

Abstract
Although endocannabinoid signaling is important for certain aspects of gastrointestinal homoeostasis, the role of the cannabinoid receptors (CB) in colorectal cancer has not been defined. Here we show that CB1 expression was silenced in human colorectal cancer due to methylation of the CB1 promoter. Our genetic and pharmacologic studies reveal that loss or inhibition of CB1 accelerated intestinal adenoma growth in ApcMin/+ mice whereas activation of CB1 attenuated intestinal tumor growth by inducing cell death via downregulation of the anti-apoptotic factor survivin. This downregulation of survivin by CB1 is mediated by a cAMP-dependent PKA signaling pathway. These results indicate that the endogenous cannabinoid system may represent a potential therapeutic target for prevention or treatment of colorectal cancer.
Keywords: Colorectal cancer, Cannabinod receptors, Apoptosis, Survivin, and Methylation
Introduction
Cannabinoids are currently used to treat chemotherapy- or radiotherapy-induced nausea, vomiting as well as for pain relief, insomnia relief, mood elevation, and appetite stimulation (1, 2). Medicinal use of these compounds is also thought to be beneficial for digestive disorders such as diarrhea and Crohn’s disease (3). Plant-derived cannabinoids and their derivatives exert a wide variety of biological effects by mimicking endogenous compounds (endocannabinoids), which primarily activate two cannabinoid-specific G-protein-coupled receptors, CB1 and CB2, encoded by the Cnr1 and Cnr2 genes, respectively. Studies have recently suggested that cannabinoids exert potential anti-tumor effects on a wide spectrum of human tumor cell lines in culture and in xenograft studies (4, 5) and have anti-inflammatory properties (6). CB1 and CB2 can activate several different cellular pathways depending on the biologic context being examined (4, 7). However, there is currently no in vivo genetic evidence demonstrating the role of the endocannabinoid system in mouse models of cancer.
The gastrointestinal tract of mice, rats and humans produce two major endocannabinoids, anandamide (AEA) and 2-arachidonoyl-glycerol (2-AG) (8–10). Interestingly, human colorectal cancers (CRCs) and adenomas produce two to three times more AEA and 2-AG than does the neighboring normal mucosa (10). In humans and mice, CB1 is expressed in normal colonic epithelium, smooth muscle, and the submucosal myenteric plexus (11, 12). CB2 is found mainly in subepithelial macrophages of normal colonic tissues (11). However, little is known regarding the expression of cannabinoid receptors in CRC.
Endocannabinoid signaling is important in regulating gastrointestinal motility, secretion, and neurotransmitter release (8, 13–15). For example, the endocannabinoid signaling has been implicated in an autoimmune intestinal disorder (16, 17); specifically, Cnr1−/− mice exhibited a stronger inflammatory response in the colon than did wild-type mice in response to treatment with pro-inflammatory agents (18), suggesting that CB1 provides intrinsic protection against colonic inflammation. Since chronic inflammation is a known risk factor for CRC, the aim of the present study was to determine the role of cannabinoid receptors in a mouse model of colon cancer and the mechanism of the receptor action.
Materials and Methods
Cell culture and reagents
Ten CRC cell lines were obtained from the American Type Culture Collection (Manassas, VA) and HCA-7 cells were a gift from Susan Kirkland (University of London). All cells were maintained in McCoy’s 5A medium with 10% fetal bovine serum. AM251 and R-1 methanandamide were purchased from Cayman Chemical (Ann Arbor, MI). 5-Aza-2-deoxycytidine (5-aza-dC) and 8-AHA-cAMP were obtained from Sigma (St Louis, MO). PD98059, LY294002, and H-89 were obtained from Calbiochem (La Jolla, CA). A negative control siRNA (siRNAC) and a CB1-selective siRNA (siRNACB1) were purchased from Ambion (Austin, TX).
Animals
CB1- and CB2-deficient mice on a C57BL/6J genetic background were generated as previously described (19, 20). CB1-deficient ApcMin/+ mice (Cnr1−/−/ApcMin/+), CB2-deficient ApcMin/+ mice (Cnr2−/−/ApcMin/+) and their controls (Cnr1+/+/Cnr2+/+/ApcMin/+) were derived from same littermates by breeding Cnr1−/− or Cnr2−/− mice with ApcMin/+ mice on a C57BL/6J genetic background (Jackson Laboratory, Bar Harbor, ME). Male mice were killed at the age of 13 weeks. For the AM251 treatment, 6-week-old male Cnr1+/+/ApcMin/+ control mice (n=28) with a genetic background identical to that of the Cnr1−/−/ApcMin/+ mice were randomly assigned to one of two groups treated with 150 μl of 0.5% carboxymethylcellulose with or without AM251 (10 mg/kg body weight) by daily gavage for 7 weeks. For the R-1 methanandamide treatment, 32 male Cnr1+/+/ApcMin/+ control mice with a genetic background identical to that of the Cnr1−/−/ApcMin/+ mice and 20 male Cnr1−/−/ApcMin/+ mice at the age of 6 weeks were assigned to one of two groups and injected intraperitoneally with 150 μl of phosphate-buffered saline with or without R-1 methanandamide (10 mg/kg body weight) daily for 8 weeks. At the end of the experiments, mice were killed and the number and size of polyps was measured as previously described (21). After tumors were counted, intestinal tissues were embedded in paraffin. For histologic analysis, 5-μm-thick sections from all groups were stained with H&E to examine polyp morphology.
