

Abstract
A central challenge of chemical biology is the development of small-molecule tools for controlling protein activity in a target-specific manner. Such tools are particularly useful if they can be systematically applied to the members of large protein families. Here we report that protein tyrosine phosphatases can be systematically “sensitized” to target-specific inhibition by a cell-permeable small molecule, Fluorescein Arsenical Hairpin Binder (FlAsH), which does not inhibit any wild-type PTP investigated to date. We show that insertion of a FlAsH-binding peptide at a conserved position in the PTP catalytic-domain's WPD loop confers novel FlAsH sensitivity upon divergent PTPs. The position of the sensitizing insertion is readily identifiable from primary sequence alignments, and we have generated FlAsH-sensitive mutants for seven different classical PTPs from six distinct subfamilies of receptor and non-receptor PTPs, including one phosphatase (PTP-PEST) whose three-dimensional catalytic-domain structure is not known. In all cases, FlAsH-mediated PTP inhibition was target-specific and potent, with inhibition constants for the seven sensitized PTPs ranging from 17 to 370 nM. Our results suggest that a substantial fraction of the PTP superfamily will be likewise sensitizable to allele-specific inhibition; FlAsH-based PTP targeting thus potentially provides a rapid, general means for selectively targeting PTP activity in cell-culture- or model-organism-based signaling studies.
Keywords: Protein tyrosine phosphatases, Allele-specific inhibitors, Inhibitor sensitization, FlAsH
1. Introduction
Engineering of protein/small-molecule interfaces has emerged as a powerful means of generating small-molecule ligands that control target-protein activity with high selectivity.1,2 Such protein “sensitization” strategies are appealing because many of the potential bottlenecks of inhibitor discovery (e.g., compound synthesis and screening) are bypassed: in principle, sensitivity to a known small molecule can simply be “programmed” into a target protein of interest through genetic mutation. Several key criteria must be met, however, for protein-sensitization strategies to be biologically and generally useful: mutagenesis must confer to the target protein small-molecule sensitivity that is not present in the wild-type protein or other members of the same protein family; the mutation(s) employed should be functionally silent (i.e., the mutant protein must retain its activity in the absence of the small-molecule ligand); and, ideally, the amino-acid residue(s) identified for sensitization in a prototype target should be present in other members of the protein family, eliminating the need to redesign a protein/inhibitor interface for each new target.3 To date, the protein kinases have proven to be the enzyme family most amenable to protein/small-molecule engineering: with a few caveats, essentially any eukaryotic protein kinase can be rendered sensitive to a known ATP-competitive inhibitor through a simple site-directed mutation in the kinase active site.4 Attempts to systematically engineer inhibitor sensitivity in other important enzyme families such as the protein methyltransferases5 and phosphatidyl-inositol-3 kinases6 have also met with some success.
