurrent collateral synapses reinforced the conclusion that the overall impact of TTX treatment was a downregulation of their collective synaptic efficacy. A plausible interpretation (Figures 4F and 4G) is that activity deprivation leads to the silencing of a substantial fraction of the recurrent synapses, but also augments Pr of the synapses that remain active. The net effect is an overall downregulation (Figure 2 and Figure 4), but the enhanced Pr causes a loss of paired-pulse facilitation of the eEPSC (Figure 3).
Changes in burst firing of hippocampal networks after chronic inactivity
Next, we asked how the disparate adaptations to chronic inactivity influenced the integrated activity of the neuronal network overall. The induction of the adaptation was performed under conditions that suppressed evoked transmission at both excitatory and inhibitory synapses. However, to study the expression of adaptation, we deliberately focused on excitatory inputs by recording EPSCs and network activity with GABAergic inputs blocked. In the presence of gabazine (10 µM), area CA3 undergoes in vitro seizures (Rutecki et al., 1985; Schwartzkroin and Prince, 1978; Wong et al., 1986), appearing as spontaneous bursts of action potentials and prolonged depolarizations in individual CA3 pyramidal neurons (Figure 5). This provided a simple framework for studying altered excitatory connections and their impact on circuit behavior under circumstances that were internally consistent. In TTX-treated slices, the frequency of spontaneous bursts was markedly enhanced (Figures 5A and 5B), in accord with in vivo infusion experiments (Galvan et al., 2000; Galvan et al., 2003). The mean burst frequency was 0.017±0.0037 Hz (n=16) in untreated slices, and 0.20±0.036 Hz (n=26) in TTX-treated slices, more than 10-fold higher (p<0.001, t-test). The increase in burst frequency conformed to general expectations for homeostatic upregulation, whereby upregulation of synaptic activity is expected after chronic inactivity.
Figure 5. Changes in CA3 burst activity in after chronic TTX.

(A) Current clamp recordings from CA3 pyramidal cells in 10 µM gabazine. Depolarizations in the traces are spontaneous bursts of action potentials. The resting membrane potentials in mV are indicated.
(B) Group data of burst frequency in TTX-treated (n=26) and untreated slices (n=16). *, p<0.001, t-test.
(C) Individual burst events on expanded scale. A burst was evoked by extracellular stimulation at the border of st. oriens and st. pyramidale in CA3.
(D) Mean width of individual bursts was shorter in TTX-treated slices (n=12) than in control (n=13). *, p<0.001, t-test.
Bursts arise from synchronized depolarization of many recurrently connected neurons and are sustained by ongoing synaptic excitation within the recurrent network (Miles and Wong, 1983; Miles et al., 1984; Scanziani et al., 1994; Swann et al., 1993; Traub et al., 1993). Accordingly, conventional homeostatic upregulation of recurrent synapses would have predicted a prolongation of bursts after chronic TTX. However, we observed the opposite change (Figures 5A, 5C and 5D). The duration of spontaneous bursts averaged 1.04 ± 0.11 s (n=5) in untreated slices and 0.57±0.11 s (n=12) in TTX-treated slices (p<0.01, t-test). Likewise, when bursts were evoked by extracellular stimulation to restrict excitation to a more precisely defined set of cells, the mean burst width averaged 1.12±0.10 s (n=13) for untreated cells and 0.44±0.053 s (n=12) for TTX-treated cells (p<0.001, t-test; Figures 5C and 5D). Thus, in both cases, activity deprivation caused a ~2-fold abbreviation of the bursts. The inactivity-induced shortening of burst width, against expectations for classical synaptic homeostasis, was all the more interesting because burst frequency was elevated in the same system, in line with the anticipated effect of a conventional homeostatic change. The combination of effects was reminiscent of the disparate changes in mEPSC frequency at recurrent and mossy fiber synapses, raising the question of whether the changes in bursting activity might be directly linked to the distinct synaptic adaptations at the two types of synapses.
Contributions of recurrent and mossy fiber synapses to the pattern of burst firing
First, we tested the effect of DCG-IV on the frequency of spontaneous bursts (Figure 6). In control slices, the burst frequency was not changed by acute application of 1 µM DCG-IV (Figures 6A and 6C), averaging 0.021±0.009 Hz before DCG-IV and 0.016±0.008 Hz after DCG-IV (n=6; p>0.1, paired t-test). In contrast, in TTX-treated cells, DCG-IV sharply reduced burst frequency from 0.30±0.055 Hz to 0.13±0.087 Hz (n=11; p<0.05, paired t-test). The eleven cells included two neurons which failed to respond to DCG-IV; if these were discounted, the DCG-IV-induced change in frequency was from 0.25±0.051 Hz to 0.0062±0.0045 Hz (n=9; p<0.005, paired t-test; Figure 6D). These results implied that the increased incidence of spontaneous bursts after chronic TTX was mostly due to increased mossy fiber transmission, consistent with changes in mEPSC frequency (Figure 2).
Figure 6. DCG-IV, an agonist of mGluR2, silenced the inactivity-induced spontaneous bursts, but did not change the burst width.

(A–B) Recording of spontaneous bursts from untreated (A) or TTX-treated (B) CA3 pyramid before and after 1 µM DCG-IV.
(C) In untreated slices (n=6), the burst frequency was not affected by DCG-IV (p>0.1, paired t-test).
(D) In most TTX-treated slices (n=9/11), the frequency of spontaneous bursts were suppressed by DCG-IV. *, p<0.005, paired t-test. Two DCG-IV resistant cells were excluded from this graph.
(E–F) Individual evoked burst events in untreated (E) or TTX-treated (F) slice before and after 1 µM DCG-IV.
(G) Seven untreated cells showed no effect of DCG-IV on the burst width (p>0.5, paired t-test).
(H) Burst widths were not changed by DCG-IV in eight TTX-treated cells (p>0.5, paired t-test).
We next asked whether changes in mossy fiber transmission also brought about the inactivity-induced decrease in burst width. Burst events arising from a defined set of cells were evoked by extracellular stimulation of CA3 cells near the recorded neuron. Unlike burst frequency, the burst widths were not affected by 1 µM DCG-IV either in untreated cells or in TTX-treated cells (Figures 6E–6H). In the untreated group, mean burst width was 1.07±0.18 s and 1.05±0.15 s before and after DCG-IV (n=7; p>0.5, paired t-test; Figures 6E and 6G). In the TTX-treated group, the mean burst width was 0.44±0.059 s and 0.44±0.047 s before and after DCG-IV (n=8; p>0.5, paired t-test; Figures 6F and 6H). Thus, the difference in burst width between untreated (n=7) and TTX-treated (n=8) cells persisted even in the presence of DCG-IV (p<0.005, t-test). Under these conditions, CA3 recurrent synapses were the only excitatory synapses to remain functional. Therefore, CA3-CA3 recurrent connections were sufficient to account for the shortening of burst width, without involvement of mossy fiber connections, supporting the idea that changes in synaptic properties at the recurrent synapses played an important role in altering burst duration.
