

Abstract
Site-specific protein labeling methods allow cell biologists to access the vast array of existing chemical probes for the study of specific proteins of interest in the live cell context. Here we describe the use of the transglutaminase enzyme from guinea pig liver (gpTGase), whose natural function is to cross-link glutamine and lysine sidechains, to covalently conjugate various small molecule probes to recombinant proteins fused to a 6- or 7-amino acid transglutaminase recognition sequence, called a Q-tag. We demonstrate labeling of Q-tag fusion proteins both in vitro and on the surface of living mammalian cells with biotin, fluorophores, and a benzophenone photoaffinity probe. To illustrate the utility of this labeling, we tagged the NF-κB p50 transcription factor with benzophenone, cross-linked with UV light, and observed increased levels of p50 homodimerization in the presence of DNA and the binding protein myotrophin
Compared to green fluorescent protein (GFP), non-genetically-encoded probes such as organic dyes and inorganic nanoparticles offer the possibility of smaller probe sizes and access to a much wider array of functionality, from photocrosslinking and photoregulation to imaging of cellular processes by non-optical methods such as electron microscopy, magnetic resonance imaging, and positron emission tomography. The major limitation to the use of these probes in cells, however, is the shortage of robust methods for targeting them to specific proteins of interest. Recently, many new methodologies for protein labeling in cells have been reported.1–3 Most of these use special peptide or protein handles, which are genetically fused to the target protein and recruit the probe of interest via a covalent or non-covalent interaction. There is great variation among these methods in terms of labeling specificity, speed, stability, tag size, toxicity, and versatility for probe structure and cell type, and no single method yet excels in all these respects. Thus new methods are still needed to facilitate the routine use of non-genetically-encoded probes in the cellular context.
Transglutaminases (TGases) are enzymes that catalyze amide bond formation between glutamine and lysine sidechains, with the loss of ammonia (Figure 1). They are ubiquitous in multicellular organisms and function in protein cross-linking events in migration, apoptosis, and wound healing, as well as in physiological disorders such as Huntington’s disease and Celiac Sprue.4–6 One of the most well-studied TGases is the guinea pig liver transglutaminase (gpTGase), which is a 77 kD monomeric protein expressed in the cytosol.5 gpTGase has unique properties which make it ideal for protein labeling applications: it exhibits high specificity for its glutamine-containing protein substrate, but wide tolerance for the structure of the amine-containing substrate.7 Instead of lysine, amines as diverse as fluorescein cadaverine8 and biotin cadaverine9 can be utilized by gpTGase. Also peptide substrates have been found which are efficiently modified by gpTGase both in isolation and when fused to recombinant proteins.10,11 gpTGase has previously been used to label Q-tagged proteins in vitro; for instance, interleukin-2, glutathione S-transferase, and insulin have been tagged with monodansyl cadaverine,10 fluorescein cadaverine,11 and an alkylamine derivative of lactose,12 respectively. However, the use of TGases for labeling specific Q-tagged proteins in cells or complex mixtures has never been reported.
Figure 1.
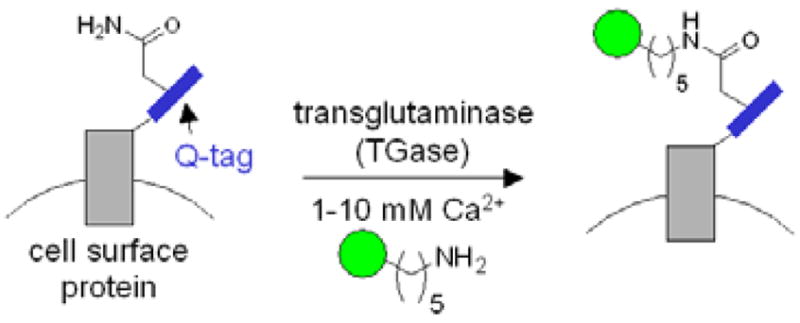
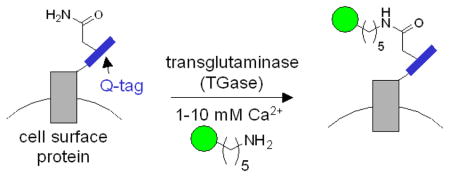
Transglutaminase-catalyzed ligation of cadaverine-functionalized probes (green circle) to Q-tag-fused recombinant cell surface proteins. TGase ligates the glutamine sidechain of the Q-tag (blue) to the amine probe, giving an amide bond and releasing ammonia. In this study, three Q-tag peptide substrates were used: Q1 (PNPQLPF), Q2 (PKPQQFM), and Q3 (GQQQLG).