Tight control of protein-phosphorylation states is, arguably, the most important signaling-regulation mechanism in mammalian biology, and the mammalian protein tyrosine phosphatases (PTPs), which catalyze the dephosphorylation of phosphotyrosine residues in protein substrates, are an attractive family of targets for small-molecule inhibitor discovery.7,8 PTPs, roughly 100 of which are encoded in the human genome, make up a large and diverse set of signaling enzymes, and small molecules that specifically inhibit individual PTPs would aid in the development of a full understanding of PTP function in complex signaling cascades. We have made significant progress in the development of a method for engineering novel inhibitor sensitivity into PTP active sites.9-11 Recent attempts at applying our active-site-targeting strategy across the PTP family, however, have suggested that active-site sensitization will be somewhat limited in scope, presumably due to significant differences in the detailed topology of individual PTP active sites.12 Moreover, active-site-directed PTP inhibitor discovery is complicated by the low cellular permeability of many known competitive PTP inhibitors, most of which are negatively charged phosphotyrosine mimetics.8
While active-site sensitization has proven extremely powerful for the study of protein-kinase function, it is possible that the kinases represent a “special case.” Many, or most, enzyme families may not be readily amenable to analogous strategies: the requirement for finding a sensitizing mutation in the active site that does not disrupt enzyme activity may simply be too constraining. However, a method for generating small-molecule-sensitized enzymes that avoids the pitfalls of active-site engineering has recently been described: insertion of a small-molecule-binding peptide into a protein domain has been used to generate sensitized variants of two enzymes, TEM-1 (a β-lactamase)13 and T-cell PTP (TCPTP).14 These studies utilized a cell-permeable small molecule, Fluorescein Arsenical Hairpin Binder (FlAsH, Figure 1A),15,16 to specifically inhibit TEM-1 and TCPTP mutants that contain the optimized FlAsH-binding peptide, CCPGCC, embedded in their respective catalytic domains.13,14 Here we show that insertion of the CCPGCC motif can be widely used for the generation of FlAsH-sensitized PTPs. Guided by PTP primary-sequence alignments, we demonstrate that site-specific insertion of CCPGCC confers novel FlAsH sensitivity upon seven classical PTPs from six distinct subfamilies. Importantly, the observed inhibition of the FlAsH-sensitized mutants is highly specific; FlAsH does not inhibit any wild-type PTP investigated to date. Our results demonstrate that a substantial fraction of the classical PTP superfamily can be readily sensitized to a single small-molecule inhibitor, and suggest that other PTPs will be likewise sensitizable to allele-specific inhibition by FlAsH.
Figure 1.
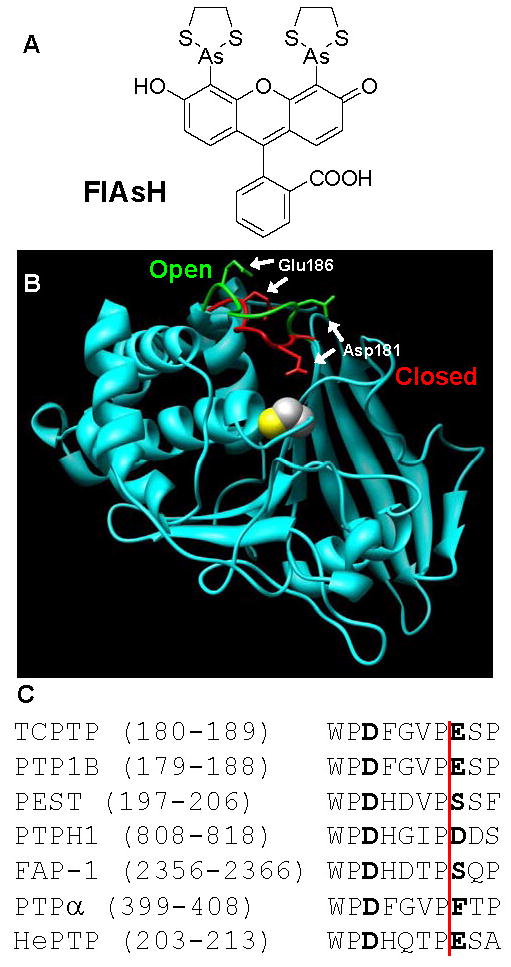
(A) Chemical structure of FlAsH. (B) Structure-based-design of CCPGCC insertion mutants. The crystal structures of PTP1B (cyan), a close homolog of TCPTP, in the “open” (PDB: 2HNQ)34 and “closed” (PDB: 1C83)35 forms were superimposed and the WPD loops of the two PTP1B structures were colored green and red, respectively. The position of the CCPGCC insertion corresponds to Glu186 in PTP1B. The side chain of the catalytically important aspartate residue, Asp181, is shown to highlight the movement of the WPD loop. For perspective, PTP1B's active-site catalytic cysteine residue (Cys215) is shown in space-filling representation. This image was generated using the Chimera software package (http://www.cgl.ucsf.edu/chimera). (C) WPD-loop alignment of the PTPs discussed in this study. (See reference 20 for a sequence alignment of all 39 human classical PTP domains.) The amino-acid residues corresponding to Asp181 and Glu186 of PTP1B are noted in bold type. CCPGCC-insertion positions are indicated by the vertical red line. The numbers of the insertion positions vary widely due to the diversity in size and structure of the PTP family outside of the conserved PTP domains.36