If mossy fibers are responsible for the inactivity-induced elevation of burst frequency, the elevation should disappear in dentateless slices. In contrast, if the shortening of bursts is wholly dependent on recurrent synapses, such shortening should persist even with dentate removal. Indeed, gabazine-disinhibited dentateless slices failed to display increased burst frequency after chronic TTX (Figures 7A and 7B). Burst frequencies were 0.022±0.011 (n=7) in untreated cells and 0.025±0.014 (n=7) in TTX-treated cells (p>0.5, t-test). In contrast, burst width was significantly shorter in TTX-treated cells (0.53±0.14 s; n=6) than in untreated cells (1.01±0.093 s; n=6)(p<0.02, t-test; Figures 7C and 7D). Thus, both aspects of burst activity conformed to our predictions. The finding that TTX treatment induced shortening of burst width even in dentateless slices confirmed that it is the CA3-CA3 network itself which is dampened by inactivity. The adaptive changes in the CA3-CA3 circuitry appeared to be a pre-programmed feature of CA3 recurrent synapses, rather than a secondary compensation for increased mossy fiber input.
Figure 7. Similar effects of dentate removal and CaM kinase inhibition on inactivity-induced changes in burst firing of CA3 pyramidal cells.

(A–D) Removal of dentate specifically prevented inactivity-induced changes in burst frequency but not burst duration.
(A) Spontaneous bursts in 10 µM gabazine from CA3 pyramidal cells in dentateless slice cultures.
(B) Chronic TTX did not increase the burst frequency in dentateless slices (n=7 each group). p>0.5, t-test.
(C) Individual bursts on expanded scale. A burst was evoked by extracellular stimulation at the border of st. oriens and st. pyramidale.
(D) Chronic TTX shortened the mean burst width (n=6 each group). *, p<0.02, t-test.
(E–H) KN93, a CaMK inhibitor, prevented inactivity-induced increase in burst frequency, but spared inactivity-induced shortening of burst.
(E) The slices were incubated with TTX+vehicle (0.3% DMSO v/v) or TTX+KN93 (3 µM, in vehicle). Compared to vehicle control, the burst frequency is lower when the cells were treated with TTX+KN93.
(F) Group data of the effect of TTX+KN93. n=8 for TTX+vehicle and n=7 for TTX+KN93. *, p<0.05, t-test. For direct comparison, data from Figure 5B were replotted with dashed bars. Vehicle itself (TTX+Veh) slightly, but insignificantly (p>0.2, t-test), reduced TTX-induced bursting frequency when compared to vehicle-naive TTX group (TTX; right dashed bar). The TTX+KN93 group showed similar burst frequency to untreated group (left dashed bar) (p>0.5, t-test).
(G) The widths of individual burst were similar when cells were treated with either TTX+vehicle or TTX+KN93.
(H) Group data shows no difference between the two groups (filled bars; p>0.4, t-test). The burst width of each group was not significantly different from that of vehicle-naive TTX group (TTX; right dashed bar) (p>0.2, t-tests). When compared to untreated cells (left dashed bar), both TTX+vehicle and TTX+KN93 group had shorter burst width (p<0.001, t-tests). The dashed bars are re-plots of Figure 5D.
Involvement of Ca2+/calmodulin-dependent protein kinase in modulating specific aspects of burst activity
As a first step toward linking changes in burst activity to possible mechanisms, we tested the effects of KN93, an inhibitor of Ca2+/calmodulin-dependent protein kinase (CaMK). Previous work in cultured hippocampal neurons has shown that elevation of βCaMKII is a key step in the signaling cascade that links inactivity to upregulation of presynaptic activity (Thiagarajan et al., 2005; Thiagarajan et al., 2002). By this logic, the synapses which exhibited inactivity-induced increases in mini frequency—mossy fiber inputs—might rely on CaMK signaling. This led, in turn, to a testable prediction at the level of bursting activity: that inhibition of CaMK might interfere with the consequences of enhanced mossy fiber input (leaving open whether such inhibition might also interfere with outcomes of altered recurrent transmission). To test this hypothesis, we performed activity-deprivation experiments in the presence of KN93 (3 µM). As predicted for mossy fiber activity, KN93 blocked the increase in burst frequency by chronic TTX (n=7; Figures 7E and 7F), greatly reducing the burst frequency in comparison to vehicle control (0.03% DMSO+TTX; n=8)(p<0.05, t-test). In control experiments with KN92, a CaMK-inactive congener of KN93, there was no significant difference in burst frequency obtained with TTX+KN92 (n=8) or TTX+vehicle (p>0.3, t-test).
In contrast to the abolition of inactivity-induced bursting, the ability of chronic TTX to reduce burst width was fully preserved in KN93 (Figures 7G and 7H). There was no significant difference between the burst widths following pretreatment with TTX (n=12 from Figure 5D), TTX+vehicle (n=7) or TTX+KN93 (n=7)(p>0.2, t-tests after ANOVA). The implication is that regulations of burst frequency and width not only involve different synapses but may also rely on different molecular mechanisms, possibly CaMK at mossy fiber synapses, but not at recurrent collateral synapses. Thus, our findings with CaMK inhibition complemented the dentateless experiments. Modulation of burst frequency was eliminated by suppression of a biochemical signaling pathway or by removal of an anatomical pathway.
Changes in intrinsic excitability after chronic inactivity
Other factors besides changes in recurrent synapses were considered as possible contributory factors to the decreased burst width. First, frequent spontaneous bursts may deplete glutamate-containing vesicles in the terminals of CA3 recurrent synapses, thus resulting in shortening of bursts by activity-dependent synaptic depression (Staley et al., 1998). This possibility was rendered unlikely by our finding that greatly slowing the incidence of spontaneous bursts with DCG-IV (Figures 6B and 6D) did not affect the burst width (Figures 6F and 6H). In addition, individual bursts were always preceded by burst-free period lasting >3 s, sufficiently long to allow recovery of synaptic efficacy as monitored by restoration of burst width to its rested-state duration (Supplemental Figure 2A)(Staley et al., 1998). A second possibility was that the number of cells immediately recruited by the stimulation might be altered by inactivity, and that this might exert a secondary effect on burst width. To test this idea, we evoked bursts with widely varying stimulus intensities in continuous recordings from individual cells, but found no significant change in burst width (n=5; Supplemental Figures 2B and 2C). This was as expected if bursts can be triggered by direct stimulation of very few primary cells, possibly even one initiator neuron (Miles and Wong, 1983; Traub and Wong, 1982; Wong et al., 1984).