Quantitative real-time PCR
Cnr1 and Cnr2 mRNAs were quantified by quantitative real-time PCR with an iCycler and iQ™ SYBR Green Supermix (both from Bio-Rad, Hercules, CA) as previously described (22). Primers for the Cnr1, Cnr2, and Actin genes were chosen by using the Beacon Designer 4 program (Premier BioSoft International, Palo Alto, CA). The sequences of the specific PCR primers were as follows (5′ to 3′): hCnr1, forward: AAGACCCTGGTCCTGATCCT; reverse: CGCAGGTCCTTACTCCTCAG; hCnr2, forward: ATCATGTGGGTCCTCTCAGC; reverse: GATTCCGGAAAAGAGGAAGG; hβ-actin, forward: AGAAAATCTGGCACCACACC; reverse: AGAGGCGTACAGGGATAGCA.
5-aza-dC treatment
The cells (1 × 106 cells per 100-mm plate) were cultured in McCoy’s 5A medium containing 10% fetal bovine serum and 5 μM of 5-aza-dC for 3 days and subjected to the assays described below.
Bisulphite genomic sequencing PCR
Sodium bisulphite modification was carried out with the CpGenome DNA Modification Kit (Chemicon, Temecula, CA, USA) according to the manufacturer’s instructions. The bisulphite sequencing PCR primers were designed with the Web software program MethPrimer (see footnotes). The sequences of the bisulphite sequencing PCR primers were as follows (5′ to 3′): forward GAAGAGGTTTGTTTTTTTTGGTTT; reverse CCCTTCCCAAACTCTTCACTAA. This primer set was used to amplify the CpG islands around the transcription start site of the Cnr1 gene (−212 to +140) in the promoter region with an expected 352-bp product. PCR reactions were performed with HotStar Taq polymerase (Qiagen, Valencia, CA). The PCR products were resolved on a 2% agarose gel, purified with a QIAquick Gel Extraction Kit (Qiagen), and sequenced with an ABI 377 automated sequencer (Applied Biosystems Inc., Foster City, CA) using the above primers.
Transfection and retroviral transduction
SW-480 cells (1.8 ×105) were transfected with either a negative control siRNA (siRNAC) or a Cnr1-selective siRNA (siRNACB1) at 100 nM by using LipofectAMINE 2000 reagent according to the manufacturer’s instructions (Life Technologies, Inc. Rockville, MA). Transfection efficiency was monitored with 100 nM of a control fluorescence-labeled RNA (Invitrogen, Carlsbad, CA) at 24 hours after transfection. Transfection efficiency was 90% with this system. Retrovirus transduction of human survivin in MIEG3 vector or vector alone into SW-480 cells was carried out as described (23). SW-480 cells stably expressing survivin were selected by FACS.
Apoptosis assays
The cells (9 × 105 per 100-mm plate) were incubated in serum-free medium for 1 day and then treated with either vehicle, R-1 methanandamide, or H-89 at indicated concentrations in serum-free medium for 1 day. For the 8-AHA-cAMP treatment, the cells were pretreated with 8-AHA-cAMP for 1 h and then treated with R-1 methanandamide in serum-free medium for 1 day after serum starvation for 24 h. For the 5-aza-dC treatment, the cells were treated with 5-aza-dC for 3 days as noted above, followed by incubation with R-1 methanandamide in medium containing 1% fetal bovine serum for 1 day. For the siRNA assays, the transfected cells were treated with R-1 methanandamide in serum-free medium for 1 day. Numbers of apoptotic cells were determined by flow cytometry with the TACS Annexin V-FITC Apoptosis Detection Kit according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). Data are expressed as the means ± SE of the percentages of apoptotic cells from three separate experiments.
Membrane protein preparation
Plasma membranes were prepared according to the procedure described previously (24). CB1 protein levels in the plasma membrane were measured by western blotting as described below.
Western blot analysis
Whole cell extracts were prepared from cells (1.5 × 106) treated with indicated concentration of R-1 methanandamide or H-89 in serum-free medium for 1 day after serum starvation for 24 h. For the 8-AHA-cAMP treatment, SW-480 cells were pretreated with 8-AHA-cAMP for 1 h and then treated with R-1 methanandamide for 1 day after serum starvation for 24 h. Western blotting was performed as described elsewhere (25). Antibodies to survivin, phospho-survivin (Thr 34), Bcl-2, PTEN, and cdc2 (all from Santa Cruz Biotechnology); cleaved caspase-3, caspase-3, and cleaved PARP (all from Cell Signaling Technology, Danvers, MA); and CB1 (Affinity Bioreagents, Golden, CO) were used in 1:500 dilutions. The blots were stripped and then reprobed with β-actin antibody (Sigma).
PKA Kinase assay
PKA kinase activity in cultured cells was measured by using a SignaTECT cAMP-Dependent Protein Kinase (PKA) Assay system (Promego, Madison, WI) according to the manufacturer’s instructions. Briefly, SW-480 cells (3 × 106) were cultured in serum-free medium for 24 h and then were treated with indicated concentration of R-1 methanandamide in serum-free medium for 24 h. The cell extracts were subjected to in vitro PKA kinase assays. Data are represented as the mean ± SE of the kinase activity from three independent experiments with duplicate.
