

Abstract
Disturbances of iron homeostasis are associated with altered susceptibility to infectious disease, but the underlying molecular mechanisms are poorly understood. To study this phenomenon, we examined innate immunity to oral Salmonella infection in Hfe knock-out (Hfe-/-) mice, a model of the human inherited disorder of iron metabolism type I hemochromatosis. Salmonella- and LPS-induced inflammatory responses were attenuated in the mutant animals, with less severe enterocolitis observed in vivo and reduced macrophage TNFα and IL-6 secretion measured in vitro. The macrophage iron exporter ferroportin was up-regulated in the Hfe-/- mice and, correspondingly, intra-macrophage iron levels were lowered. Consistent with the functional importance of these changes, the abnormal cytokine production of the mutant macrophages could be reproduced in wild-type cells by iron chelation, and in a macrophage cell line by over-expression of ferroportin. The results of analyzing specific steps in the biosynthesis of TNFα and IL-6, including intracellular concentrations, post-translational stability and transcript levels, were consistent with reduced translation of cytokine mRNAs in Hfe-/- macrophages. Polyribosome profile analysis confirmed that elevated macrophage ferroportin expression and low intracellular iron impaired the translation of specific inflammatory cytokine transcripts. Our results provide molecular insight into immune function in type I hemochromatosis and other disorders of iron homeostasis, and reveal a novel role for iron in the regulation of the inflammatory response.
Keywords: Macrophages, bacterial infection, inflammation, gene regulation
INTRODUCTION
Iron plays important roles in both pathogen virulence and host anti-microbial resistance (1). As a consequence, disturbances of iron homeostasis in humans lead to changes in susceptibility to infectious disease. Iron overload from dietary sources, excessive hemolysis, or inherited disorders of metabolism predisposes to salmonellosis, tuberculosis and other infections (2-6). Correspondingly, iron deficiency is associated with relative resistance to infection, while iron supplements reverse this effect (7-9). Studies in experimental animals have generally substantiated these clinical observations, although iron depletion has been found to enhance susceptibility to some pathogens (10-13). Abnormalities in immune response, as well as direct effects on microbial growth, have been proposed as explanations for the influence of altered iron homeostasis on the course of infection (1), but the underlying mechanisms have not been well characterized at the molecular level.
Hereditary hemochromatosis is a group of genetically-determined disorders characterized by abnormal accumulation of iron in different tissues (14). The most common form of the disease (type I) is caused by variations in HFE, the hemochromatosis gene, which encodes a class I major histocompatibility complex-like cell surface protein expressed on hepatocytes, macrophages and intestinal crypt cells (15). The HFE protein functions to sense iron status and regulate the expression of hepcidin, a secreted hepatocyte peptide that is the principal regulator of iron homeostasis (16-18). Hepcidin production increases with iron loading and inflammation, and decreases with iron deficiency, anemia, and hypoxia (16, 17). It binds to ferroportin (FPN)3, an iron export protein expressed on the surface of macrophages and on the basolateral membrane of duodenal enterocytes, and induces its internalization and degradation (19). Therefore, hepcidin interrupts cellular iron export at two sites: the intestinal epithelium and tissue macrophages. The HFE-dependent synthesis of hepcidin and the hepcidin-dependent down-modulation of FPN constitute an important regulatory loop that helps to maintain iron homeostasis. In the absence of functional HFE, inappropriately low circulating levels of hepcidin lead to high FPN expression, consequent increases in iron absorption from the gut and release from phagocytes, and ultimate deposition of the metal in sites such as the liver, pancreas and heart (14). Hfe knock-out mice reproduce several features of type I hemochromatosis, including low hepcidin and high liver iron levels (20-23).
Like other disturbances of iron metabolism, type I hemochromatosis is associated with altered responses to infection. Individuals with this disorder have an increased risk of infection with pathogens such as Yersinia enterocolitica and Vibrio vulnificus (24-26). However, it has also been suggested that type I hemochromatosis may confer protection against intra-macrophage pathogens, thereby providing a survival advantage to the host during epidemics of such organisms. This idea has been used to explain the unusually high frequency of HFE gene variants in European populations (27), and has received support from recent cell culture studies carried out by our group (28) and other investigators (29-31). These experiments showed that elevated macrophage FPN expression, an abnormality that occurs in type I hemochromatosis, can inhibit the growth of Salmonella typhimurium and other pathogens inside these cells by lowering intracellular iron concentrations. While these observations illustrate the influence of local iron availability on microbial growth, the potential effects of altered iron levels on host immunity also need to be considered to fully understand how altered iron homeostasis can modulate the course of infectious disease. We address this issue in the present work by examining innate immune responses to Salmonella infection in Hfe-deficient mice.
MATERIALS AND METHODS
Animals
Wild-type C57BL/6 mice were purchased from the Jackson Laboratory. Hfe knock-out mice on the C57BL/6 background were kindly provided by Dr. Nancy C. Andrews (Children’s Hospital, Boston, MA) (22). All mice were bred and housed in a specific pathogen-free facility at the Massachusetts General Hospital. Animals were given water and standard laboratory chow ad libitum and used at 7–12 weeks of age. All animal experiments were approved by the institutional Sub-committee on Research Animal Care.
Salmonella enterocolitis model
The protocol described by Barthel et al. (32) was followed. In brief, mice were given 20 mg of streptomycin in water by gavage with a 21-gauge feeding needle. Twenty-four h later, they were infected orally with 108 cfu of the streptomycin-resistant, wild-type, invasion-competent SL1344 strain of S. typhimurium, and then sacrificed 48 h after infection. Intestinal inflammation was assessed at necropsy based on gross appearance and length of the cecum, histopathology, and quantitative RT-PCR-based measurement of TNFα and IL-6 as described in detail earlier (33). TNFα and IL-6 mRNA levels were normalized to the transcript encoding the housekeeping ribosomal 36B4 protein. Primer sequences for TNFα, IL-6, and 36B4 were as follows: TNF-α sense, 5’-ATGAGCACAGAAAGCATGATC-3, and anti-sense, 5’-TACAGGCTTGTCACTCGAATT-3; IL-6 sense, 5’-TAGTCCTTCCTACCCCAATTTCC -3’, and anti-sense, 5’-TTGGTCCTTAGCCACTCCTTC-3’; 36B4 sense, 5’-AGATGCAGCAGATCCGCAT-3’, and anti-sense, 5’- GTTCTTGCCCATCAGCACC-3’. Bacterial burden in Salmonella-infected animals was assessed, as described previously (33), by homogenizing weighed portions of tissue or fecal pellets in sterile 1% Triton X-100 and plating serial dilutions of the homogenates on LB agar containing 50 μg/ml of streptomycin.
Macrophage activation
Thioglycollate-elicited peritoneal macrophages were prepared and infected with Salmonella (multiplicity of infection approximately 10:1) or treated with LPS (Ultra-pure, List Biological Laboratories, 100 ng/ml) in triplicate wells of a 24-well tissue culture plate as previously described (34). Infections were allowed to proceed for 1 h, after which the cells were washed and placed in medium containing 100 μg/ml of gentamicin to kill extracellular bacteria. Cell supernatants were collected at 3 h after the start of infection or LPS treatment (unless specified otherwise) and analyzed by ELISA for TNFα and IL-6, using antibody pairs obtained from R&D Systems or Pharmingen. To assess intracellular cytokine levels, control and Salmonella-infected macrophages were treated with 10 μg/ml of brefeldin A to block secretion. Protein extracts were prepared and used for the ELISA. Cytokine concentrations were normalized to the protein concentrations of the corresponding cell lysates in order to correct for any well-to-well variations in cell number. Total RNA was prepared using Trizol reagent (Invitrogen) and quantitative RT-PCR carried out to determine TNFα, IL-6 and 36B4 mRNA levels, following methods described previously (33, 34). Numbers of intracellular Salmonella surviving at 3 and 24 h post-infection were determined by gentamicin protection assay, as detailed in earlier publications (33, 34). To evaluate cell viability, the amount of lactate dehydrogenase released into supernatants was assayed using the Cytotox 96 kit from Promega.