2. Results and discussion
2.1. Design strategy
Recently, we demonstrated that insertion of a tetra-cysteine-containing hexapeptide (CCPGCC) in the catalytic domain of TCPTP is sufficient for sensitization of that phosphatase to inhibition by FlAsH.14 We found that, for TCPTP, the most highly sensitizing site of insertion (187 in human TCPTP numbering) is near a canonical PTP loop that is known to close upon PTP-substrate binding (the WPD loop, Figure 1B).17,18 FlAsH binding presumably precludes proper closing (or, possibly, opening) of the mutant's WPD loop and specifically attenuates the activity of the engineered TCPTP. It has been surmised from the basis of sequence homology and structural evidence that almost every classical PTP similarly utilizes the conserved WPD region as a critical activity determinant (39 classical PTPs are encoded in the human genome).19,20 Therefore, we hypothesized that insertion of CCPGCC at the position corresponding to 187 of TCPTP could represent a general strategy for the site-specific incorporation of inhibition sites in many different PTPs (Figure 1C).
To test the scope of FlAsH-based PTP targeting, we selected six biologically important PTPs (in addition to TCPTP) from six distinct sub-families that are well spread across the human family of both non-transmembrane (NT) and receptor-like (R) classical PTPs: PTP1B, PTP-PEST, PTPH1, FAP-1, HePTP, and PTPα.20 PTP1B, subtype NT1, is a close homolog of TCPTP (69% PTP-domain identity) and an important type-II-diabetes drug target.21 PTP-PEST (subtype NT4, 34% PTP-domain identity with TCPTP) is a wide-ranging and ubiquitously expressed signaling molecule, which is involved in the regulation of the actin cytoskeleton and has recently been shown to be cleaved by caspase-3 during apoptosis.22,23 PTPH1 and FAP-1 (subtypes NT5 and NT7, 41% and 37% PTP-domain identity with TCPTP, respectively) have been found mutated in human colorectal cancers.24 PTPα (subtype R4, 38% PTP-domain identity with TCPTP) is a receptor PTP that dephosphorylates and activates Src-family kinases, affecting a host of downstream pathways, including cell proliferation and neuronal differentiation.25 And, HePTP (subtype R7, 34% PTP-domain identity with TCPTP) is a T-cell-specific phosphatase that can be overexpressed in preleukemic myeloproliferative diseases.26
When the PTP-domain primary sequences of the six PTPs described above are aligned with that of TCPTP, the position corresponding to TCPTP's residue 187 is readily identifiable, as it lies immediately to the C-terminal side of an invariant proline residue that marks the end of the conserved portion of the WPD loop (Figure 1C). To test the hypothesis that a FlAsH-binding peptide at the 187 position could sensitize multiple PTPs to inhibition, we cloned, expressed, and purified the catalytic domains of the six PTPs described above, as well as their corresponding CCPGCC insertion mutants: PTP1B-186, PTP-PEST-204, PTPH1-816, FAP1-2364, PTPα-406, and HePTP-211 (insertion position is indicated by the red line in Figure 1C; numbering is according to the human enzymes and corresponds to the position of the first cysteine residue in the insert).