Statistical analysis
A post-hoc analysis of variance (ANOVA) was used to calculate P values for the experiments testing the effect of CB1 deletion or treatment with a CB1 agonist or antagonist on intestinal polyp formation (Figs. 3 and 4).
Fig. 3.

The effect of Cnr1 deletion on intestinal polyp burden. (A-B) Male mice with different genotypes were killed at 13 weeks of age and polyp numbers and sizes were measured in the small intestine (A) and colon (B). Data are expressed as means ± SEM (*P < 0.05, Bonferroni test). (C) Representative H&E-stained sections of intestines from Cnr1−/−/ApcMin/+ and Cnr1+/+/ApcMin/+ mice are shown (scale bar = 500 μm).
Fig. 4.

The effect of CB1 antagonist and agonist on intestinal polyp growth. (A-B) In the CB1 antagonist treatments, 6-week-old male Cnr1+/+/ApcMin/+ mice were treated with 0.5% CMC with or without AM251. (C-D) In the CB1 agonist experiments, 6-week-old male Cnr1+/+/ApcMin/+ (C) and Cnr1−/−/ApcMin/+ (D) mice were treated with PBS with or without R-1. At the end of the experiments, the number and size of polyps in the small intestine (A, C, and D) and colon (B, C, and D) were quantified.
Results
Loss of CB1 expression in colorectal carcinomas
To understand the role of endocannabinoid signaling in CRC, we examined the expression pattern of CB1 and CB2 in human grade II-III colon carcinomas and human CRC cell lines. Quantitative real-time PCR analysis revealed greatly reduced expression of CB1 in 18 of 19 cancer specimens as compared with that in adjacent normal mucosa (Fig. 1A) and in 9 of 10 cell lines (Fig. 1B). In contrast, no recognizable pattern of CB2 expression was found in tumor tissues (see Supplementary Fig. S1A) or in the CRC cell lines examined (Supplementary Fig. S1B). Similarly, CB1 protein levels were lost in 15 of 16 cancer specimens in paired samples as measured by western blotting since CB1 protein levels were barely detected in samples 17–19, consistent with very low levels of Cnr1 mRNA (Fig. 1C and A). Overall, CB1 mRNA and protein levels correlate well in these human biopsies. These results led us to consider that loss of CB1 expression could be associated with CRC progression.
Fig. 1.

CB1 and CB2 expression in human colorectal tumors. (A-B) Cnr1 mRNA expression in 19 pairs of human tumors with matched normal tissues (A) and in 10 CRC cell lines (B) was measured as described the Method. In (A), the relative expression of CB1 is the average of triplicate samples normalized against the transcript levels of h-β-actin; in (B), data are the means + SE of the relative expression from three independent experiments. (C) CB1 protein levels in these human samples were determined by western blotting. (D) Treatment with 5-aza-dC restored the expression of Cnr1 mRNA (left panel) and CB1 protein (right panel) in CRC cells lines as measured by quantitative real-time PCR and western blotting.
Inactivation of tumor suppressor genes in cancer result from epigenetic silencing as frequently as that due to genetic mutations (26). Therefore, we first examined whether epigenetic silencing (DNA methylation and histone modifications) of Cnr1 contributes to loss of its transcription. Treatment with the demethylating agent 5-aza-2-deoxycytidine (5-aza-dC) restored Cnr1 mRNA expression in seven of eight CRC cell lines tested (Fig. 1D, left panel) and CB1 protein expression in three cell lines (Fig. 1D, right panel), whereas treatment with the histone deacetylase inhibitor sodium butyrate did not significantly affect Cnr1 mRNA expression (data not shown). These results suggest that aberrant methylation of CpG islands within the promoter results in transcriptional silencing of Cnr1.
We next determined the methylation status of 39 CpG sites flanking the transcription start site of Cnr1 (−212 to +140) using bisulphite sequencing in four CRC cell lines and in the first 13 sets of human paired samples with high levels of CB1 mRNA in normal tissues. The CpG sites of this region were either fully or partially methylated in these tumor samples (Fig. 2). When the threshold for methylation was set at 30% (the upper limit of normal), the hypermethylation of this region was found in three low-Cnr1-expressing cell lines (HCT-116, HT-29, and LS-174T) and in 8 of 13 human tumor tissues (T3, T5, T7, T8, T9, T10, T12 and T13) (62%). Multiple CpG sites were methylated in 10 of 13 tumor tissues (77%) but not in the matched normal tissues. None of the above 39 CpG sites assessed were methylated in the high-Cnr1-expressing cell line SW-480 or in the normal tissues (N1, N5, N8, and N13) showing high Cnr1 expression (Fig. 1). Overall, methylation of Cnr1 correlated well with loss of its transcription in these samples (Figs. 1 and 2). The most methylated CpG sites are located in the transcription factor binding sites and the transcription start site (Supplementary Table S1). Moreover, after 5-aza-dC treatment, the Cnr1 promoter changed from fully methylated to un-methylated at any site in HCT-116 cells and from fully to partially methylated in LS-174T and HT-29 cells (Supplementary Fig. S2), providing evidence that silencing of Cnr1 in human CRCs is due to methylation of its promoter. These findings uncover a new and previously unrecognized regulatory mechanism responsible for CB1 expression in CRC and help to explain one mechanism reason for the loss of CB1 in CRC.
Fig. 2.