Determination of TNFα and IL-6 protein stability
Peritoneal macrophages were treated with 100 ng/ml of LPS for 2 hours in the presence of 10 μg/ml of brefeldin A, followed by addition of 10 μg/ml of cycloheximide to stop translation. Cell lysates were prepared immediately or 4 h after cycloheximide treatment and intracellular cytokine levels were determined by ELISA.
Splenic membrane preparation and western blotting
Spleens from wild-type and Hfe knock-out mice were homogenized in 20 mM HEPES, pH 7.4, 100 mM KCl, 85 mM sucrose, 20 μM EGTA containing protease inhibitors (Roche, Complete Mini). After centrifugation at 10,000 g at 4°C for 10 min, supernatants were collected and centrifuged at 100,000 g at 4°C for 10 min. The resulting pellets were washed and resuspended in RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1.0% NP40, 0.5% deoxycholate, 0.1% SDS, 2 mM EDTA) containing protease inhibitors. The protein concentration of the detergent extracts was determined by Bradford assay. One hundred μg of total protein per lane were separated on a 10% SDS-polyacrylamide gel, and immunoblotted with a polyclonal anti-FPN antibody provided by Dr. David Haile, South Texas Veterans Health Care System. Ponceau staining of the blot was used to confirm equal loading of lanes.
Calcein assay for intracellular iron concentrations
Thioglycollate-elicited peritoneal exudate cells were stained with phycoerythrin-conjugated F4/80 antibody (eBioscience) and 0.5 μM calcein-AM (Molecular Probes) before being subjected to flow cytometry analysis with a Becton–Dickinson FACScan using CELLQUEST software. The fluorescein channel was employed to detect calcein fluorescence after using forward and side scatter characteristics, as well as F4/80 positivity, to gate on macrophages. In some experiments, the membrane permeable iron chelator salicylaldehyde isonicotinoyl hydrazone (SIH, kindly provided by Dr. Prem Ponka, McGill University) was added to the cells at 5 or 10 μM 30 minutes before staining with calcein.
Polyribosome profile analysis
Polyribosomes were separated from monosomes following a published protocol (35). In brief, J774 cells were stimulated with LPS for the times indicated, with 10 μg/ml of cyloheximide being added for the last 10 minutes. The cells were lysed in a Dounce homogenizer in 20 mM HEPES, pH 7.6, 5 mM MgCl2, 100 mM KCl, 1 mM DTT, 100 μg/ml cycloheximide, 0.5% NP-40, 1 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and RNase inhibitor. After spinning out nuclei, the cell extracts were layered over a continuous 10-50% sucrose gradient and centrifuged at 40,000 rpm for 3 hours in an SW40Ti rotor. At the end of the centrifugation, 1 ml fractions were collected from the top of the tube and the absorbance at 260 nm of each fraction was measured. Total RNA was prepared from all the fractions and used for quantitative RT-PCR analysis of IL-6 and β-actin. The amount of the transcript in each fraction was expressed as a percentage of the total present in all fractions. Primer sequences for IL-6 were specified above. Those for β-actin were sense, 5’-TGGAATCCTGTGGCATCCATGAAAC -3’, and anti-sense, 5’-TAAAACGCAGCTCAGTAACAGTCCG -3’, and for IL-1β were sense, 5’-CAACCAACAAGTGATATTCTCCATG-3’, and anti-sense, 5’-GATCCACACTCTCCAGCTGCA-3’.
Statistical analysis
Two-tailed student’s t test was used to compare results. A p value of < 0.05 was considered significant. The figures depict means and standard errors where appropriate.
RESULTS
Salmonella-induced intestinal inflammation is attenuated in Hfe-/- mice
In order to examine the effects of Hfe deficiency on anti-bacterial innate immunity, we compared Salmonella typhimurium-induced enterocolitis (32) in Hfe-/- and wild-type C57BL6 mice. In this experimental system, mice are given a single 20 mg dose of streptomycin orally and then infected 24 h later by oral gavage with the streptomycin-resistant, wild-type, virulent SL1344 strain of S. typhimurium. A robust inflammation of the large intestine develops that peaks at about 48 h post-infection and that is most apparent in the cecum. Using this model, we found that Salmonella-induced intestinal inflammation was clearly attenuated in the Hfe-/- mice as indicated by several parameters. Firstly, the ceca of the infected wild-type animals were pale, thick-walled, shrunken and devoid of stool, in keeping with the development of a vigorous intestinal inflammatory response. In contrast, the organs in the knock-out mice were large, stool-filled and thin-walled (Figure 1A). This difference in macroscopic appearance was quantitated by measurement of cecal length, which indicated that the knock-out ceca had undergone significantly less shrinkage (Figure 1B). On microscopic examination, ceca from the wild-type mice displayed appreciable submucosal edema and a prominent inflammatory infiltrate, whereas both these features were reduced in the Hfe-deficient animals (Figure 1C). Consistent with the lower numbers of neutrophils and monocytes recruited to the intestine of the knock-out mice, quantitative RT-PCR analysis indicated that cecal mRNA levels of the inflammatory mediators TNFα and IL-6 were significantly decreased in these animals (Figure 1D). The reduced intestinal inflammation in the Hfe-/- mice is unlikely to be related to decreased pathogen colonization of the intestine or other tissues since we recovered similar numbers of Salmonella from the mesenteric lymph nodes and livers of the two groups of animals, while the numbers in the feces and spleens were higher in the knock-outs (Figure 1E).
Figure 1. Salmonella-induced intestinal inflammation is attenuated in Hfe-/- mice.
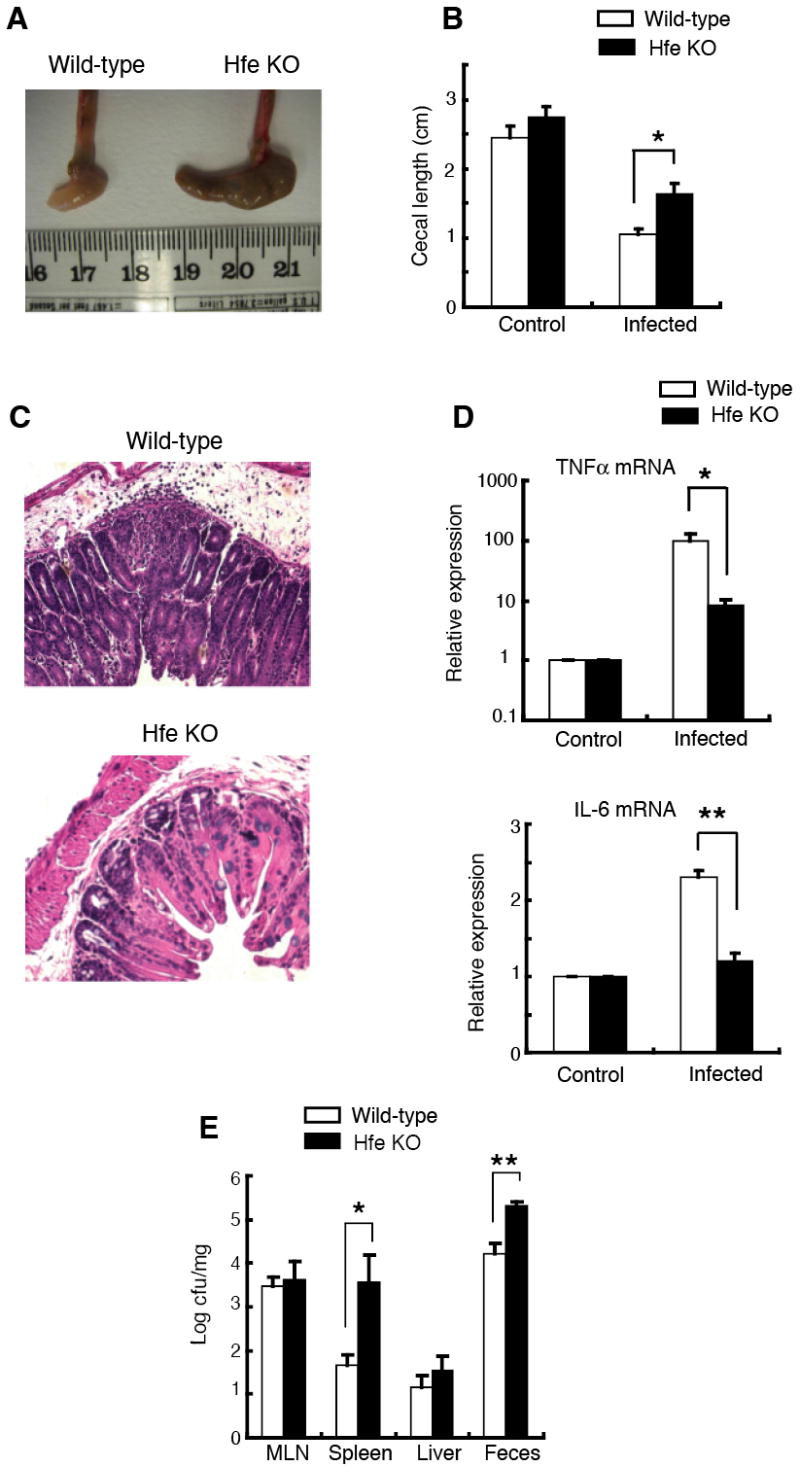
(A) Gross appearance of ceca from wild-type and Hfe knock-out (KO) mice 48 h after infection with Salmonella.