A third possibility was that the shorter bursts arose in part from changes in the firing patterns of CA3 cells, beyond the observed effects on presynaptic properties recurrent collaterals. Accordingly, we probed the intrinsic excitability of CA3 pyramidal cells in whole-cell recordings. Action potentials were elicited by injecting step currents ranging from +200 to +800 pA (Figure 8). With injection of +600 and +800 pA, fewer action potentials were generated in TTX-treated cells than in untreated cells (Figures 8A and 8B). Comparing untreated cells (n=11) with TTX-treated cells (n=12), the number of spikes fell from 25.9±1.5 to 14.9±1.5 for a +800 pA, 1.5 s stimulus, and from 18.0±1.4 to 11.5±1.1 for +600 pA (both p<0.001, Bonferroni t-test after two-way ANOVA). With weaker stimuli, the number of action potentials was not significantly different between the two groups. The reduced firing activity of TTX-treated CA3 cells (for strong depolarizations at least) may have contributed to the earlier termination of bursting activity in the recurrent network. Analysis of spike parameters (Supplemental Figure 3) revealed a significant decrease in the steepness of the action potential upstroke, possibly consistent with the reduced firing.
Figure 8. Intrinsic excitability of CA3 pyramidal cells (A–B) or dentate granule cells (C–D).

(A) Action potentials from an untreated or a TTX-treated cell in 50 µM D-AP5, 10 µM NBQX and 10 µM gabazine. Step currents were injected for 1.5 s varying from +200 pA to +800 pA in 200 pA increments. Only the recordings of +400 and +800 pA injection are shown.
(B) Group data of the number of action potentials during 1.5 s current injection. *, p<0.001, Bonferroni t-test between untreated (n=11) and TTX-treated (n=12) groups, after two-way ANOVA.
(C) Similar experiments as in (A) but from dentate granule cells. Only recordings of +200 and +600 pA injection are shown.
(D) Group data from dentate granule cells. *, p<0.05, Bonferroni t-test between untreated (n=6) and TTX-treated (n=5) groups, after two-way ANOVA.
(E) Changes in resting membrane potential after chronic TTX in CA3 pyramidal cells and dentate granule cells. Measured in AP5, NBQX and gabazine. In CA3, n=11 and 12 for untreated and TTX-treated cells, respectively, and in dentate gyrus, n=7 and 6 for untreated and TTX-treated cells, respectively. *, p<0.05, t-test.
We also looked for changes in the excitability of dentate granule cells. In this case, a more complex response to activity deprivation was observed, with changes in opposite directions at low and high current intensities. Injection of +200 pA generated significantly more spikes in activity-deprived cells (26.9±2.5; n=5) than in control (9.17±4.6; n=6)(Figures 8C and 8D). On the other hand, with +600 and +800 pA pulses, the TTX-treated neurons displayed significantly fewer spikes. The increased excitability during small depolarizations was more relevant to basal firing of spontaneous spikes, and was likely to have contributed to the stronger mossy fiber input and the increased burst frequency. We examined the repolarization of single spikes with an eye toward possible downregulation of K+ channel activity (Desai et al., 1999). If anything, repolarization in dentate gyrus neurons was slightly slowed following inactivity, but the changes failed to reach statistical significance (Supplemental Figures 3M–3O).
The striking differences in induced firing of CA3 pyramidal cells and dentate granule cells could not be attributed to a region-specific adaptation of the major ionic currents (INa and IK) that support the rising and falling phases of the action potential. The overall pattern of changes in spike parameters was very similar (Supplemental Figure 3). In contrast, resting membrane potentials underwent significant inactivity-induced changes that were region-specific and well-aligned with changes in firing pattern (Figure 8E). Resting potentials were measured in the absence of glutamatergic and GABAergic inputs (10 µM NBQX, 50 µM D-AP5 and 10 µM gabazine) during the baseline period before current injection (Figures 8A and 8C). Strikingly, TTX treatment caused oppositely directed changes in the resting potentials of CA3 pyramids and dentate granule cells. In CA3 pyramids, resting potential averaged −68.4±0.43 mV (n=11) in untreated neurons and −73.1±1.1 mV (n=12) in TTX-treated cells (p<0.0005, t-test; Figure 8E). In contrast, in dentate granule cells, the resting potential was quite negative in untreated neurons (Fricke and Prince, 1984), −81.6±2.5 mV (n=7), but became less negative in TTX-treated cells, −73.3±1.9 mV (n=6) (p<0.05, t-test). The polarity of the changes was entirely consistent with the overall changes in network activity following inactivity. Resting depolarization of dentate granule cells would favor enhanced synaptic input by the mossy fiber pathway, while the more negatively polarized CA3 neurons would be less inclined to display a long burst. Inactivity-dependent enlargement of mEPSCs onto dentate granule cells (Bausch et al., 2006) would also help strengthen mossy fiber input. Thus, the elevated rate of CA3 bursting may be supported by a combination of a more active dentate gyrus and a stronger connection from dentate gyrus to CA3.
Evaluating the impact of changes in intrinsic excitability of individual neurons
We previously established that burst frequency is mostly driven by mossy fiber inputs, not by spontaneous firing of CA3 pyramidal cells, because DCG-IV silenced the spontaneous bursts almost completely. This left open whether inactivity-dependent abbreviation of burst width, a feature resistant to DCG-IV, was governed by activity of recurrent synapses or by intrinsic electrical properties of the CA3 neuron itself. To distinguish between these possibilities, we tested how burst duration responded to manipulations of intrinsic voltage-dependent conductances or recurrent synaptic inputs.
To assess possible contributions of changes in intrinsic conductances, we applied current steps in the midst of burst firing (Figures 9A and 9B), reasoning that the impact of voltage-gated channels would be revealed by the current-induced voltage change. The holding current was stepped to negative (−600 to −1000 pA) or positive values (+800 to +1200 pA), 200 ms after the burst onset, causing changes in membrane potential spanning a ~75 mV range. Nonetheless, burst width, measured as the time elapsed from burst onset to the last trough, was not significantly affected (n=4; p>0.2, one-way repeated measures ANOVA). This excluded the notion that burst duration was predominated by voltage-gated channels in individual bursting cells. However, the possibility remained that voltage-gated channels in the surrounding population of presynaptic neurons exerted an influence that was conveyed synaptically.