Methylation status of the CB1 promoter in human colorectal tumors. Bisulphite sequencing PCR was used to determine the methylation status of all 39 of the CpG sites in the Cnr1 promoter region (−212 to +140) in first 13 sets of paired human samples used in Fig. 1 and in four CRC cell lines. Numbers represent CpG sites relative to the transcription start site. The black regions indicate percentages of methylation exceeding 30%. The threshold for methylation was set at 30% (the upper limit of normal).
CB1 signaling and intestinal tumor growth in ApcMin/+ mice
To further explore the in vivo significance of endocannabinoid signaling in CRC progression, we examined the consequences of either silencing or enhancing cannabinoid signaling in ApcMin/+ mice by deletion of cannabinoid receptor genes or by treatment with a selective CB antagonist or agonist. ApcMin/+ mice have been widely used to study CRC progression because they contain a germline mutation in the APC gene and (like humans) spontaneously develop multiple polyps in the intestine. Moreover, the reduction of CB1 expression was not observed in intestinal adenomas as compared to the adjacent normal tissues in Cnr1+/+/ApcMin/+ mice (Supplementary Fig. S1C). As a first step, we investigated the impact of the loss of Cnr1 or Cnr2 in ApcMin/+ mice. Thirteen-week-old male CB1-deficient ApcMin/+ mice exhibited 2.5- to 3.8-fold increases in small intestinal (panel A) and colonic (panel B) polyp burden relative to littermate control mice (Cnr1+/+/ApcMin/+) (Fig. 3). Disruption of Cnr1 in particular resulted in a greater than 10-fold increase in the number of large polyps (> 2 mm) in the small intestine. In contrast, deletion of Cnr2 had no effect on intestinal polyp burden (Fig. 3A and B). Histologic analysis revealed that large, medium, and small polyps from mice of different genotypes were all adenomas (Fig. 3C). These results provide the first genetic evidence that CB1 is involved in regulating polyp growth in vivo.
To determine whether a CB1 antagonist would mimic the phenotypic changes found in the Cnr1−/−/ApcMin/+ mice, male Cnr1+/+/ApcMin/+ mice with a genetic background identical to that of the Cnr1−/−/ApcMin/+ mice were treated with carboxymethylcellulose (CMC) with or without a CB1-selective antagonist (AM251) for 7 weeks. The mice treated with AM251 exhibited 2- to 6-fold increase in small intestinal and colonic tumor burden relative to controls (Fig. 4A and B). Notably, AM251 treatment mainly increased the number of polyps larger than 1 mm in the small intestine, whereas AM251 stimulated the growth of colon polyps of all sizes. Interestingly, ApcMin/+ mice treated with CMC itself had fewer and smaller polyps than the untreated mice (Fig. 3A and 4A), suggesting that the CMC has some inhibitory influence on tumor growth in this model. However, this effect was not observed in the large intestine. We further explored the role of CB1 in regulating tumor growth by evaluating the effect of a CB1 agonist in ApcMin/+ mice. Treating Cnr1+/+/ApcMin/+ mice with a CB1 agonist R-1 methanandamide (R-1) in PBS for 8 weeks resulted in half to one-sixth as many tumors in the small intestine and colon compared to control mice (Fig. 4C). Again, this effect was consistently observed mostly in terms of fewer large polyps. This finding is interesting because larger polyps are known to have a higher risk of progressing to carcinomas. Similarly, the higher average polyp number of the control group in panel C than that in panel A is due to the effect of CMC and one-week difference of treatment between two experiments. Unlike the results seen in Cnr1+/+/ApcMin/+ mice, administration of R-1 failed to affect small and large intestinal polyp burden in male Cnr1−/−/ApcMin/+ mice, demonstrating that CB1 is required for the tumor-inhibiting effects of R-1 (Fig. 4D). Collectively, these genetic and pharmacologic results demonstrate that silencing of endocannabinoid signaling via CB1 accelerates intestinal adenoma growth and that activation of CB1 attenuates intestinal tumor growth, suggesting that CB1 has tumor suppressor effects.
CB1 activation and tumor cell apoptosis via downregulation of survivin
Cannabinoids were previously shown to induce apoptotic cell death and to inhibit proliferation in cancer cell lines (4). To determine how CB1 signaling inhibits tumor growth, we examined the ability of the CB1 agonist, R-1 methanandamide (R-1), to induce apoptosis in cultured CRC cells. As expected, SW-480 cells expressing high CB1 levels were sensitive to R-1-induced apoptosis, whereas LS-174T cells with low CB1 were more resistant to R-1 (Fig. 5A). Importantly, knockdown of Cnr1 mRNA with a selective small interfering RNA (siRNA) (Fig. 5B, left panel) prevented CB1 agonist-induced apoptosis of SW-480 cells (Fig. 5B, right panel), confirming that CB1 mediates the pro-apoptotic effect of a CB1 agonist (R-1). Because treatment with 5-aza-dC led to increased CB1 expression in HCT-116 cells (Fig. 1D), we next examined the pro-apoptotic effect of R-1 on HCT-116 cells treated with 5-aza-dC. Similar to the results seen in SW-480 and LS-174T cells, 5-aza-dC-treated HCT-116 cells were more sensitive to R-1-induced apoptosis than controls (Supplementary Fig. S3A), demonstrating that restoration of CB1 expression can reprogram CB1 ligand-resistant cells to become responsive to a CB1 agonist. These results demonstrate that CB1 mediates the pro-apoptotic effect of R-1 methanandamide and that the anti-tumor effect of CB1 relies, at least in part, on the ability of CB1 to induce tumor cell death.