(B) Cecal lengths of wild-type and Hfe-deficient mice 48 h after infection with Salmonella. Ceca from control and infected mice were excised and the maximum length of the organ was recorded. N = 3 (control), N = 6 (infected). *p = 0.019.
(C) Cecal histopathology in wild-type and Hfe knock-out mice 48 h after infection with Salmonella (nominal magnification, 100X).
(D) Cecal TNFα and IL-6 mRNA levels in wild-type and Hfe-/- mice. Total cecal RNA from wild-type and Hfe-/- mice were subjected to quantitative RT-PCR analysis with primers specific for TNFα and IL-6. The mRNA levels of each cytokine were normalized to the housekeeping 36B4 transcript and expressed relative to control. N = 3 (control), N = 6 (infected). *p = 0.008, **p = 0.002.
(E) Numbers of Salmonella recovered from mesenteric lymph nodes (MLN), spleen, liver and stool of wild-type and Hfe-deficient mice 48 h after infection. N = 9. *p = 0.039, **p = 0.044.
Hfe-/- macrophages exhibit altered inflammatory cytokine biosynthesis
Further experiments were directed at finding a mechanistic explanation for the attenuated Salmonella enterocolitis in the Hfe-/- mice. Salmonella-induced intestinal inflammation in mice is independent of T and B lymphocytes and is mediated largely by innate immune mechanisms (32, 36). Epithelial cells, neutrophils, macrophages, dendritic cells and natural killer cells are major contributors to innate immunity in the intestine and gut-associated lymphoid tissue following oral Salmonella infection (32, 36-38). Furthermore, neutrophils and macrophages are important sources of inflammatory mediators such as TNFα (37). Of these cell types, only the macrophage expresses FPN in appreciable amounts (FPN expression in intestinal epithelial cells is largely confined to the duodenum), making it most likely to be influenced by the low hepcidin levels associated with Hfe deficiency (14, 20). We hypothesized, therefore, that altered macrophage function could play a role in the attenuated Salmonella enterocolitis in the Hfe-/- mice.
To examine this possibility, we prepared thioglycollate-elicited peritoneal macrophages from wild-type and Hfe-/- mice and infected them with S. typhimurium strain SL1344. Three h after infection, the amounts of the cytokines TNFα and IL-6 secreted by the mutant macrophages were significantly less than by the wild-type cells (Figure 2A). Differences in the number of bacteria providing the activating stimulus are unlikely to explain the lower response of the mutant cells: the number of bacteria present in the Hfe-/- macrophages was only modestly reduced compared to the wild-type at 3 h after infection and very similar at 24 h post-infection (Figure 2B). Furthermore, we also found significantly reduced secretion of TNFα and IL-6 by the Hfe-deficient macrophages following 3 h of treatment with 100 ng/ml of LPS (Figure 2C), the agonist for Toll-like receptor 4, a key pattern recognition receptor involved in Salmonella-macrophage interactions (34, 39, 40). The levels of Salmonella-induced TNFα and IL-6 proteins that accumulated intracellularly after blocking secretion with brefeldin A were significantly lower in the Hfe-/- macrophages (Figure 3A), suggesting that changes in cytokine secretion do not contribute to the attenuated response of these cells. Cell viability was similar in the wild-type and knock-out macrophages, both under basal conditions and following stimulation (data not shown), making it unlikely that an increase in Salmonella- or LPS-induced cell death in the latter could account for the difference in cytokine production.
Figure 2. Macrophages from Hfe-/- mice express reduced levels of TNFα and IL-6 proteins in response to Salmonella and LPS.
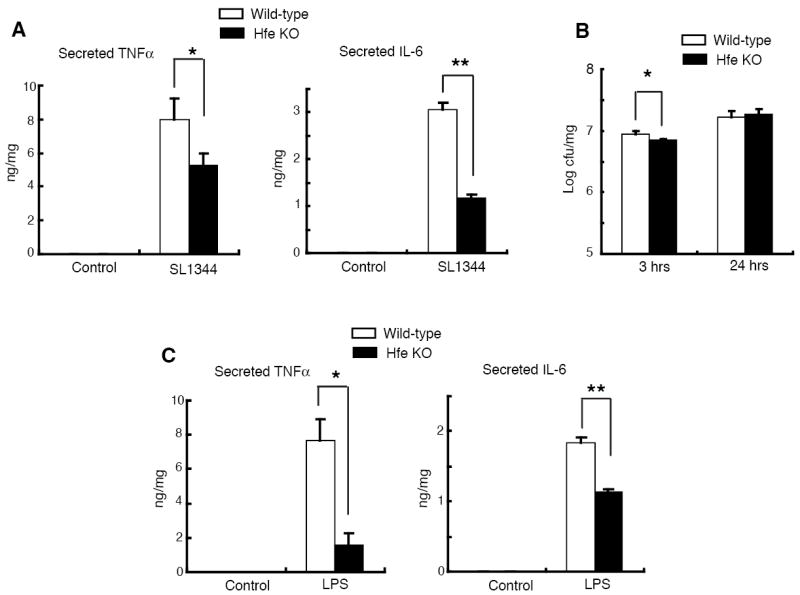
(A)Salmonella (SL1344)-induced TNFα and IL-6 protein secretion in wild-type and Hfe knock-out macrophages at 3 h post-infection. Supernatant cytokine levels were measured by ELISA and normalized to total protein concentrations of cell lysates. *N = 6, p = 0.007; **N = 9, p = 0.009.
(B) Numbers of intracellular Salmonella recovered from wild-type and Hfe knock-out macrophages at 3 or 24 h post-infection. *N = 6, p = 0.03.
(C) TNFα and IL-6 protein secretion in wild-type and Hfe knock-out macrophages induced by 3 h of treatment with 100 ng/ml of LPS, determined as described in (A). *N = 6, p = 0.003; **N = 6, p = 2×10-5.
Figure 3. Macrophages from Hfe-/- mice have normal up-regulation of cytokine mRNAs in response to Salmonella or LPS despite reduced intracellular levels of the proteins.