Figure 9. Synaptic activity, and not voltage-gated activity intrinsic to the recorded cell, is an essential determinant of burst width in CA3.

(A) To displace the membrane potential, the holding current (dotted lines) was shifted to negative (−600 to −1000 pA) or positive (+800 to +1200 pA) values in the middle of a burst firing (200 ms from the onset). The traces are from a single cell.
(B) At the last trough of oscillatory bursting, membrane potential and time from burst onset were measured to examine their relationships. Burst width (i.e., time from burst onset to the last trough) was not significantly affected by changes in membrane potential (n=4; p>0.2, one-way repeated measures ANOVA). Each connected line represents one cell. Circle, no current; triangle, negative current; square, positive current.
(C) Low dose (0.2 µM) of NBQX shortened burst width in a reversible manner. The traces are from one cell.
(D) The mean reduction of burst width is 65.5±7.9% (n=4; **, p<0.01, paired t-test). In three cells, reversibility of NBQX was examined (94.6±10.3% of pre-NBQX value; p>0.5, t-test between Control and Wash). The recovery of burst width was significant (*, p<0.05, t-test between NBQX and Wash). Each connected line represents one cell.
To interfere with recurrent synaptic inputs, we applied a low dose of NBQX (0.2 µM) while evoking burst firing (Figure 9C and 9D). As reported (Scanziani et al., 1994), attenuation of AMPA receptor activity reduced burst width, consistent with the notion that burst duration is strongly influenced by synaptic transmission, that spreads the excitation from primary neurons to follower cells (Miles et al., 1984; Staley et al., 1998; Swann et al., 1993). Relative to its pre-NBQX value, burst width fell to 65.5±7.9% in low NBQX (n=4; p<0.01, paired t-test), and recovered to 94.6±10.3% after NBQX removal (p>0.5, paired t-test) (n=3 out of 3 attempts). These results show that the synaptic activity, rather than activity of voltage-gated ion channels in the recorded cell, is a major determinant of burst width.
DISCUSSION
Using anatomical and pharmacological distinctions to isolate specific circuit components in hippocampal slice cultures, we found three distinct patterns of response to activity blockade: (1) an augmented mini amplitude at Schaffer collateral synapses, (2) an unexpected fall in mini frequency at recurrent collateral synapses, and (3) an inferred increase in mini frequency at mossy fiber synapses (Table 1). The synapse-specific adaptations in area CA3 helped reconcile two seemingly contradictory objectives, controlling throughput gain but opposing network reverberation. Gain control could be accomplished by inactivity-driven upregulation of mossy fiber inputs, while dampening of network reverberation could be achieved through regulation of CA3 recurrent synapses. Indeed, recordings of CA3 burst firing highlighted the disparate impact of the synapse-specific adaptations. Inactivity sharply enhanced epileptoform burst frequency, due to increased feedforward synaptic input, but also shortened burst duration, by attenuating recurrent synaptic activity.
Table 1. Summary of synapse-specific adaptations to chronic inactivity.
Experimental observations for mEPSC amplitude and decay are described in the text. Decay of mEPSC was documented as 90-10% decay time.
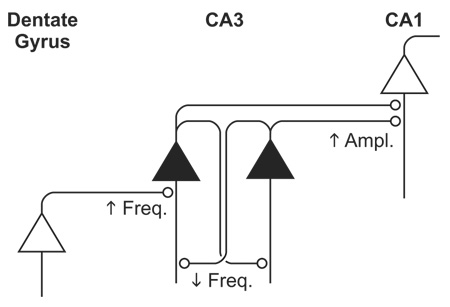 | |||
|---|---|---|---|
| Mossy Fiber (DG-CA3) | Recurrent Collateral (CA3-CA3) | Schaffer Collateral (CA1-CA1) | |
| mEPSC Amplitude | - | - | ↑ |
| mEPSC Frequency | ↑ | ↓ | - |
| mEPSC Decay | Faster* | - | - |
| eEPSC STP | Depress | Depress | N/E |
| Burst Width | - | ↓ | N/A |
| Burst Frequency | ↑ | - | N/A |
In CA3 recordings without DCG-IV, mEPSC decay time was significantly briefer in TTX-treated slices (n=8) than in untreated ones (n=8) (p<0.01, t-test). Because no such difference was observed in DCG-IV (p>0.3, t-test), we inferred that it was the DCG-IV-sensitive mossy fiber mEPSCs that had undergone a faster decay, not the recurrent collateral mEPSCs. STP, short-term plasticity. N/E, not examined. N/A, not applicable.
Our experiments utilized slice cultures at a late stage (21–25 DIV) when they are mature with respect to synaptic density and function (Buchs et al., 1993; De Simoni et al., 2003; Muller et al., 1993a; Robain et al., 1994). We checked for systematic changes in synaptic properties over the 21–25 DIV period, but found no significant trend in either mini frequency or amplitude for any recording condition (p>0.3 in every case, linear regression). Thus, our findings are most relevant to adaptation of developmentally mature circuits rather than early circuit development (Buckby et al., 2006; Galvan et al., 2003; Lauri et al., 2003; Maffei et al., 2006; Maffei et al., 2004). This sets our data apart from evidence that responses to inactivity vary widely depending on the maturity of the neural network (Burrone et al., 2002; Hartman et al., 2006; Karmarkar and Buonomano, 2006; Kirov et al., 2004).
Afferent-specific and target-specific adaptation to inactivity
Comparison of mossy fiber and recurrent synapses onto the same CA3 pyramid indicated oppositely directed changes in mEPSC frequency. Thus, adaptation to inactivity can be afferent-specific. Furthermore, comparison of the various outputs of CA3 neurons demonstrated that inactivity-induced changes in minis can be target-specific: the change in mEPSC amplitude was expressed specifically at Schaffer collateral synapses, not at recurrent synapses. Thus, the machinery for AMPA receptor scaling seems not to be recruited for adaptation to inactivity even though it is clearly present at CA3-CA3 synapses (Montgomery et al., 2001). In contrast, the alteration in mEPSC frequency was specific to recurrent synapses, not CA3-CA1 synapses. These manifestations of target-specificity are unprecedented for synaptic adaptation to inactivity in the hippocampus. In visual cortex, layer IV pyramidal neurons also display target specificity in response to activity deprivation, strengthening their outputs to inhibitory neurons but not to other pyramids (Maffei et al., 2006). Possible mechanisms include target-specific differences in post-to-presynaptic signaling, or differences in the presynaptic terminals themselves. Inactivity can change levels of presynaptic molecules such as synapsin 1 and vesicular glutamate transporter 1 (Buckby et al., 2006) but how this happens is unclear. Hints of possible mechanisms can be gleaned from our recordings of spontaneous mEPSCs, eEPSCs, and evoked asynchronous mEPSCs. Increased Pr at active recurrent synapses, indicated by accentuated synaptic depression, was seen in combination with an overriding decrease in the proportion of active synapses. The data suggest the participation of all-or-none synaptic silencing (Moulder et al., 2004), and call for further study at the single synapse level.