Fig. 5.

Activation of CB1 induces apoptosis and downregulates survivin. (A) SW-480 and LS-174T cells were treated with the R-1 for 24 hours and percentages of apoptotic cells were determined by flow cytometry as described in the Methods. (B) SW-480 cells transfected with either a negative control (siRNAC) or a CB1-selective siRNA (siRNACB1) were treated with R-1 and the relative expression of Cnr1 mRNA (left panel) in these cells was determined by quantitative real-time PCR as described in Fig. 1B, and the apoptotic rate (right panel) was measured as noted above. (C) SW-480 cells were treated with R-1 and levels of indicated protein targets were detected by western blotting as described in the Methods (top panel). Survivin levels were also determined from the experiments in panel B by western blotting (lower panel). (D) SW-480 cells overexpressing survivin or containing vector were treated with R-1 and the apoptotic rate in these cells was measured as noted above. The panel C is a representative of three different experiments with similar results.
To investigate the molecular mechanism(s) by which activation of CB1 induces tumor cell apoptosis, we tested whether activation of CB1 regulates genes known to control cell death. Bcl-2 and inhibitor of apoptosis protein (IAP) represent members of a large family of genes that regulate different aspects of apoptosis. IAP genes encode proteins that inhibit cellular apoptosis by binding to caspases and inhibiting their activity (27). Survivin is unique among the IAP gene family in that it is overexpressed in almost every human tumor studied but is barely detectable in most normal adult tissues (28). Overexpression of survivin is associated with a poor clinical outcome and reduced tumor cell apoptosis in patients with CRC (29, 30). Therefore, we examined the ability of a CB1 agonist to regulate these apoptotic genes and found that treatment of SW-480 cells with R-1 decreased survivin expression but had no effect on Bcl-2 or PTEN expression (Fig. 5C). R-1 treatment also led to increased caspase-3 activity, which in turn resulted in cleavage of its target poly ADP-ribose polymerase (PARP) in SW-480 cells (Fig. 5C). In contrast, inhibition of CB1 expression with siRNA prevented the R-1-induced downregulation of survivin in SW-480 cells (Fig. 5C), demonstrating that CB1 mediates the downregulation of survivin. Consistent with its effects on apoptosis, R-1 failed to downregulate survivin expression in LS-174T cells, which lack the CB1 receptor (Supplementary Fig. S3B). Similarly, treatment with 5-aza-dC, which increased CB1 expression, restored the ability of R-1 to reduce survivin expression in HCT-116 cells (Supplementary Fig. S3C). Overexpression of survivin in SW-480 cells inhibited R-1-induced apoptosis (Fig. 5D), demonstrating that survivin mediates the effects of CB1 agonist on inducing apoptosis. Because the phosphorylation of survivin at Thr34 by cdc2 increases protein stability (31), we also examined whether treatment of R-1 would diminish survivin phosphorylation at Thr34 and reduce cdc2 expression. Indeed, reductions in both survivin phosphorylation and cdc2 expression were observed in response to R-1 in SW-480 cells (Supplementary Fig. S3D), indicating that activation of CB1 enhances survivin degradation by dephosphorylation via downregulation of cdc2. Taken together, these results provide the first evidence that survivin is a target of CB1 to regulate programmed cell death in CRC cells.
Since activation of CB1 regulates multiple cellular pathways, including stimulation of MAPK and PI3K-Akt signaling and inhibition of cAMP-dependent PKA activity, we examined whether these pathways are involved in CB1 downregulation of survivin. As shown in Fig. 6, treatment of SW480 cells with R-1 decreased PKA kinase activity (panel A) and a PKA inhibitor (H-89) mimicked the effects of CB1 on downregulation of survivin and promotion of cell death (panel B). In contrast, a PKA activator (8-AHA-cAMP) inhibited the effects of CB1 (panel C). These results suggest that the effects of CB1 are mediated, at least in part, via a cAMP-dependent PKA pathway since inhibition of MAPK and PI3K pathways failed to affect the CB1 regulation of survivin (Fig. 6D).
Fig. 6.