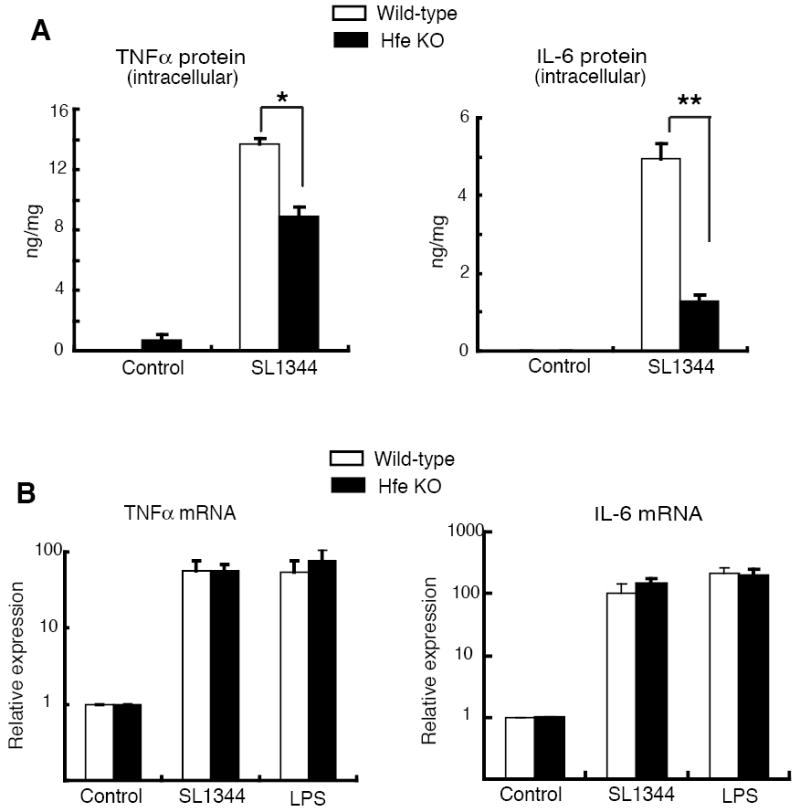
(A) Salmonella-induced intracellular TNFα and IL-6 protein levels at 3 h post-infection after blocking secretion with 10 μg/ml of brefeldin A. Cytokine levels in cell lysates were determined as described in Figure 2A. *N = 3, p = 0.002; **N = 3, p = 0.0009.
(B) Salmonella- and LPS-induced TNFα and IL-6 mRNA levels in wild-type and Hfe-/- macrophages at 3 h post-infection or treatment. Total RNA was subjected to quantitative RT-PCR analysis with primers specific for TNFα or IL-6. The TNFα and IL-6 mRNA levels were normalized to 36B4 and expressed relative to control. N = 3.
To further elucidate the mechanism responsible for decreased inflammatory cytokine production in Hfe-/- macrophages, we examined levels of TNFα and IL-6 mRNA induced by activation with Salmonella or LPS. We found that the transcript levels were similar in the wild-type and mutant cells at 3 h after the start of activation (Figures 3B), even though the amounts of the proteins at this time were clearly lower in the Hfe-/- macrophages (Figures 2A, C and 3A). The discrepancy between the effects of Hfe deficiency on the amounts of TNFα and IL-6 protein versus mRNA suggests that the abnormality in cytokine biosynthesis in Hfe-/- macrophages may be at the level of translation or post-translational protein stability. We found that the stabilities of intracellular TNFα and IL-6 were not significantly altered in the mutant macrophages, as indicated by the rates of decline of the protein levels following inhibition of translation with cycloheximide and blocking of secretion with brefeldin A (Figure 4). Thus, taken together, our results suggest that there is a reduction of cytokine mRNA translation in the Hfe-/- macrophages.
Figure 4. Stability of TNFα and IL-6 proteins in wild-type (WT) and Hfe-/- macrophages.

Cells were treated with 100 ng/ml of LPS for 2 h in the presence of 10 μg/ml of brefeldin A, and then 10 μg/ml of cycloheximide (CHX) was added to stop translation as indicated by the arrow. Cell lysates were prepared immediately or after an additional 4 h, and intracellular TNFα and IL-6 measured by ELISA. Cytokine concentrations were normalized to total protein concentrations of cell lysates. N = 6 (TNFα) or 3 (IL-6).
Increased macrophage FPN expression reduces inflammatory cytokine production via effects on translation
Type I hemochromatosis is characterized by low levels of hepcidin, resulting in increased macrophage FPN expression and relatively low iron concentrations in these cells (14). We carried out experiments to determine whether FPN levels and intracellular iron contributed to the reduced cytokine production by the Hfe-/- macrophages. First, we examined FPN expression in the mutant mice. Although the available anti-FPN antibody was unable to detect expression of the protein on isolated peritoneal macrophages, western blot analysis of membrane fractions prepared from total splenic homogenates did confirm elevated FPN expression in the Hfe-deficient mice (2-3 fold higher than wild-type based on densitometry) (Figure 5A).
Figure 5. Increased expression of FPN contributes to the reduced cytokine production by Hfe-/- macrophages.
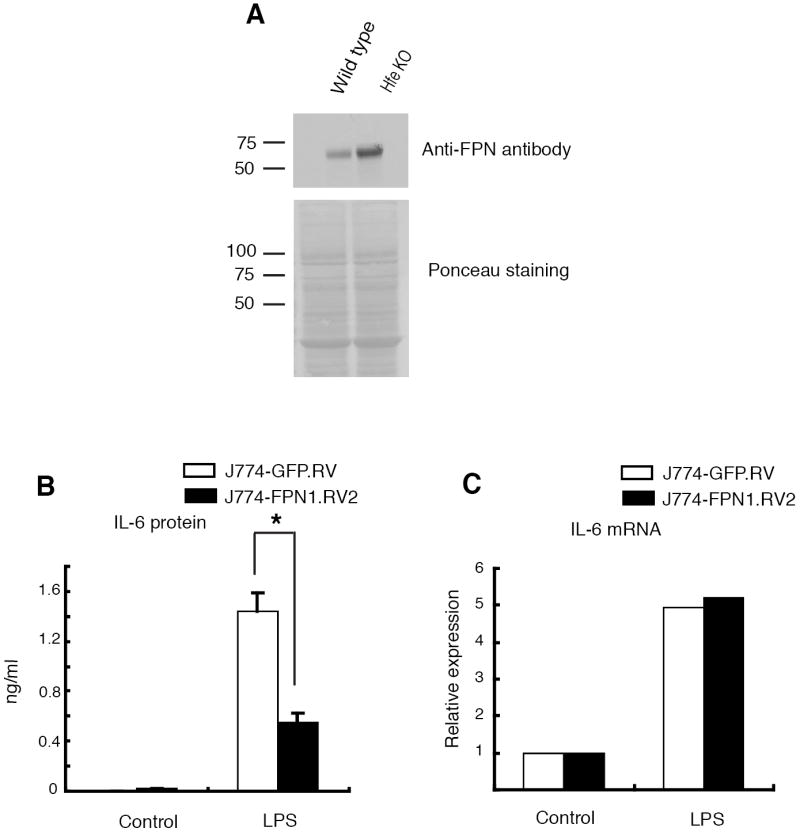
(A) Increased FPN expression in splenic membrane preparation from wild-type and Hfe-/- mice. Splenic membranes were analyzed by western blotting with an anti-FPN antibody. Ponceau staining of the blot confirmed equal lane loading. The numbers indicate molecular weights in kDa.
(B) LPS-induced IL-6 protein secretion in control J774-GFP.RV and FPN over-expressing J774-FPN1.RV2 cells. The cells were stimulated for 24 h with 100 ng/ml of LPS. IL-6 was measured in the supernatants by ELISA. *N = 3, p = 0.014.
(C) LPS-induced IL-6 mRNA levels in control J774-GFP.RV and FPN over-expressing J774-FPN1.RV2 cells. The cells were stimulated for 24 h with 100 ng/ml of LPS. Total RNA was prepared and quantitative RT-PCR was carried out with IL-6 specific primers. IL-6 mRNA levels were normalized to 36B4 and expressed relative to control.