Adaptation to chronic inactivity and hyperactivity: heterogeneous mechanisms?
Since the adaptation of recurrent synapses was a mixture of increased Pr and decreased synaptic density, it is of interest to ask if those synapses are reciprocally adjusted by chronic hyperactivity. Prolonged elevation of activity by GABAA blockers in organotypic hippocampal cultures leads to a set of downregulatory changes that include spine loss (Drakew et al., 1996; Muller, 1993; Muller et al., 1993b), decreased levels of NR2A, NR2B, GluR1, GluR2 (Gerfin-Moser et al., 1995), smaller evoked excitatory postsynaptic potentials upon stimulation of dentate gyrus (Muller et al., 1993b), and shorter evoked bursts (Muller, 1993). Thus, opposing interventions (inactivity and hyperactivity) do not produce mirror-image responses, as expected for a single unifying mechanism. The responses to hyperactivity could be classified as conventional homeostasis, but this cannot be so for the response to inactivity described here. The absence of a unifying explanation leaves room for wholly different mechanisms of expression. What the adaptations clearly share is the ability to promote network stability.
Functional significance
In a purely feedforward network, synaptic strengthening by chronic inactivity makes good sense functionally. If synapses were scaled uniformly during homeostatic adjustment, and synaptic gain were retuned at every stage, information stored as relative synaptic strengths could well be preserved. To a first approximation, this hypothetical scenario seems to apply to the multistage hippocampal pathway from dentate gyrus to CA3, and from CA3 to CA1. Indeed, we found that both sets of synapses were upregulated in response to inactivity, albeit in different ways.
Consideration of recurrent connections complicates the picture. If synaptic up-scaling applied uniformly across the board and extended to recurrent synapses, the CA3 self-excitation loop would tilt the excitation-inhibition balance toward excitation and the network would run the risk of runaway excitation (Rutecki et al., 1985; Schwartzkroin and Prince, 1978; Wong et al., 1986). In fact, certain forms of epileptogenesis have been attributed to synaptic homeostasis (Houweling et al., 2005; Trasande and Ramirez, 2007), contrary to its proposed role in network stabilization (Burrone and Murthy, 2003; Turrigiano et al., 1998; Turrigiano and Nelson, 2004). Thus, a recurrent system would face the challenge of accommodating two opposing goals: boosting synaptic gain while lessening epileptogenesis. Our data show that such accommodation can be achieved by synapse-specific adaptation. We found that recurrent connections between CA3 neurons did not conform to the prevailing view of synaptic homeostasis: active recurrent terminals underwent an increase in Pr, associated with greater depression during repetitive transmission, but the synapses also underwent a substantial downregulation overall, seen as a reduction of mini frequency, likely due to a binary shutdown of synapses (Moulder et al., 2004). The intensified depression and the lowered proportion of functional synapses would both contribute to downregulating recurrent feedback synaptic excitation during burst activity, working along with dampened CA3 neuron excitability. In combination, these effects were well-suited for limiting network instability, even when pitted against a frank upregulation of feedforward signaling. Indeed, our observations directly indicated that the pro-convulsive effect of increased mossy fiber input (more frequent bursts) was counteracted by the anti-convulsive effects on recurrent synapses (shortened bursts). Thus, differing changes in the efficacy of specific kinds of synapses were expressed at the level of network activity. If both sets of synapses had been uniformly strengthened after chronic inactivity, bursts would have become longer, not just more frequent, increasing the chances of uncontrollably prolonged depolarization, excess Ca2+ entry, and cytotoxic effects.
Neuronal activity can be chronically perturbed in many clinical situations, such as neurodegeneration, trauma and coma. The adaptive phenomena described here may be relevant to changes in CNS function during and following such disorders. While the heterogeneous pattern of adaptive changes could offer benefit in a pathological setting, it may also come at a price for information storage. If a memory trace were stored or processed by multiple sets of synapses, their differential adaptation would alter the overall pattern of synaptic activity and thereby affect the memory trace. Associational memory is heavily dependent on recurrent connections between CA3 pyramids (Bennett et al., 1994; Nakazawa et al., 2002) and would thus be particularly vulnerable to perturbation after chronic inactivity.
EXPERIMENTAL PROCEDURES
Preparation of Slices
Hippocampal organotypic slice cultures were derived from 6–7 day old Sprague-Dawley rats (Charles-River Laboratories, Wilmington, MA). With a tissue chopper, 500 µm hippocampal slices were cut in ice-cold normal saline consisting of (in mM) 122 NaCl, 3 KCl, 26 NaHCO3, 1 NaH2PO4, 2 CaCl2, 2 MgCl2, 20 glucose (300–305 mOsm), bubbled with 95% O2/5% CO2. After washing with warm culture medium, the slices were placed on culture membrane (Millipore, Billerica, MA) at the interface of culture medium and air (Stoppini et al., 1991). The medium was composed of 50% Hank’s-based minimum essential media, 25% Hank’s solution, 25% horse serum, 2 mM L-glutamine, 50 unit/mL penicillin, 50 µg/mL streptomycin and 10 mM HEPES, supplemented with 5 mM glucose (Gibco, Carlsbad, CA, except for Hank’s solution). Slices were kept at 36°C and medium was exchanged every 2–3 days. In some experiments, dentate gyrus was removed at1 DIV by cuts along the hippocampal fissure and at the CA3-dentate gyrus border. The protocols were approved by the Institutional Animal Care and Use Committee of Stanford University.