2.2. PTP activity of insertion mutants
It is, of course, quite possible that insertion of a hexapeptide into a structured loop could seriously disrupt the fold and/or activity of an enzyme. In previous work on TCPTP, however, we found that its PTP domain is remarkably tolerant to peptide insertions. In particular, the TCPTP-187 mutant does retain inherent PTP activity, albeit with a catalytic efficiency (kcat/KM) that is an order of magnitude lower than that of wild-type TCPTP.14
In good agreement with the TCPTP-187 precedent, all six of the analogous PTP insertion mutants could be expressed from E. coli, and they were readily purified as affinity-tagged proteins (either His6 or GST, see Experimental). When assayed for activity, four of the six insertion mutants demonstrated significant reductions in inherent catalytic efficiency when assayed in the absence of FlAsH (Table 1). These diminutions in PTP activity, compared to the corresponding wild-type PTP, were similar in scale to that of TCPTP-187 (TCPTP-187: 7-fold14; PTP1B-186: 19-fold; HePTP: 9-fold; FAP1-2364: 14-fold; and PEST-204: 7-fold). We cannot predict a priori what level of decrease in PTP enzymatic efficiency is tolerable for a mutant to complement the function of the wild-type. And, it is possible that the reductions in catalytic activity noted above are of sufficient magnitude to limit the cellular-complementation ability of the CCPGCC-insertion mutants. There does exist, however, a potential mitigation of this obstacle: we have recently shown that much less radical mutations in the WPD loop (as few as two cysteine point mutations, with no insertions) can be used to confer strong FlAsH sensitivity upon TCPTP.27 These more subtly sensitized TCPTP mutants retain wild-type-like activity in the absence of FlAsH, and the homology of the WPD loop (Figure 1C) suggests that similar “rescuing” strategies may work for other PTPs—such as PTP1B, HePTP, FAP-1, and PTP-PEST—that lose some activity upon hexapeptide insertion.
Table 1.
Kinetic constants of wild-type and CCPGCC-insertion PTPs assayed with pNPP
| enzyme | kcat (s-1) | KM (mM) | kcat/KM (mM-1s-1) |
|---|---|---|---|
|
| |||
| PTP1B | 5.5±0.24 | 2.4±0.25 | 2.3±0.15 |
| PTP1B-186 | 0.11±0.0021 | 0.94±0.042 | 0.12±0.0050 |
|
| |||
| HePTP | 3.1±0.045 | 6.9±0.19 | 0.45±0.014 |
| HePTP-211 | 0.22±0.0081 | 4.4±0.24 | 0.049±0.00084 |
|
| |||
| FAP-1 | 0.82±0.0036 | 0.94±0.032 | 0.87±0.026 |
| FAP-1-2364 | 0.24±0.0033 | 3.8±0.10 | 0.062±0.0011 |
|
| |||
| PTP-PEST | 0.59±0.018 | 3.9±0.072 | 0.15±0.0057 |
| PTP-PEST-204 | 0.081±0.0012 | 3.5±0.13 | 0.023±0.00061 |
|
| |||
| PTPH1 | 5.2±0.18 | 0.49±0.038 | 11±0.47 |
| PTPH1-816 | 5.3±0.28 | 0.74±0.064 | 7.1±0.24 |
|
| |||
| PTPα | 1.7±0.15 | 8.0±0.90 | 0.21±0.0052 |
| PTPα-406 | 2.2±0.28 | 2.7±0.60 | 0.83±0.075 |
Remarkably, two of the CCPGCC-insertion mutants, PTPH1-816 and PTPα-406, retain their full catalytic activities in the absence of FlAsH (Table 1). There is no significant difference in the catalytic-rate constants of either of these proteins with respect to their corresponding wild-type PTPs, PTPH1 and PTPα. These results suggest that a substantial fraction of PTPs (2 out 7 in the current panel) will be fully enzymatically tolerant of the CCPGCC insertion and need no rescuing as described above.