To independently evaluate the effects of elevated FPN expression on macrophage responses, we made the murine macrophage cell line, J774-FPN1.RV2, that stably over-expresses about 3 fold higher levels of FPN than a control transductant cell line, J774-GFP.RV (41). Both cell lines were stimulated with LPS for 24 h and their expression of IL-6 protein and mRNA was analyzed (we found that production of TNFα by these lines was very low and could not be usefully compared). J774-FPN1.RV2 cells produced significantly lower amounts of IL-6 protein than J774-GFP.RV cells, although the levels of the mRNA were comparable (Figures 5B and 5C). Similar results were obtained with 8 h of LPS stimulation (data not shown). These observations resemble those made in the wild-type and Hfe-/- macrophages, and suggest that increased expression of FPN contributes to the reduced cytokine response of the latter.
The results with both the primary macrophages (Figures 2 and 3) and the J774 cells lines (Figures 5B and 5C) raise the possibility of a FPN-dependent alteration in cytokine translation. To test this idea, polyribosome profile analysis was carried out to examine the translational status of IL-6 mRNA in the J774-FPN1.RV2 and J774-GFP.RV cells. Cytoplasmic extracts prepared after 24 h of LPS treatment were fractionated over sucrose gradients and evaluated for differences in distribution of IL-6, IL-1β and β-actin mRNAs (Figure 6). About 70% of the total RNA present in the unfractionated extract was recovered after fractionation. We found that most of the IL-6 transcript was present in the heavy polyribosome-containing fractions in the control J774-GFP.RV cells, indicating that it was being actively translated (Figure 6A). In the FPN over-expressing J774-FPN1.RV2 cells, there was a shift in the distribution of IL-6 mRNA to the lighter fractions (Figure 6A). There was no difference between the cell lines in the distribution of IL-1β or β-actin mRNAs (Figures 6B and 6C). There was also no difference in the distribution of total RNA across the gradient, indicating that the differences in the distribution of IL-6 are unlikely to be the result of selective recovery of certain fractions from the J774-GFP.RV cells versus the J774-FPN1.RV2 cells (Figure 6D). These results provide support for the idea that increased FPN expression leads to reduced translation of specific inflammatory cytokine transcripts, and substantiate the observations in the primary macrophages.
Figure 6. Polyribosome profile analysis demonstrates impaired IL-6 translation in FPN over-expressing J774 cells.

Cytoplasmic extracts of control J774-GFP.RV (white diamonds) and FPN over-expressing J774-FPN1.RV2 cells (black squares) after LPS treatment (100 ng/ml, 24 h) were subjected to sucrose density gradient centrifugation and the relative proportions of (A) IL-6, (B) IL-1β, and (C) β-actin mRNAs in each fraction were determined. (D) The optical density (OD) at 260 nm of each fraction was used as a measure of total RNA.
Decreased intracellular iron inhibits macrophage inflammatory cytokine translation
Phagocytes in type I hemochromatosis have relatively low intracellular iron levels because of increased FPN expression (14). We assessed the intracellular iron concentration in Hfe-/- macrophages using a flow cytometric assay based on the fluorescence of calcein, which is quenched by free iron (42, 43). The results, presented as calcein fluorescence histograms of F4/80-positive peritoneal macrophages, showed that the fluorescence of the Hfe-deficient macrophages was greater than that of the wild-type (Figure 7, left panel), indicating less quenching and therefore a lower level of intracellular iron in the mutant cells. This finding is consistent with elevated FPN expression.
Figure 7. Effects of Hfe deficiency and SIH on intracellular iron levels in macrophages.

Left panel, Calcein fluorescence histograms of F4/80-positive wild-type and Hfe knock-out macrophages. The histograms in light dashed and dotted lines indicate the fluorescence of wild-type and knock-out macrophages, respectively, in the absence of calcein.
Right panel, Calcein fluorescence histograms of F4/80-positive wild-type macrophages in the absence of SIH (control), or in the presence of different concentrations of the iron chelator. The histogram in light dashed lines indicates the fluorescence in the absence of calcein. The FACS data for this experiment were acquired in parallel with those in the left panel. The histograms marked “control” and “no calcein” are identical to the corresponding wild-type histograms in the left panel.
To determine if the low intracellular iron in the Hfe-/- macrophages contributed to their attenuated cytokine production, we made use of the membrane permeable iron chelator SIH (44). Treatment of wild-type macrophages with SIH resulted in a dose-dependent increase in calcein fluorescence (Figure 7, right panel), indicating a decrease in intracellular iron. SIH treatment also led to a significant reduction in the amount of TNFα secreted in response to 3 h of LPS stimulation (Figure 8A), with a less dramatic influence on the level of the corresponding mRNA (Figure 8B). The doses of SIH used in these experiments had no effect on cell viability (data not shown). Furthermore, the inhibitory effect of SIH on TNFα secretion was reversed in a dose-dependent fashion by addition of ferrous sulphate to the medium (Figure 8C), indicating that the SIH was acting by chelating iron. Consistent with the decrease in TNFα production caused by iron chelation, addition of ferrous sulphate alone to the medium resulted in an increase in LPS-induced secretion of the cytokine (Figure 8C). SIH treatment also inhibited the LPS-induced secretion of IL-6, with a less pronounced inhibition of expression of the mRNA (Figures 8D and 8E). Thus, the effects of SIH treatment were similar to those of Hfe deficiency, suggesting that lowered intracellular iron concentrations contributed to the impaired cytokine biosynthesis by the mutant macrophages. The discordance between the effects of SIH on the cytokine protein and transcript levels is consistent with the notion that lowered intracellular iron inhibits translation of the mRNAs. To substantiate this idea, we used polyribosome profile analysis to examine the effect of SIH on the translation of IL-6 mRNA in the J774-GFP.RV cell line. Iron chelation caused a shift in the distribution of the IL-6 transcript to less dense fractions (Figure 9A), indicating a decrease in the proportion of actively translated message and mimicking the effect of FPN over-expression (Figure 6A). The distribution of ß-actin mRNA and total RNA revealed no major differences between control and SIH treated cells (Figures 9B and 9C). These results indicate that decreased intracellular iron selectively impairs translation of IL-6 mRNA.
Figure 8. Decreased intracellular iron contributes to attenuated cytokine production by Hfe-/- macrophages.
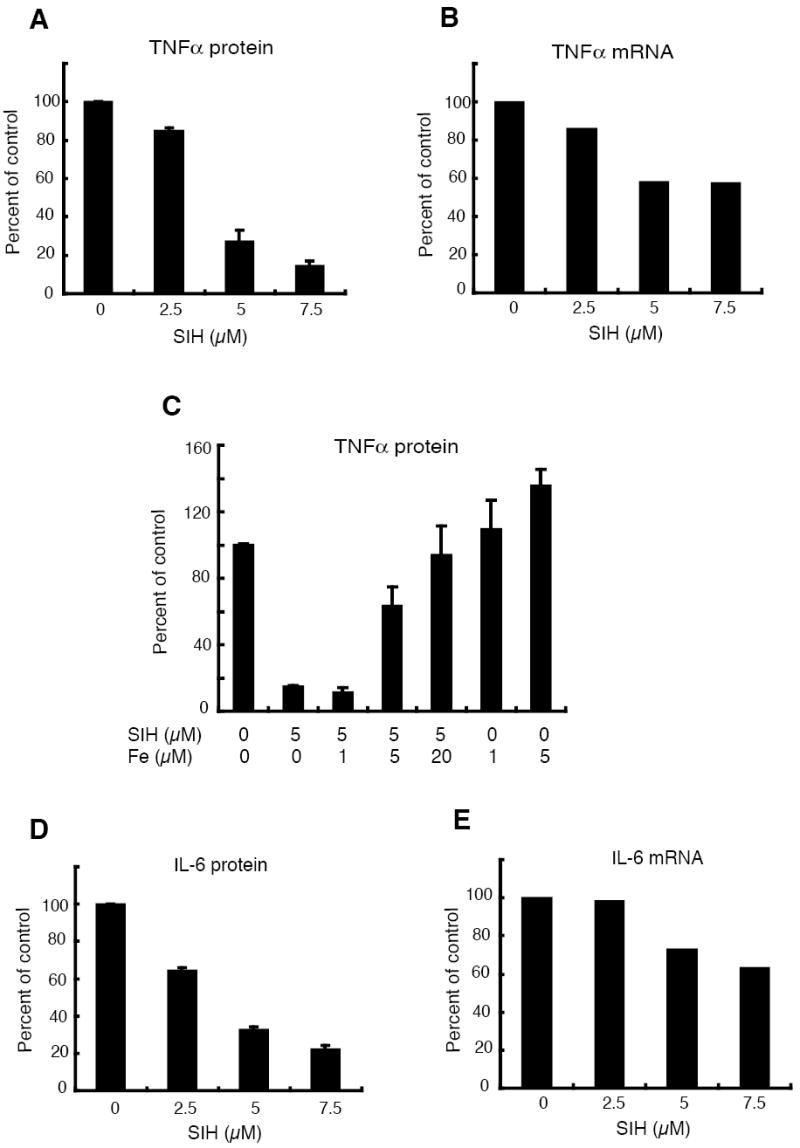
(A) Effects of SIH on TNFα protein secreted by wild-type macrophages in response to LPS. The macrophages were treated with 100 ng/ml of LPS for 3 h in the presence or absence of different amounts of SIH as indicated. TNFα concentrations in the supernatants were determined by ELISA and expressed as a percentage of that of the control cells (not treated with SIH). N = 3.