Electrophysiology
Recordings were made from slice culture at 21–25 DIV. To chronically suppress neuronal activity, we exposed slices to 1–1.5 µM TTX for 3–4 days, starting as early as 18 DIV. Once TTX was added, the medium was exchanged every other day. Slices were removed from TTX-containing solution for 30–60 min before starting recordings. Whole-cell voltage or current clamp recordings from pyramidal cells or dentate granule cells (one cell per slice) were carried out at 22–24°C while the recording chamber was perfused with bath solution at 1–1.5 mL/min. Data from TTX-treated slices were compared with those from untreated sister slices. Electrode resistance in the normal saline was 3–4 MΩ and series resistance (<25 MΩ) was stable within <15%. Data were collected using Axopatch 1C (Axon Instruments, Union City, CA) or PC-501A amplifier (Warner Instruments, Hamden, CT), filtered at 1–2 kHz and digitized at 20 kHz using Digidata 1200 and Clampex 9 program (Axon Instruments). The bath solution (normal saline, see above for composition) contained 10 µM gabazine in all experiments and was continuously bubbled with 95% O2/5% CO2. The pipette solution for current clamp contained (in mM) 130 K-gluconate, 7 KCl, 2 NaCl, 1 MgCl2, 0.4 EGTA, 2 ATP-Mg, 0.3 GTP-tris and 10 HEPES (pH = 7.20 with KOH, 295 mOsm with sucrose). For mEPSC recordings under voltage clamp, the pipette solution contained (in mM) 120 CsCH3SO3, 8 CsCl, 1 MgCl2, 0.4 EGTA, 2 ATP-Mg, 0.3 GTP-tris, 10 HEPES, 5 QX-314 bromide and 10 phosphocreatine (pH=7.20 with CsOH, 295 mOsm with sucrose). For eEPSC recordings, pipettes contained 130 CsCH3SO3, 15 TEA-Cl, 1 MgCl2, 0.3 EGTA, 3 ATP-Mg, 0.3 GTP-tris, 10 HEPES and 5 QX-314 chloride (pH=7.20 with CsOH, 295 mOsm with sucrose).
mEPSCs were recorded at −70 mV with 50 µM D-AP5 and 1 µM TTX (plus gabazine) in the bath. The series resistances in mEPSC experiments were 10–19 MΩ (Figure 1 and Figure 2) or 9–14 MΩ (Figure 4). eEPSCs were evoked every 6–15 s with bath solution-filled θ glass. The peak amplitude of eEPSC was measured over 0.5 ms window from individual traces, and then the peak values from 19–80 traces were averaged for a given condition per cell.
In current clamp experiments, membrane current was clamped at 0 pA (except for Figure 8 and Figure 9) and leaky cells (resting potential below −60mV) were discarded. When epileptic bursts were evoked, a 0.2–0.6 mA pulse of 50 or 100 µs duration was delivered every 15–60 s with a concentric bipolar tungsten electrode (WPI, Sarasota, FL), placed extracellularly at the border of st. pyramidale and st. oriens ~100 µm apart from the recording site. To test the effect of KN93 (Figures 7E–7H), we incubated the slices with both TTX and KN93 (or TTX and vehicle) for 3–4 days. TTX, DCG-IV, D-AP5, NBQX, KN93 and gabazine were from Tocris (Ellisville, MO). Other chemicals were from Sigma (St. Louis, MO) unless noted.
Data Analysis
mEPSC were detected with the Clampfit program (Axon Instruments) with a detection threshold of −5 pA and a template made from our own data. To ensure that each cell contributed equally to statistical analysis, we used 100 mEPSCs per cell. For statistics, amplitudes or interevent intervals from all the mEPSC (100 mEPSC per cell) were pooled and subjected to K–S test with a confidence level of p<10−4. A burst is defined as a prolonged (>100 ms) depolarization starting with 1 or 2 action potentials followed by a broad (50–100 ms) plateau-like spike and then, usually multiple, oscillatory potentials composed of action potential plus a broad plateau spike. The “peak amplitude of burst” is defined as the amplitude of the first plateau spike. A burst event is considered to be terminated when the membrane potential falls below 20% of the “peak amplitude of burst”. A burst width is defined (except Figure 9B) as the duration from the fastest rising point of the first action potential to the time when the last spike falls below 50% of peak amplitude of the burst. Frequency of spontaneous bursts was measured from recordings of 90–260 s. For burst width or number of spikes, measurements from 2 or 3 traces were averaged.
Supplementary Material
Acknowledgments
This work was supported by a MERIT Award from NIMH and by grants from NINDS and NIGMS (R.W.T.). We thank Drs. Rachel Groth, Charles Harata, Yulong Li, Tara Thiagarajan and Bradley Alger for comments on the manuscript, members of the Madison lab for advice on slice cultures and discussions, and members of the Tsien lab for helpful discussions throughout the project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abdul-Ghani MA, Valiante TA, Pennefather PS. Sr2+ and quantal events at excitatory synapses between mouse hippocampal neurons in culture. J. Physiol. 1996;495:113–125. doi: 10.1113/jphysiol.1996.sp021578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aptowicz CO, Kunkler PE, Kraig RP. Homeostatic plasticity in hippocampal slice cultures involves changes in voltage-gated Na+ channel expression. Brain Res. 2004;998:155–163. doi: 10.1016/j.brainres.2003.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch SB, He S, Petrova Y, Wang XM, McNamara JO. Plasticity of both excitatory and inhibitory synapses is associated with seizures induced by removal of chronic blockade of activity in cultured hippocampus. J. Neurophysiol. 2006;96:2151–2167. doi: 10.1152/jn.00355.2006. [DOI] [PubMed] [Google Scholar]
- Beggs JM, Plenz D. Neuronal avalanches in neocortical circuits. J. Neurosci. 2003;23:11167–11177. doi: 10.1523/JNEUROSCI.23-35-11167.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Clements JD. Quantal amplitude and quantal variance of strontium-induced asynchronous EPSCs in rat dentate granule neurons. J. Physiol. 1999;516:227–248. doi: 10.1111/j.1469-7793.1999.227aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR, Gibson WG, Robinson J. Dynamics of the CA3 pyramidal neuron autoassociative memory network in the hippocampus. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1994;343:167–187. doi: 10.1098/rstb.1994.0019. [DOI] [PubMed] [Google Scholar]
- Buchs PA, Stoppini L, Muller D. Structural modifications associated with synaptic development in area CA1 of rat hippocampal organotypic cultures. Brain Res. Dev. Brain Res. 1993;71:81–91. doi: 10.1016/0165-3806(93)90108-m. [DOI] [PubMed] [Google Scholar]