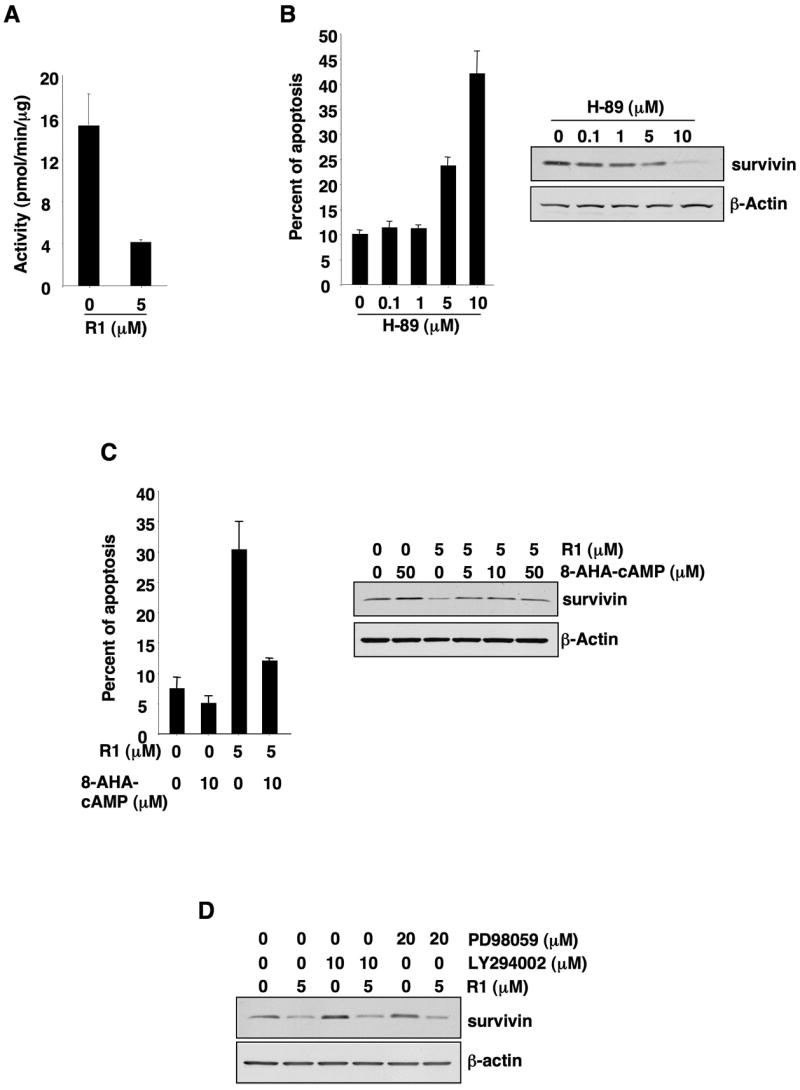
A cAMP-dependent PKA pathway mediates pro-apoptotic effects of CB1. (A) The levels of PKA kinase activity in SW-480 cells treated with R-1 were determined as described in the Methods. (B) SW-480 cells treated with H-89 were subjected to examine apoptosis rate (left panel) and survivin expression (right panel) described in the Method. (C) SW-480 cells were pretreated with 8-AHA-cAMP and then treated with R-1; and the apoptotic rate (left panel) and survivin expression (right panel) were determined as described the Method. (D) SW-480 cells were pretreated with a PI3K inhibitor (LY294002) or a MAPK inhibitor (PD98059) for 1 h and then treated with R-1 in serum-free medium for 1 day after serum starvation 24 h. The survivin expression was determined by western blotting.
Discussion
Study of the potential application of cannabinoids as anti-tumor drugs is an exciting consideration. Several studies have already suggested that cannabinoids exert potential anti-tumor effects, but they were performed in cultured cell lines or in xenograft models. However, one recent study showed that increased endocannabinoid levels by a FAAH inhibitor reduce the development of precancerous lesions in an AOM-treated mouse model (32). Our results further demonstrate that CB1 receptor mediates the anti-tumor effects of endocannabinoids in vivo.
Epigenetic silencing plays a role in carcinogenesis and most CRCs show some epigenetic abnormalities (33). Aberrant DNA methylation occurs in normal mucosa at very early pre-neoplastic stages of cancer development as well as during the later stages of cancer progression, but seems to be a defining event in about half of all sporadic colon tumors (34). Hypermethylation of CpG islands is one mechanism for silencing tumor suppressor and DNA repair genes such as the cyclin-dependent kinase inhibitor P16/INK4α, P14/ARF, APC, hMLH1, and MGMT (33). However, these genes are methylated in relatively few cases (10%–30%) of CRC. We found the frequency of Cnr1 methylation to be quite high (77%), indicating that loss of CB1 could be involved in many, if not most, CRCs. These results also suggest that increased levels of endogenous cannabinoids in humans may not affect tumor growth because loss of the CB1 in most CRCs would make cells resistant to these ligands. Our findings that CB1 downregulation is due to methylation of the CB1 promoter in CRC may represent a general mechanism in other cancer types and may provide a new therapeutic strategy for treatment and/or prevention of CRC. For example, an initial treatment with a demethylating agent to boost CB1 levels followed by administration of a CB1 agonist to achieve optimal stimulation of programmed cell death might be effective. Moreover, downregulation of the CB1 is also correlated with a number of neurodegenerative diseases, including Huntington’s disease, Alzheimer’s disease, and multiple sclerosis (35). Further investigation is required for examining whether CB1 expression is reduced in other cancer types as well. Since CB1 antagonists are known to inhibit appetite and induce weight loss (36), a CB1 antagonist, rimonabant (SR141716), is approved in Europe and Latin American for the treatment of obesity and associated dyslipidemias. However, our findings that treatment with a CB1 antagonist accelerated intestinal adenoma growth raise concerns about developing CB1 antagonists for human use, especially in people who are at high risk of developing CRC.