(B) Effect of SIH on TNFα mRNA levels in wild-type macrophages in response to LPS. The macrophages were treated with 100 ng/ml of LPS for 3 h in the presence or absence of different amounts of SIH as indicated. Total RNA was prepared and TNFα mRNA levels determined by quantitative RT-PCR. After normalization to 36B4 mRNA, the cytokine transcript levels were expressed as a percentage of that of the control cells (not treated with SIH).
(C) Effect of iron on LPS-induced TNFα secretion in wild-type macrophages. The macrophages were treated with 100 ng/ml of LPS for 3 h in the presence or absence of 5 μM SIH as indicated, with or without addition of the indicated amounts of iron in the form of ferrous sulphate. TNFα concentrations in the supernatants were determined by ELISA and expressed as a percentage of that of the control cells (treated with LPS alone). N = 6.
(D) Effects of SIH on IL-6 protein secreted by wild-type macrophages in response to LPS. The macrophages were treated with 100 ng/ml of LPS for 3 h in the presence or absence of different amounts of SIH as indicated. IL-6 concentrations in the supernatants were determined by ELISA and expressed as a percentage of that of the control cells (not treated with SIH). N = 3.
(E) Effect of SIH on IL-6 mRNA levels in wild-type macrophages in response to LPS. The macrophages were treated with 100 ng/ml of LPS for 3 h in the presence or absence of different amounts of SIH as indicated. Total RNA was prepared and IL-6 mRNA levels determined by quantitative RT-PCR. After normalization to 36B4 mRNA, the cytokine transcript levels were expressed as a percentage of that of the control cells (not treated with SIH).
Figure 9. Polyribosome profile analysis demonstrates impaired IL-6 translation in J774 cells following iron chelation with SIH.

Cytoplasmic extracts of the J774-GFP.RV cells not treated (white diamonds) or treated with 5 μM SIH (black squares) and subjected to LPS stimulation (100 ng/ml, 8 h) were analyzed by sucrose density gradient centrifugation and the relative proportions of (A) IL-6 and (B) β-actin mRNAs in each fraction were determined. (C) The optical density at 260 nm of each fraction was used as a measure of total RNA.
DISCUSSION
Our observations demonstrate that a mouse model of type I hemochromatosis is associated with an attenuated inflammatory response to Salmonella infection, both in vivo and in isolated macrophages. They indicate further that the underlying mechanism involves reduced translation of pro-inflammatory cytokine mRNAs. The altered cytokine biosynthesis appears to be caused by the lowered intracellular iron levels that occurs in macrophages in type I hemochromatosis because of elevated FPN expression. Besides shedding light on the immunological consequences of a relatively common disorder of iron metabolism, our findings reveal a novel role for iron in the regulation of cytokine mRNA translation.
Iron is known to regulate the expression of several genes at the transcriptional level, most prominently via the generation of reactive oxygen species and their effects on the activity of NF-κB and other transcription factors (45). It also has a well-documented function in controlling the stability and translation of transcripts that are involved in iron homeostasis (46). However, to our knowledge, there are no previous reports of iron-dependent translational control of cytokine biosynthesis. The known post-transcriptional effects of iron are mediated via iron response elements (IREs) present in the untranslated regions of mRNAs, which are bound by iron regulatory proteins (IRPs) under conditions of low iron concentration. There are no canonical IREs in the untranslated regions of the TNFα or IL-6 messages. However, IRP-dependent iron responsiveness can be conferred by atypical IREs that may be missed by standard in silico analysis (47), raising the possibility that such cryptic elements may be involved in the iron-dependent regulation of TNFα and IL-6 translation. Alternately, iron may exert its effects via the AU-rich sequences present in the 3’ untranslated regions of many cytokine transcripts, including TNFα and IL-6 (48). Several proteins have been shown to bind to these elements, with effects on both mRNA stability and translation (35, 48), and it is possible that changes in intracellular iron may influence these interactions, either directly or indirectly.
Although further work will be required to determine precisely how intracellular iron regulates the translation of TNFα and IL-6 mRNAs, our data indicate that this regulatory mechanism has important consequences for host-pathogen interactions in the experimental model of type I hemochromatosis. TNFα and IL-6 play important roles in inflammatory cell recruitment induced by Salmonella, and in innate resistance to this pathogen (37, 38, 49-51). It is not surprising, therefore, that decreased production of these cytokines results in the attenuation of Salmonella-induced intestinal inflammation observed in the Hfe-/- mice.
It is worth mentioning here that earlier work from our laboratory as well those of other investigators has shown that increased macrophage FPN expression inhibits the growth of several intracellular pathogens, including Salmonella (28-31). It is therefore somewhat surprising that Hfe deficiency, with its associated increase in macrophage FPN expression, enhanced recovery of Salmonella from infected tissues in vivo (Figure 1E), and only modestly reduced growth of the pathogen in macrophages in vitro (Figure 2B). The increase in tissue bacterial burden in the Hfe-/- mice in vivo is probably explained by the attenuated inflammatory response in these animals, which would lead to reduced recruitment of phagocytic cells as well as impairment of macrophage-intrinsic microbicidal mechanisms (52). The lack of a more robust inhibitory effect on Salmonella growth in the Hfe-deficient macrophages in vitro is more puzzling, and we do not have a definite explanation for the discrepancy with the earlier results. Differences in experimental methodology could be contributing factors, and it is also possible that the level of intracellular iron in the Hfe-/- macrophages is simply not low enough to have direct effects on the multiplication of Salmonella. Of note, variations in macrophage iron status may also help to explain some of the experiment-to-experiment variability in cytokine responses observed in the present study.
Humans with type I hemochromatosis are unusually susceptible to certain bacterial infections (24-26). Although increased iron availability to the infecting pathogen may be one explanation for this susceptibility, our findings raise the possibility that alterations in the host immune response could also be involved. Interestingly, it has been reported that monocytes from hemochromatosis patients have decreased LPS-induced TNFα production, although the mechanism has not been elucidated previously (53). This observation highlights the clinical relevance of our findings in the Hfe-deficient mice, and is potentially explained by the iron-dependent translational control of cytokine biosynthesis that our studies have revealed. An attenuated inflammatory response could also contribute to the phenomenon of epidemic pathogen selection that has been used to explain the high frequency of HFE gene variants in Caucasian populations (27). In support of this idea, it has been shown recently that decreased inflammation in a mouse model of pneumonic plague is associated with improved host survival (54). It is an intriguing possibility that individuals with HFE deficiencies may have had a survival advantage during the European plague epidemics because they expressed reduced amounts of inflammatory cytokines.