We selected three Q-tag substrates of gpTGase, and appended them to the N-terminus of cyan fluorescent protein (CFP). The Q1 (PNPQLPF),13 Q2 (PKPQQFM),10,11 and Q3 (GQQQLG)14 fusions to CFP were each labeled efficiently (~70–80% labeling extent, data not shown) and specifically (alanine mutations within the Q-tags suppressed labeling by 3.3–16.7 fold) by purified gpTGase with fluorescein cadaverine in vitro (Figure S1). To test gpTGase for labeling of cell surface proteins (Figure 1), we expressed each Q-tag CFP fusion on the surface of HeLa (Figure 2) and labeled, in the presence of 1–10 mM Ca2+, with either biotin cadaverine (followed by detection with streptavidin-Alexa 568 conjugate) or Alexa 568 cadaverine directly. Both probes were successfully conjugated to all three Q-tags, and Figure 2 shows that Alexa 568 conjugation to Q2 was both site-specific (signal intensity ratio compared to labeling of the alanine mutant 2.4–5.7) and enzyme-dependent. Biotin cadaverine conjugation to Q3-tagged epidermal growth factor receptor (EGFR) was also successful (Figure S2), and labeling did not impair receptor response to EGF (data not shown). As an additional test of the potential cytotoxicity of the labeling conditions, we monitored the intracellular Ca2+ levels using Calcium Green-1™,15 and observed no change during labeling (data not shown). HeLa growth rate was also unaffected 24 hr after labeling.
Figure 2.
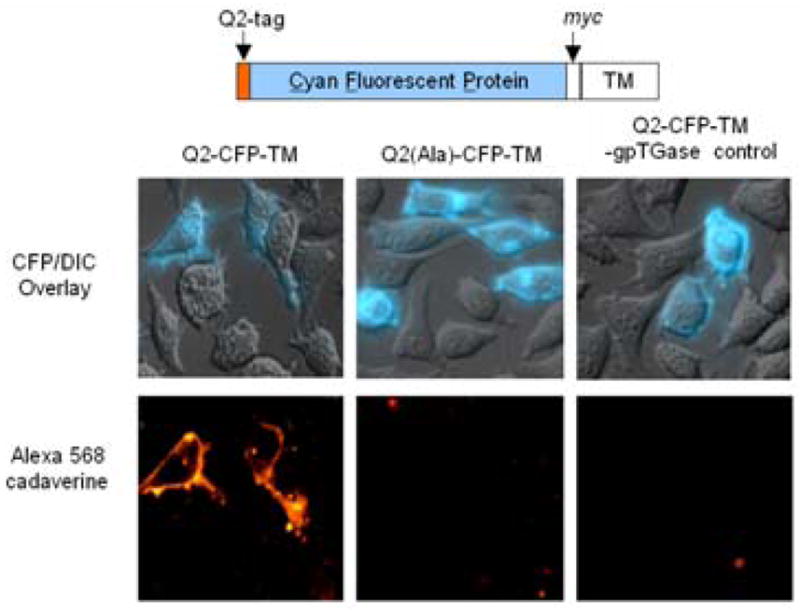
TGase-catalyzed labeling of Q-tagged cell surface protein. The domain structure of Q2-CFP-TM is shown, from N- to C-terminus. TM is the transmembrane domain of the PDGF receptor, which targets Q2-CFP to the cell surface. HeLa cells expressing Q2-CFP-TM were labeled with Alexa 568 cadaverine by incubating with the probe, gpTGase, and CaCl2 for 25 min at 4 °C. The top row shows the CFP images superimposed on the DIC images. The bottom row shows the Alexa 568 fluorescence. Negative controls are shown with the alanine mutant Q2(Ala)-CFP-TM and with gpTGase enzyme left out.
We also assessed the specificity of labeling by immunoblot (Figure S3). Living cells expressing Q2-CFP targeted to the cell surface (Q2-CFP-TM, Figure 2) were labeled with biotin cadaverine and then lysed. The lysate was run on SDS-PAGE and blotted with streptavidin. A single biotinylated band was observed at the molecular weight of Q2-CFP-TM (37 kD). The band disappeared when the alanine mutant of Q2-CFP-TM was used instead, or when gpTGase was omitted from the labeling reaction.