Dysregulation of apoptosis, proliferation, and angiogenesis all participate in cancer progression. In addition to stimulating apoptosis, cannabinoids have been shown to have anti-proliferative effects on the CRC cell lines Caco-2 and DLD-1 (10) and anti-angiogenic effects in gliomas (37). However, whether the inhibitory effect of cannabinoids on proliferation and angiogenesis depends on the presence of CB receptors is not clear, because Caco-2 and DLD-1 cells express very low levels of CB1 (Fig. 1B) and some biological effects of cannabinoids are known to be independent of CB receptor binding (38). Further investigation is needed to determine whether cannabinoids have anti-angiogenic effects in CRC and whether CB receptors mediate these effects. In addition, we examined several potential signaling pathways of CB1, including MAPK, PI3K, and cAMP-dependent PKA pathways. We found that primarily a cAMP-dependent PKA intracellular signaling mediates the effects of activated CB1 on downregulation of survivin and induction of programmed cell death. It has been established that PKA could regulate cdc2 activity and expression via two cdc2 kinase regulators, cdc25 phosphatase and Wee 1 kinase in oocytes (39). Our results showed that activation of CB1 downregulated cdc2 expression. Consistent with our results, a recent study showed that treatment of breast cancer cells with Δ9-tetrahytocannabino (THC) resulted in a decrease in cdc2 and survivin protein expression with enhanced Wee 1 and reduced cdc25C protein expression (40), supporting the hypothesis that PKA regulates survivin expression via Wee 1/cdc25C-cdc2 cascade.
In conclusion, our studies reveal the molecular mechanism by which cannabinoids inhibit tumor growth. We found that aberrant methylation of CB1 represents a clear mechanism for loss of expression in CRC. Using multiple approaches, we provide in vivo evidence demonstrating that endocannabinoid signaling via CB1 plays a key role in regulating intestinal tumor growth. Importantly, we elucidated the signaling pathway that mediates the pro-apoptotic effects of CB1. Moreover, our results may provide a rationale for the development of CB1 agonists that do not cross the blood-brain barrier for cancer prevention or treatment in combination with a demethylating agent.
Supplementary Material
Acknowledgments
Supported by National Institutes of Health grants R01-DK-62112, P01-CA-77839, R37-DK-47297, and P30-DK-58404 (R.N.D.); the National Colorectal Cancer Research Alliance (R.N.D.); and by NIH grants R37-DA06668 and R37-HD12304 (S.K.D.).
The abbreviations used are
- CB
Cannabinoid receptors
- APC
adenomatous polyposis coli
- CRC
Colorectal cancer
- CMC
carboxymethylcellulose
- R-1
R-1 methanandamide
Footnotes
Web software program MethPrimer: http://www.urogene.org/methprimer/index1.html
References
- 1.Hall W, Christie M, Currow D. Cannabinoids and cancer: causation, remediation, and palliation. Lancet Oncol. 2005;6:35–42. doi: 10.1016/S1470-2045(04)01711-5. [DOI] [PubMed] [Google Scholar]
- 2.Walsh D, Nelson KA, Mahmoud FA. Established and potential therapeutic applications of cannabinoids in oncology. Support Care Cancer. 2003;11:137–43. doi: 10.1007/s00520-002-0387-7. [DOI] [PubMed] [Google Scholar]
- 3.Massa F, Monory K. Endocannabinoids and the gastrointestinal tract. J Endocrinol Invest. 2006;29:47–57. [PubMed] [Google Scholar]
- 4.Guzman M. Cannabinoids: potential anticancer agents. Nat Rev Cancer. 2003;3:745–55. doi: 10.1038/nrc1188. [DOI] [PubMed] [Google Scholar]
- 5.Patsos HA, Hicks DJ, Greenhough A, Williams AC, Paraskeva C. Cannabinoids and cancer: potential for colorectal cancer therapy. Biochem Soc Trans. 2005;33:712–4. doi: 10.1042/BST0330712. [DOI] [PubMed] [Google Scholar]
- 6.Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400–11. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- 7.Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3:771–84. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- 8.Izzo AA, Fezza F, Capasso R, et al. Cannabinoid CB1-receptor mediated regulation of gastrointestinal motility in mice in a model of intestinal inflammation. Br J Pharmacol. 2001;134:563–70. doi: 10.1038/sj.bjp.0704293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McVey DC, Schmid PC, Schmid HH, Vigna SR. Endocannabinoids induce ileitis in rats via the capsaicin receptor (VR1) J Pharmacol Exp Ther. 2003;304:713–22. doi: 10.1124/jpet.102.043893. [DOI] [PubMed] [Google Scholar]
- 10.Ligresti A, Bisogno T, Matias I, et al. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology. 2003;125:677–87. doi: 10.1016/s0016-5085(03)00881-3. [DOI] [PubMed] [Google Scholar]
- 11.Wright K, Rooney N, Feeney M, et al. Differential expression of cannabinoid receptors in the human colon: cannabinoids promote epithelial wound healing. Gastroenterology. 2005;129:437–53. doi: 10.1016/j.gastro.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 12.Casu MA, Porcella A, Ruiu S, et al. Differential distribution of functional cannabinoid CB1 receptors in the mouse gastroenteric tract. Eur J Pharmacol. 2003;459:97–105. doi: 10.1016/s0014-2999(02)02830-3. [DOI] [PubMed] [Google Scholar]
- 13.Pinto L, Izzo AA, Cascio MG, et al. Endocannabinoids as physiological regulators of colonic propulsion in mice. Gastroenterology. 2002;123:227–34. doi: 10.1053/gast.2002.34242. [DOI] [PubMed] [Google Scholar]