Do our findings have implications for disorders of iron homeostasis other than type I hemochromatosis? Based on our results, decreased inflammatory responses may be expected in hemochromatosis types II and III, where low circulating levels of hepcidin would lead to up-regulation of macrophage FPN and decreased levels of iron in these cells (14). This mechanism may also apply to those forms of type IV hemochromatosis in which FPN mutations render the protein insensitive to hepcidin-mediated down-regulation (55). A similar situation may prevail in thalassemia, where it has been shown recently that hepcidin expression is suppressed by high levels of growth differentiation factor 15 (56). Attenuated inflammatory responses occurring secondary to low intra-macrophage iron could contribute to the increased risk of infection associated with hemolytic disorders (4, 6). Experiments to examine these possibilities are clearly warranted, and will help to shed additional light on the interconnections between iron homeostasis and innate immunity.
Acknowledgments
We are grateful to Dr. Nancy Andrews for providing the Hfe knock-out mice, to Dr. David Haile for the anti-FPN antibody, and to Dr. Prem Ponka for his gift of SIH.
This work was supported by a pilot feasibility project grant from the Harvard Clinical Nutrition Research Center to BJC, by an unrestricted educational grant from Wyeth Nutrition to LW, and by NIH grants R21AI06461 (BJC), and R01ES014638 and R01DK064750 (MW-R).
Footnotes
Abbreviations used: FPN, ferroportin; IRE, iron response element; IRP, iron regulatory protein; SIH, salicylaldehyde isonicotinoyl hydrazone.
DISCLOSURES The authors have no conflicting financial interests.
References
- 1.Schaible UE, Kaufmann SH. Iron and microbial infection. Nat Rev Microbiol. 2004;2:946–953. doi: 10.1038/nrmicro1046. [DOI] [PubMed] [Google Scholar]
- 2.Gangaidzo IT, Moyo VM, Mvundura E, Aggrey G, Murphree NL, Khumalo H, Saungwenre T, Kasvosve II, Gomo ZA, Rouault T, Boelaert JR, Gordeuk VR. Association of pulmonary tuberculosis with increased dietary iron. J Infect Dis. 2001;184:936–939. doi: 10.1086/323203. [DOI] [PubMed] [Google Scholar]
- 3.Gordeuk VR, McLaren CE, MacPhail AP, Deichsel G, Bothwell TH. Associations of iron overload in Africa with hepatocellular carcinoma and tuberculosis: Strachan’s 1929 thesis revisited. Blood. 1996;87:3470–3476. [PubMed] [Google Scholar]
- 4.Magnus SA, Hambleton IR, Moosdeen F, Serjeant GR. Recurrent infections in homozygous sickle cell disease. Arch Dis Child. 1999;80:537–541. doi: 10.1136/adc.80.6.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moyo VM, Gangaidzo IT, Gordeuk VR, Kiire CF, Macphail AP. Tuberculosis and iron overload in Africa: a review. Centr Afr Med J. 1997;43:334–339. [PubMed] [Google Scholar]
- 6.Wanachiwanawin W. Infections in E-beta thalassemia. J Pediatr Hematol Oncol. 2000;22:581–587. doi: 10.1097/00043426-200011000-00027. [DOI] [PubMed] [Google Scholar]
- 7.Murray MJ, Murray AB, Murray MB, Murray CJ. The adverse effect of iron repletion on the course of certain infections. Br Med J. 1978;2:1113–1115. doi: 10.1136/bmj.2.6145.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nyakeriga AM, Troye-Blomberg M, Dorfman JR, Alexander ND, Back R, Kortoak M, Chemtai AK, Marsh K, Williams TN. Iron deficiency and malaria among children living on the coast of Kenya. J Infect Dis. 2004;190:439–447. doi: 10.1086/422331. [DOI] [PubMed] [Google Scholar]
- 9.Sazawal S, Black RE, Ramsan M, Chwaya HM, Stoltzfus RJ, Dutta A, Dhingra U, Kabole I, Deb S, Othman MK, Kabole FM. Effects of routine prophylactic supplementation with iron and folic acid on admission to hospital and mortality in preschool children in a high malaria transmission setting: community-based, randomized, placebo-controlled trial. Lancet. 2006;367:133–143. doi: 10.1016/S0140-6736(06)67962-2. [DOI] [PubMed] [Google Scholar]
- 10.Alford CE, King TE, Jr, Campbell PA. Role of transferrin, transferrin receptors, and iron in macrophage listericidal activity. J Exp Med. 1991;174:459–466. doi: 10.1084/jem.174.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins HL, Kaufmann SHE, Schaible UE. Iron chelation via deferoxamine exacerbates experimental salmonellosis via inhibition of the nicotinamide adenine dinucleotide phosphate oxidase-dependent respiratory burst. J Immunol. 2002;168:3458–3463. doi: 10.4049/jimmunol.168.7.3458. [DOI] [PubMed] [Google Scholar]
- 12.Puschmann M, Ganzoni AM. Increased resistance of iron-deficient mice to Salmonella infection. Infect Immun. 1977;17:663–664. doi: 10.1128/iai.17.3.663-664.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roy M-F, Riendeau N, Bedard C, Helie P, Min-Oo G, Turcotte K, Gros P, Canonne-Hergaux F, Malo D. Pyruvate kinase deficiency confers susceptibility to Salmonella typhimurium infection in mice. J Exp Med. 2007;204:2949–2961. doi: 10.1084/jem.20062606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pietrangelo A. Hereditary hemochromatosis. Annu Rev Nutr. 2006;26:251–270. doi: 10.1146/annurev.nutr.26.061505.111226. [DOI] [PubMed] [Google Scholar]
- 15.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R, Jr, Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Bacon BR, Wolff RK. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 16.Andrews NC. Iron homeostasis. Annu Rev Physiol. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- 17.Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr. 2006;26:323–342. doi: 10.1146/annurev.nutr.26.061505.111303. [DOI] [PubMed] [Google Scholar]
- 18.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281:28494–28498. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 19.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding ferroportin and inducing its degradation. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 20.Ahmad KA, Ahmann JR, Migas MC, Waheed A, Britton RS, Bacon BR, Sly WS, Fleming RE. Decreased liver hepcidin expression in the Hfe knock-out mouse. Blood Cells Mol Dis. 2002;29:361–366. doi: 10.1006/bcmd.2002.0575. [DOI] [PubMed] [Google Scholar]
- 21.Bahram S, Gilfillan S, Kuhn LC, Moret R, Schulze JB, Lebeau A, Schumann K. Experimental hemochromatosis due to MHC class I HFE deficiency: immune status and iron metabolism. Proc Natl Acad Sci USA. 1999;96:13312–13317. doi: 10.1073/pnas.96.23.13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levy JE, Montross LK, Cohen DE, Fleming MD, Andrews NC. The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood. 1999;94:9–11. [PubMed] [Google Scholar]
- 23.Zhou XY, Tomatsu S, Fleming RE, Parkkila S, Waheed A, Jiang J, Fei Y, Brundt EM, Ruddy DA, Prass CE, Schatzman RC, O’Neill R, Britton RS, Bacon BR, Sly WS. HFE gene knock-out produces a mouse model of hereditary hemochromatosis. Proc Natl Acad Sci USA. 1998;95:2492–2497. doi: 10.1073/pnas.95.5.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bullen JJ, Spalding PB, Ward CG, Gutteridge JM. Hemochromatosis, iron and septicemia caused by Vibrio vulnificus. Arch Intern Med. 1991;151:1606–1609. [PubMed] [Google Scholar]
- 25.Doherty CP. Host-pathogen interactions: the role of iron. J Nutr. 2007;137:1341–1344. doi: 10.1093/jn/137.5.1341. [DOI] [PubMed] [Google Scholar]
- 26.Gerhard GS, Levin KA, Price GJ, Wojnar MM, Chorney MJ, Belchis DA. Vibrio vulnificus septicemia in a patient with the hemochromatosis HFE C282Y mutation. Arch Pathol Lab Med. 2001;125:1107–1109. doi: 10.5858/2001-125-1107-VVSIAP. [DOI] [PubMed] [Google Scholar]