To demonstrate the utility of TGase-mediated site-specific labeling, we investigated the homodimerization of the NF-κB transcription factor p50, which binds to DNA and acts as a transcriptional repressor in its homodimeric form.16 The most popular assay for determining the quantity of p50 homodimer in vitro and in cell lysates is the electrophoretic mobility shift assay (EMSA).17 However, this assay can be both tedious and inaccurate, because the DNA probe used for EMSA can itself change the quantity of homodimer, and because p50 can dissociate from DNA within the gel. We synthesized a benzophenone spermine photoaffinity probe (Figure S4) and ligated it to p50-Q2 through incubation with gpTGase and calcium for 1.5 hr at 37 °C. Excess probe was removed by nickel affinity purification. After irradiation with UV light, p50-Q2 monomer and covalently cross-linked homodimer were separated on SDS-PAGE and detected with anti-p50 antibody (Figure 3). We found that formation of the p50 homodimer increased in the presence of DNA, consistent with previous reports.18,19 We also tested the effect of the protein myotrophin, which is similar in structure to IκBα and has previously been shown to promote p50 homodimerization.20 We observed that myotrophin alone, without DNA, was sufficient to substantially increase the concentration of p50 homodimer. This illustrates one advantage of the photocrosslinking assay compared to EMSA, namely that it is possible to probe the effect of additives on homodimer levels in the absence of DNA.
Figure 3.

Labeling p50-Q2 with benzophenone and photoinduced cross-linking. p50-Q2 protein was labeled with benzophenone spermine using gpTGase and CaCl2 for 1.5 hr at 37 °C. After Ni-NTA purification to remove excess benzophenone, the protein was irradiated with UV light for 7 min at 4 °C, in the presence or absence of DNA (0.18 μM) or myotrophin (360 μM; 90 equivalents over p50).26 The products were separated on 10 % SDS-PAGE and blotted with anti-p50 antibody. Formation of p50 dimer (MW 78.5 kD) is seen in lanes 8–10, but not in others where the alanine mutant was used, gpTGase was omitted, or the samples were not UV-irradiated.
A trend that has emerged from existing labeling techniques is that there is a tradeoff between labeling specificity and tag size. Methods that use peptide tags to direct probes, such as FlAsH,21 are generally less specific than methods that use protein tags, such as FKBP.22 One way to circumvent this problem is to use enzymes to mediate the tag-probe interaction, as we have previously done with biotin ligase.23,24 The excellent peptide specificity of this labeling technique derives from the natural specificity of biotin ligase. However, in its present form, the versatility of biotin ligase labeling is limited because the enzyme is extremely specific for the biotin structure and major active site re-engineering is required to incorporate probes with little structural similarity to biotin. The TGase labeling method described here, which is highly versatile for probe structure, is superior to biotin ligase labeling in this respect.
It is unlikely that TGase labeling can be extended to tagging of intracellular proteins, because of competition from endogenous TGase substrates, and because intracellular calcium concentrations under basal conditions are too low for TGase activity. In addition, gpTGase’s specificity for the Q-tag is not extremely high, so that under forcing conditions, labeling of non-Q-tagged proteins is observed. This contrasts with the exceptional peptide specificity of biotin ligase.23,24 However, through optimization of labeling conditions, we were able to achieve highly specific labeling of the Q-tag both in vitro and on live cell surfaces, within relatively short timescales. In future work, we plan to investigate the use of factor XIIIa transglutaminase, which is known to exhibit higher sequence specificity than gpTGase.25 We developed an assay for detection of NF-κB homodimers that provides some advantages over the commonly used EMSA assay. Future efforts will focus on refining the NF-κB assay, and exploring the use of TGase labeling for the study of protein trafficking and cell-cell interactions.
Supplementary Material
Experimental procedures for the preparation of all Q-tag constructs, labeling conditions in vitro and on living cells, benzophenone spermine synthesis, and the photocrosslinking assay for p50. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by MIT, NIH (1 P20 GM072029-01), ONR (N00014-03-1-0456), the Sloan Foundation, and the Dreyfus Foundation (New Faculty Award). C.-W. L. was supported by a Merck/MIT fellowship. We thank Jeremy A. Ryan for technical assistance, Marta Fernandez-Suarez for myotrophin and helpful suggestions, and Jeff Keillor for the Q1-tag sequence.