- Buckby LE, Jensen TP, Smith PJ, Empson RM. Network stability through homeostatic scaling of excitatory and inhibitory synapses following inactivity in CA3 of rat organotypic hippocampal slice cultures. Mol. Cell. Neurosci. 2006;31:805–816. doi: 10.1016/j.mcn.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Burrone J, Murthy VN. Synaptic gain control and homeostasis. Curr. Opin. Neurobiol. 2003;13:560–567. doi: 10.1016/j.conb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 2002;420:414–418. doi: 10.1038/nature01242. [DOI] [PubMed] [Google Scholar]
- Dailey ME, Buchanan J, Bergles DE, Smith SJ. Mossy fiber growth and synaptogenesis in rat hippocampal slices in vitro. J. Neurosci. 1994;14:1060–1078. doi: 10.1523/JNEUROSCI.14-03-01060.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu. Rev. Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- De Simoni A, Griesinger CB, Edwards FA. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J. Physiol. 2003;550:135–147. doi: 10.1113/jphysiol.2003.039099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat. Neurosci. 1999;2:515–520. doi: 10.1038/9165. [DOI] [PubMed] [Google Scholar]
- Drakew A, Muller M, Gahwiler BH, Thompson SM, Frotscher M. Spine loss in experimental epilepsy: quantitative light and electron microscopic analysis of intracellularly stained CA3 pyramidal cells in hippocampal slice cultures. Neuroscience. 1996;70:31–45. doi: 10.1016/0306-4522(95)00379-w. [DOI] [PubMed] [Google Scholar]
- Echlin FA, Battista A. Epileptiform seizures from chronic isolated cortex. Arch. Neurol. 1963;9:154–170. doi: 10.1001/archneur.1963.00460080064009. [DOI] [PubMed] [Google Scholar]
- Fricke RA, Prince DA. Electrophysiology of dentate gyrus granule cells. J. Neurophysiol. 1984;51:195–209. doi: 10.1152/jn.1984.51.2.195. [DOI] [PubMed] [Google Scholar]
- Furshpan EJ, Potter DD. Seizure-like activity and cellular damage in rat hippocampal neurons in cell culture. Neuron. 1989;3:199–207. doi: 10.1016/0896-6273(89)90033-0. [DOI] [PubMed] [Google Scholar]
- Galvan CD, Hrachovy RA, Smith KL, Swann JW. Blockade of neuronal activity during hippocampal development produces a chronic focal epilepsy in the rat. J. Neurosci. 2000;20:2904–2916. doi: 10.1523/JNEUROSCI.20-08-02904.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan CD, Wenzel JH, Dineley KT, Lam TT, Schwartzkroin PA, Sweatt JD, Swann JW. Postsynaptic contributions to hippocampal network hyperexcitability induced by chronic activity blockade in vivo. Eur. J. Neurosci. 2003;18:1861–1872. doi: 10.1046/j.1460-9568.2003.02920.x. [DOI] [PubMed] [Google Scholar]
- Gerfin-Moser A, Grogg F, Rietschin L, Thompson SM, Streit P. Alterations in glutamate but not GABAA receptor subunit expression as a consequence of epileptiform activity in vitro. Neuroscience. 1995;67:849–865. doi: 10.1016/0306-4522(95)00130-b. [DOI] [PubMed] [Google Scholar]
- Hartman KN, Pal SK, Burrone J, Murthy VN. Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nat. Neurosci. 2006;9:642–649. doi: 10.1038/nn1677. [DOI] [PubMed] [Google Scholar]
- Houweling AR, Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Homeostatic synaptic plasticity can explain post-traumatic epileptogenesis in chronically isolated neocortex. Cereb. Cortex. 2005;15:834–845. doi: 10.1093/cercor/bhh184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat. Neurosci. 2004;7:244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- Kakegawa W, Tsuzuki K, Yoshida Y, Kameyama K, Ozawa S. Input- and subunit-specific AMPA receptor trafficking underlying long-term potentiation at hippocampal CA3 synapses. Eur. J. Neurosci. 2004;20:101–110. doi: 10.1111/j.1460-9568.2004.03461.x. [DOI] [PubMed] [Google Scholar]
- Kamiya H, Ozawa S. Dual mechanism for presynaptic modulation by axonal metabotropic glutamate receptor at the mouse mossy fibre-CA3 synapse. J. Physiol. 1999;518:497–506. doi: 10.1111/j.1469-7793.1999.0497p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Shinozaki H, Yamamoto C. Activation of metabotropic glutamate receptor type 2/3 suppresses transmission at rat hippocampal mossy fibre synapses. J. Physiol. 1996;493:447–455. doi: 10.1113/jphysiol.1996.sp021395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmarkar UR, Buonomano DV. Different forms of homeostatic plasticity are engaged with distinct temporal profiles. Eur. J. Neurosci. 2006;23:1575–1584. doi: 10.1111/j.1460-9568.2006.04692.x. [DOI] [PubMed] [Google Scholar]
- Kirov SA, Goddard CA, Harris KM. Age-dependence in the homeostatic upregulation of hippocampal dendritic spine number during blocked synaptic transmission. Neuropharmacology. 2004;47:640–648. doi: 10.1016/j.neuropharm.2004.07.039. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Lamsa K, Pavlov I, Riekki R, Johnson BE, Molnar E, Rauvala H, Taira T. Activity blockade increases the number of functional synapses in the hippocampus of newborn rats. Mol. Cell. Neurosci. 2003;22:107–117. doi: 10.1016/s1044-7431(02)00012-x. [DOI] [PubMed] [Google Scholar]
- Liu G, Tsien RW. Properties of synaptic transmission at single hippocampal synaptic boutons. Nature. 1995;375:404–408. doi: 10.1038/375404a0. [DOI] [PubMed] [Google Scholar]
- Maffei A, Nataraj K, Nelson SB, Turrigiano GG. Potentiation of cortical inhibition by visual deprivation. Nature. 2006;443:81–84. doi: 10.1038/nature05079. [DOI] [PubMed] [Google Scholar]
- Maffei A, Nelson SB, Turrigiano GG. Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat. Neurosci. 2004;7:1353–1359. doi: 10.1038/nn1351. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Debanne D, Gahwiler BH, Thompson SM. Lesion-induced axonal sprouting and hyperexcitability in the hippocampus in vitro: implications for the genesis of posttraumatic epilepsy. Nat. Med. 1997;3:990–996. doi: 10.1038/nm0997-990. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong RK. Single neurones can initiate synchronized population discharge in the hippocampus. Nature. 1983;306:371–373. doi: 10.1038/306371a0. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong RK, Traub RD. Synchronized afterdischarges in the hippocampus: contribution of local synaptic interactions. Neuroscience. 1984;12:1179–1189. doi: 10.1016/0306-4522(84)90012-5. [DOI] [PubMed] [Google Scholar]