- 14.Izzo AA, Capasso F, Costagliola A, et al. An endogenous cannabinoid tone attenuates cholera toxin-induced fluid accumulation in mice. Gastroenterology. 2003;125:765–74. doi: 10.1016/s0016-5085(03)00892-8. [DOI] [PubMed] [Google Scholar]
- 15.MacNaughton WK, Van Sickle MD, Keenan CM, Cushing K, Mackie K, Sharkey KA. Distribution and function of the cannabinoid-1 receptor in the modulation of ion transport in the guinea pig ileum: relationship to capsaicin-sensitive nerves. Am J Physiol Gastrointest Liver Physiol. 2004;286:G863–71. doi: 10.1152/ajpgi.00482.2003. [DOI] [PubMed] [Google Scholar]
- 16.Di Marzo V, Izzo AA. Endocannabinoid overactivity and intestinal inflammation. Gut. 2006;55:1373–6. doi: 10.1136/gut.2005.090472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Argenio G, Petrosino S, Gianfrani C, et al. Overactivity of the intestinal endocannabinoid system in celiac disease and in methotrexate-treated rats. J Mol Med. 2007;85:523–30. doi: 10.1007/s00109-007-0192-3. [DOI] [PubMed] [Google Scholar]
- 18.Massa F, Marsicano G, Hermann H, et al. The endogenous cannabinoid system protects against colonic inflammation. J Clin Invest. 2004;113:1202–9. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5. doi: 10.1073/pnas.96.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarai Z, Wagner JA, Varga K, et al. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc Natl Acad Sci U S A. 1999;96:14136–41. doi: 10.1073/pnas.96.24.14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang D, Wang H, Shi Q, et al. Prostaglandin E(2) promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor delta. Cancer Cell. 2004;6:285–95. doi: 10.1016/j.ccr.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 22.Wang D, Wang H, Brown J, et al. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med. 2006;203:941–51. doi: 10.1084/jem.20052124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Fukuda S, Pelus LM. Survivin regulates the p53 tumor suppressor gene family. Oncogene. 2004;23:8146–53. doi: 10.1038/sj.onc.1207992. [DOI] [PubMed] [Google Scholar]
- 24.Yang ZM, Paria BC, Dey SK. Activation of brain-type cannabinoid receptors interferes with preimplantation mouse embryo development. Biol Reprod. 1996;55:756–61. doi: 10.1095/biolreprod55.4.756. [DOI] [PubMed] [Google Scholar]
- 25.Wang D, Buchanan FG, Wang H, Dey SK, DuBois RN. Prostaglandin E2 enhances intestinal adenoma growth via activation of the Ras-mitogen-activated protein kinase cascade. Cancer Res. 2005;65:1822–9. doi: 10.1158/0008-5472.CAN-04-3671. [DOI] [PubMed] [Google Scholar]
- 26.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 27.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;3:401–10. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 28.Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 29.Kawasaki H, Altieri DC, Lu CD, Toyoda M, Tenjo T, Tanigawa N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res. 1998;58:5071–4. [PubMed] [Google Scholar]
- 30.Sarela AI, Scott N, Ramsdale J, Markham AF, Guillou PJ. Immunohistochemical detection of the anti-apoptosis protein, survivin, predicts survival after curative resection of stage II colorectal carcinomas. Ann Surg Oncol. 2001;8:305–10. doi: 10.1007/s10434-001-0305-0. [DOI] [PubMed] [Google Scholar]
- 31.O’Connor DS, Wall NR, Porter AC, Altieri DC. A p34(cdc2) survival checkpoint in cancer. Cancer Cell. 2002;2:43–54. doi: 10.1016/s1535-6108(02)00084-3. [DOI] [PubMed] [Google Scholar]
- 32.Izzo AA, Aviello G, Petrosino S, et al. Increased endocannabinoid levels reduce the development of precancerous lesions in the mouse colon. J Mol Med. 2008;86:89–98. doi: 10.1007/s00109-007-0248-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004;23:29–39. doi: 10.1023/a:1025806911782. [DOI] [PubMed] [Google Scholar]
- 34.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–7. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 35.Micale V, Mazzola C, Drago F. Endocannabinoids and neurodegenerative diseases. Pharmacol Res. 2007;56:382–92. doi: 10.1016/j.phrs.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 36.Cota D, Marsicano G, Lutz B, et al. Endogenous cannabinoid system as a modulator of food intake. Int J Obes Relat Metab Disord. 2003;27:289–301. doi: 10.1038/sj.ijo.0802250. [DOI] [PubMed] [Google Scholar]
- 37.Blazquez C, Gonzalez-Feria L, Alvarez L, Haro A, Casanova ML, Guzman M. Cannabinoids inhibit the vascular endothelial growth factor pathway in gliomas. Cancer Res. 2004;64:5617–23. doi: 10.1158/0008-5472.CAN-03-3927. [DOI] [PubMed] [Google Scholar]
- 38.Wilkinson JD, Williamson EM. Cannabinoids inhibit human keratinocyte proliferation through a non-CB1/CB2 mechanism and have a potential therapeutic value in the treatment of psoriasis. J Dermatol Sci. 2007;45:87–92. doi: 10.1016/j.jdermsci.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 39.Han SJ, Conti M. New pathways from PKA to the Cdc2/cyclin B complex in oocytes: Wee1B as a potential PKA substrate. Cell Cycle. 2006;5:227–31. doi: 10.4161/cc.5.3.2395. [DOI] [PubMed] [Google Scholar]
- 40.Caffarel MM, Sarrio D, Palacios J, Guzman M, Sanchez C. Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006;66:6615–21. doi: 10.1158/0008-5472.CAN-05-4566. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.