References
- 1.Chen I, Ting AY. Curr Opin Biotechnol. 2005;16:35–40. doi: 10.1016/j.copbio.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Miller LW, Sable J, Goelet P, Sheetz MR, Cornish VW. Angew Chem Int Ed. 2004;43:1672–1675. doi: 10.1002/anie.200352852. [DOI] [PubMed] [Google Scholar]
- 3.Johnsson N, George N, Johnsson K. Chembiochem. 2005;6:47–52. doi: 10.1002/cbic.200400290. [DOI] [PubMed] [Google Scholar]
- 4.Bruce SE, Bjarnason I, Peters TJ. Clin Sci. 1985;68:573–579. doi: 10.1042/cs0680573. [DOI] [PubMed] [Google Scholar]
- 5.Greenberg CS, Birckbichler PJ, Rice RH. FASEB J. 1991;5:3071–3077. doi: 10.1096/fasebj.5.15.1683845. [DOI] [PubMed] [Google Scholar]
- 6.Lorand L, Graham RM. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 7.Lorand L, Parameswaran KN, Stenberg P, Tong YS, Velasco PT, Jonsson NA, Mikiver L, Moses P. Biochemistry. 1979;18:1756–1765. doi: 10.1021/bi00576a019. [DOI] [PubMed] [Google Scholar]
- 8.Wolff C, Lai CS. Biochemistry. 1988;27:3483–3487. doi: 10.1021/bi00409a053. [DOI] [PubMed] [Google Scholar]
- 9.Slaughter TF, Achyuthan KE, Lai TS, Greenberg CS. Anal Biochem. 1992;205:166–171. doi: 10.1016/0003-2697(92)90594-w. [DOI] [PubMed] [Google Scholar]
- 10.Sato H, Ikeda M, Suzuki K, Hirayama K. Biochemistry. 1996;35:13072–13080. doi: 10.1021/bi952616k. [DOI] [PubMed] [Google Scholar]
- 11.Taki M, Shiota M, Taira K. Protein Eng Des Sel. 2004;17:119–126. doi: 10.1093/protein/gzh015. [DOI] [PubMed] [Google Scholar]
- 12.Sato M, Sadamoto R, Niikura K, Monde K, Kondo H, Nishimura SI. Angew Chem Int Ed. 2004;43:1516–1520. doi: 10.1002/anie.200353058. [DOI] [PubMed] [Google Scholar]
- 13.Personal Communication with J. Keillor.
- 14.Hu BH, Messersmith PB. J Am Chem Soc. 2003;125:14298–14299. doi: 10.1021/ja038593b. [DOI] [PubMed] [Google Scholar]
- 15.Mahoney MG, Randall CJ, Linderman JJ, Gross DJ, Slakey LL. Mol Biol Cell. 1992;3:493–505. doi: 10.1091/mbc.3.5.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen LF, Greene WC. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 17.Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, Knuechel R, Baeuerle PA, Scholmerich J, Gross V. Gastroenterology. 1998;115:357–369. doi: 10.1016/s0016-5085(98)70202-1. [DOI] [PubMed] [Google Scholar]
- 18.Phelps CB, Sengchanthalangsy LL, Malek S, Ghosh G. J Biol Chem. 2000;275:24392–24399. doi: 10.1074/jbc.M003784200. [DOI] [PubMed] [Google Scholar]
- 19.Siebenlist U, Franzoso G, Brown K. Annu Rev Cell Biol. 1994;10:405–455. doi: 10.1146/annurev.cb.10.110194.002201. [DOI] [PubMed] [Google Scholar]
- 20.Sivasubramanian N, Adhikary G, Sil PC, Sen S. J Biol Chem. 1996;271:2812–2816. doi: 10.1074/jbc.271.5.2812. [DOI] [PubMed] [Google Scholar]
- 21.Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. J Am Chem Soc. 2002;124:6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 22.Marks KM, Braun PD, Nolan GP. Proc Natl Acad Sci USA. 2004;101:9982–9987. doi: 10.1073/pnas.0401609101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen I, Howarth M, Lin WY, Ting AY. Nat Methods. 2005;2:99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 24.Howarth M, Takao K, Hayashi Y, Ting AY. Proc Natl Acad Sci U S A. 2005;102:7583–7588. doi: 10.1073/pnas.0503125102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorman JJ, Folk JE. J Biol Chem. 1981;256:2712–2715. [PubMed] [Google Scholar]
- 26.Knuefermann P, Chen P, Misra A, Shi SP, Abdellatif M, Sivasubramanian N. J Biol Chem. 2002;277:23888–23897. doi: 10.1074/jbc.M202937200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures for the preparation of all Q-tag constructs, labeling conditions in vitro and on living cells, benzophenone spermine synthesis, and the photocrosslinking assay for p50. This material is available free of charge via the Internet at http://pubs.acs.org.