- 27.Moalem S, Weinberg ED, Percy ME. Hemochromatosis and the enigma of misplaced iron: implications for infectious disease and survival. Biometals. 2004;17:135–139. doi: 10.1023/b:biom.0000018375.20026.b3. [DOI] [PubMed] [Google Scholar]
- 28.Chlosta S, Fishman DS, Harrington L, Johnson EE, Knutson MD, Wessling-Resnick M, Cherayil BJ. The iron efflux protein ferroportin regulates the intracellular growth of Salmonella enterica. Infect Immun. 2006;74:3065–3067. doi: 10.1128/IAI.74.5.3065-3067.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nairz M, Theuri I, Ludwiczek S, Theuri M, Mair SM, Fritsche G, Weiss G. The coordinated regulation of iron homeostasis in murine macrophages limits the availability of iron for intracellular Salmonella typhimurium. Cell Microbiol. 2007;9:2126–2140. doi: 10.1111/j.1462-5822.2007.00942.x. [DOI] [PubMed] [Google Scholar]
- 30.Olakanmi O, Schlesinger LS, Britigan BE. Hereditary hemochromatosis results in decreased iron acquisition and growth by Mycobacterium tuberculosis within human macrophages. J Leukoc Biol. 2007;81:195–204. doi: 10.1189/jlb.0606405. [DOI] [PubMed] [Google Scholar]
- 31.Paradkar P, De Domenico I, Durchfort N, Zohn I, Kaplan J, Ward DM. Iron-depletion limits intracellular bacterial growth in macrophages. Blood. 2008 doi: 10.1182/blood-2007-12-126854. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, Hardt WD. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun. 2003;71:2839–2858. doi: 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhee SJ, Walker WA, Cherayil BJ. Developmentally regulated intestinal expression of IFNγ and its target genes and the age-specific response to enteric Salmonella infection. J Immunol. 2005;175:1127–1136. doi: 10.4049/jimmunol.175.2.1127. [DOI] [PubMed] [Google Scholar]
- 34.Li Q, Cherayil BJ. Role of Toll-like receptor 4 in macrophage activation and tolerance during Salmonella enterica serovar Typhimurium infection. Infect Immun. 2003;71:4873–4882. doi: 10.1128/IAI.71.9.4873-4882.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu C, York B, Wang S, Feng Q, Xu J, O’Malley BW. An essential function of the SRC-3 co-activator in suppression of cytokine mRNA translation and inflammatory response. Mol Cell. 2007;25:765–778. doi: 10.1016/j.molcel.2007.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harrington L, Srikanth CV, Antony R, Shi HN, Cherayil BJ. A role for natural killer cells in intestinal inflammation caused by infection with Salmonella enterica serovar Typhimurium. FEMS Immunol Med Microbiol. 2007;51:372–380. doi: 10.1111/j.1574-695X.2007.00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rydström A, Wick MJ. Monocyte recruitment, activation, and function in the gut-associated lymphoid tissue during oral Salmonella infection. J Immunol. 2007;178:5789–5801. doi: 10.4049/jimmunol.178.9.5789. [DOI] [PubMed] [Google Scholar]
- 38.Wick MJ. Living in the danger zone: innate immunity to Salmonella. Curr Opin Microbiol. 2004;7:51–57. doi: 10.1016/j.mib.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 39.Royle MC, Tötemeyer S, Alldridge LC, Maskell DJ, Bryant CE. Stimulation of Toll-like receptor 4 by lipopolysaccharide during cellular invasion by live Salmonella typhimurium is a critical but not exclusive event leading to macrophage responses. J Immunol. 2003;170:5445–5454. doi: 10.4049/jimmunol.170.11.5445. [DOI] [PubMed] [Google Scholar]
- 40.Weiss DS, Raupach B, Takeda K, Akira S, Zychlinsky S. Toll-like receptors are temporally involved in host defense. J Immunol. 2004;172:4463–4469. doi: 10.4049/jimmunol.172.7.4463. [DOI] [PubMed] [Google Scholar]
- 41.Knutson MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci USA. 2005;102:1324–1328. doi: 10.1073/pnas.0409409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown JX, Buckett PD, Wessling-Resnick M. Identification of small molecule inhibitors that distinguish between transferrin bound iron uptake and transferrin mediated iron transport. Chem Biol. 2004;11:407–416. doi: 10.1016/j.chembiol.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 43.Kakhlon O, Cabantchik ZL. The labile iron pool: characterization, measurement and participation in cellular processes. Free Radic Biol Med. 2002;33:1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 44.Simunek T, Boer C, Bouwman RA, Vlasblom R, Versteilen AM, Sterba M, Gersl V, Hrdina R, Ponka P, de Lange JJ, Paulus WJ, Musters RJ. SIH – a novel lipophilic iron chelator – protects H9c2 cardiomyoblasts from oxidative stress-induced mitochondrial injury and cell death. J Mol Cell Cardiol. 2005;39:345–354. doi: 10.1016/j.yjmcc.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 45.Templeton DM, Liu Y. Genetic regulation of cell function in response to iron overload or chelation. Biochim Biophys Acta. 2003;1619:113–124. doi: 10.1016/s0304-4165(02)00497-x. [DOI] [PubMed] [Google Scholar]
- 46.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 47.Sanchez M, Galy B, Muckenthaler MU, Hentze MW. Iron-regulatory proteins limit hypoxia-inducible factor-2α expression in iron deficiency. Nat Struct Mol Biol. 2007;14:420–426. doi: 10.1038/nsmb1222. [DOI] [PubMed] [Google Scholar]
- 48.Anderson P. Post-transcriptional control of cytokine production. Nat Immunol. 2008;9:353–359. doi: 10.1038/ni1584. [DOI] [PubMed] [Google Scholar]
- 49.Arnold JW, Niesel DW, Annable CR, Hess CB, Asuncion M, Cho YJ, Peterson JW, Klimpel GR. TNFα mediates the early pathology in Salmonella infection of the gastrointestinal tract. Microb Pathog. 1993;14:217–227. doi: 10.1006/mpat.1993.1021. [DOI] [PubMed] [Google Scholar]
- 50.Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175:3463–3468. doi: 10.4049/jimmunol.175.6.3463. [DOI] [PubMed] [Google Scholar]
- 51.Nadeau WJ, Pistole TG, McCormick BA. Polymorphonuclear leukocyte migration across model intestinal epithelia enhances Salmonella typhimurium killing via the epithelial derived cytokine, IL-6. Microbes Infect. 2002;4:1379–1387. doi: 10.1016/s1286-4579(02)00020-5. [DOI] [PubMed] [Google Scholar]
- 52.Vazquez-Torres A, Fantuzzi G, Edwards CK, 3rd, Dinarello CA, Fang FC. Defective localization of the NADPH phagocyte oxidase to Salmonella-containing phagosomes in TNF p55 receptor-deficient mice. Proc Natl Acad Sci USA. 2001;98:2561–2565. doi: 10.1073/pnas.041618998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gordeuk VR, Ballou S, Lozanski G, rittenham GM. Decreased concentrations of TNFα in supernatants of monocytes from homozygotes for hereditary hemochromatosis. Blood. 1992;79:1855–1860. [PubMed] [Google Scholar]
- 54.Lathem WA, Price PA, Miller VL, Goldman WE. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science. 2007;315:509–513. doi: 10.1126/science.1137195. [DOI] [PubMed] [Google Scholar]
- 55.Drakesmith H, Schimanski LM, Ormerod E, Merryweather-Clarke AT, Viprakasit V, Edwards JP, Sweetland E, Bastin JM, Cowley D, Chinthammitr Y, Robson KJ, Townsend AR. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood. 2005;106:1092–1097. doi: 10.1182/blood-2005-02-0561. [DOI] [PubMed] [Google Scholar]
- 56.Tanno T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, Moroney JW, Reed CH, Luban NL, Wang RH, Eling TE, Childs R, Ganz T, Leitman SF, Fucharoen S, Miller JL. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096–1101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]