- Montgomery JM, Pavlidis P, Madison DV. Pair recordings reveal all-silent synaptic connections and the postsynaptic expression of long-term potentiation. Neuron. 2001;29:691–701. doi: 10.1016/s0896-6273(01)00244-6. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Olney JW, Mennerick S. Physiological activity depresses synaptic function through an effect on vesicle priming. J. Neurosci. 2006;26:6618–6626. doi: 10.1523/JNEUROSCI.5498-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Meeks JP, Shute AA, Hamilton CK, de Erausquin G, Mennerick S. Plastic elimination of functional glutamate release sites by depolarization. Neuron. 2004;42:423–435. doi: 10.1016/s0896-6273(04)00184-9. [DOI] [PubMed] [Google Scholar]
- Muller D, Buchs PA, Stoppini L. Time course of synaptic development in hippocampal organotypic cultures. Brain Res. Dev. Brain Res. 1993a;71:93–100. doi: 10.1016/0165-3806(93)90109-n. [DOI] [PubMed] [Google Scholar]
- Muller M. Morphological and functional consequences of chronic epilepsy in rat hippocampal slice cultures. Pflugers Arch. 1993;422:418–423. doi: 10.1007/BF00374304. [DOI] [PubMed] [Google Scholar]
- Muller M, Gahwiler BH, Rietschin L, Thompson SM. Reversible loss of dendritic spines and altered excitability after chronic epilepsy in hippocampal slice cultures. Proc. Natl. Acad. Sci. USA. 1993b;90:257–261. doi: 10.1073/pnas.90.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Quirk MC, Chitwood RA, Watanabe M, Yeckel MF, Sun LD, Kato A, Carr CA, Johnston D, Wilson MA, Tonegawa S. Requirement for hippocampal CA3 NMDA receptors in associative memory recall. Science. 2002;297:211–218. doi: 10.1126/science.1071795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Petralia RS, Wang YX, Niedzielski AS, Wenthold RJ. The metabotropic glutamate receptors, mGluR2 and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neuroscience. 1996;71:949–976. doi: 10.1016/0306-4522(95)00533-1. [DOI] [PubMed] [Google Scholar]
- Robain O, Barbin G, Billette de Villemeur T, Jardin L, Jahchan T, Ben-Ari Y. Development of mossy fiber synapses in hippocampal slice culture. Brain Res. Dev. Brain Res. 1994;80:244–250. doi: 10.1016/0165-3806(94)90109-0. [DOI] [PubMed] [Google Scholar]
- Rutecki PA, Lebeda FJ, Johnston D. Epileptiform activity induced by changes in extracellular potassium in hippocampus. J. Neurophysiol. 1985;54:1363–1374. doi: 10.1152/jn.1985.54.5.1363. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Debanne D, Muller M, Gahwiler BH, Thompson SM. Role of excitatory amino acid and GABAB receptors in the generation of epileptiform activity in disinhibited hippocampal slice cultures. Neuroscience. 1994;61:823–832. doi: 10.1016/0306-4522(94)90405-7. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler BH, Thompson SM. Presynaptic inhibition of excitatory synaptic transmission by muscarinic and metabotropic glutamate receptor activation in the hippocampus: are Ca2+ channels involved? Neuropharmacology. 1995;34:1549–1557. doi: 10.1016/0028-3908(95)00119-q. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Prince DA. Cellular and field potential properties of epileptogenic hippocampal slices. Brain Res. 1978;147:117–130. doi: 10.1016/0006-8993(78)90776-x. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J. Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Longacher M, Bains JS, Yee A. Presynaptic modulation of CA3 network activity. Nat. Neurosci. 1998;1:201–209. doi: 10.1038/651. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Swann JW, Smith KL, Brady RJ. Localized excitatory synaptic interactions mediate the sustained depolarization of electrographic seizures in developing hippocampus. J. Neurosci. 1993;13:4680–4689. doi: 10.1523/JNEUROSCI.13-11-04680.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW. Adaptation to synaptic inactivity in hippocampal neurons. Neuron. 2005;47:725–737. doi: 10.1016/j.neuron.2005.06.037. [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW. α- and βCaMKII. Inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron. 2002;36:1103–1114. doi: 10.1016/s0896-6273(02)01049-8. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Haas HL, Gahwiler BH. Comparison of the actions of adenosine at pre- and postsynaptic receptors in the rat hippocampus in vitro. J. Physiol. 1992;451:347–363. doi: 10.1113/jphysiol.1992.sp019168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Trasande CA, Ramirez JM. Activity deprivation leads to seizures in hippocampal slice cultures: is epilepsy the consequence of homeostatic plasticity? J. Clin. Neurophysiol. 2007;24:154–164. doi: 10.1097/WNP.0b013e318033787f. [DOI] [PubMed] [Google Scholar]
- Traub RD, Miles R, Jefferys JG. Synaptic and intrinsic conductances shape picrotoxin-induced synchronized after-discharges in the guinea-pig hippocampal slice. J. Physiol. 1993;461:525–547. doi: 10.1113/jphysiol.1993.sp019527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Wong RK. Cellular mechanism of neuronal synchronization in epilepsy. Science. 1982;216:745–747. doi: 10.1126/science.7079735. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Tyler WJ, Pozzo-Miller L. Miniature synaptic transmission and BDNF modulate dendritic spine growth and form in rat CA1 neurones. J. Physiol. 2003;553:497–509. doi: 10.1113/jphysiol.2003.052639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RK, Miles R, Traub RD. Local circuit interactions in synchronization of cortical neurones. J. Exp. Biol. 1984;112:169–178. doi: 10.1242/jeb.112.1.169. [DOI] [PubMed] [Google Scholar]
- Wong RK, Traub RD, Miles R. Cellular basis of neuronal synchrony in epilepsy. Adv. Neurol. 1986;44:583–592. [PubMed] [Google Scholar]
- Yoon KW, Rothman SM. Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus. J. Neurosci. 1991;11:1375–1380. doi: 10.1523/JNEUROSCI.11-05-01375.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino M, Sawada S, Yamamoto C, Kamiya H. A metabotropic glutamate receptor agonist DCG-IV suppresses synaptic transmission at mossy fiber pathway of the guinea pig hippocampus. Neurosci. Lett. 1996;207:70–72. doi: 10.1016/0304-3940(96)12486-1. [DOI] [PubMed] [Google Scholar]
- Zimmer J, Gahwiler BH. Cellular and connective organization of slice cultures of the rat hippocampus and fascia dentata. J. Comp. Neurol. 1984;228:432–446. doi: 10.1002/cne.902280310. [DOI] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu. Rev. Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.