2.3. Novel FlAsH sensitivity of insertion mutants
We next investigated the effects of FlAsH addition on our panel of wild-type and insertion-mutant PTPs. Importantly, the wild-type activities of all seven PTPs were essentially unaffected by the addition of FlAsH (Figure 2 and Table 2). [The DMSO vehicle had little to no effect on any PTP's activity (compare “(−)” entries on Table 2 with the corresponding Table 1 entries).] These data demonstrate that the FlAsH ligand has no significant non-specific affinity for PTP domains, implying that FlAsH could be used in a cellular context to target a single engineered PTP, without substantial off-target PTP inhibition. Consistent with this hypothesis, all of the CCPGCC-insertion mutants are strongly inhibited by FlAsH (Figure 2 and Table 2). Upon incubation with a 4-fold molar excess of FlAsH, PTP1B-186 catalytic efficiency drops 43-fold (0.13 to 0.0030 mM-1 s-1); HePTP-211 activity decreases 4-fold (0.031 to 0.0076 mM-1 s-1); FAP-1-2364 activity drops 8-fold (0.050 to 0.0062 mM-1 s-1); PTP-PEST-204 activity drops 6-fold (0.018 to 0.0028 mM-1 s-1); PTPH1-816 activity drops 12-fold (6.4 to 0.53 mM-1 s-1); and PTPα-406 activity drops 38-fold (0.79 to 0.021 mM-1 s-1). The magnitude of these FlAsH-induced reductions in sensitized-PTP activity ranges from roughly 3-fold lower to 3-fold higher than that of the previously reported 12-fold drop observed for TCPTP-187.14 And, as with TCPTP-187, the FlAsH-induced inhibition of the current mutants appears to be noncompetitive in nature, as the losses of catalytic efficiency are due predominately to decreases in kcat (Table 2). In sum, the FlAsH-mediated inhibition of PTP1B-186, PTP-PEST-204, PTPH1-816, FAP1-2364, PTPα-406, and HePTP-211 is comparable in scale and kind to that observed with TCPTP-187, implying a conserved mechanism of action across the classical PTP family.
Figure 2.
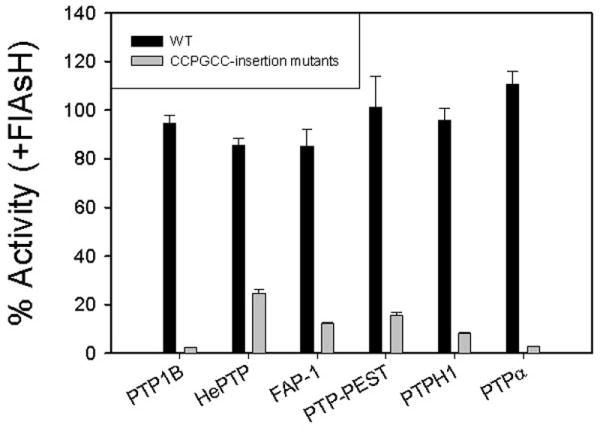
FlAsH-dependent inhibition of PTP-insertion mutants. The indicated PTP enzymes (2.5 μM) were incubated in the absence or presence of FlAsH (10 μM), diluted and assayed for activity with the artificial PTP substrate para-nitrophenyl phosphate (pNPP) at pH 7.0. “% Activity” represents the PTP catalytic efficiency (kcat/KM) in the presence of FlAsH divided by the control (vehicle only) catalytic efficiency of the same enzyme.
Table 2.
Kinetic constants of wild-type and CCPGCC-insertion PTPs in the absence (−) and presence (+) of FlAsH when assayed with pNPP
| enzyme | kcat (s-1) | KM (mM) | kcat/KM (mM-1s-1) |
|---|---|---|---|
|
| |||
| PTP1B (−) | 4.4±0.16 | 2.4±0.16 | 1.9±0.060 |
| PTP1B (+) | 4.4±0.16 | 2.4±0.12 | 1.8±0.020 |
|
| |||
| PTP1B 4C-186 (−) | 0.12±0.0062 | 0.91±0.080 | 0.13±0.0057 |
| PTP1B 4C-186 (+) | 0.0038±0.000061 | 1.3±0.043 | 0.0030±0.000082 |
|
| |||
| HePTP (−) | 3.0±0.10 | 6.2±0.37 | 0.49±0.013 |
| HePTP (+) | 2.8±0.029 | 6.6±0.16 | 0.42±0.0055 |
|
| |||
| HePTP-211 (−) | 0.16±0.020 | 5.3±0.92 | 0.031±0.0017 |
| HePTP-211 (+) | 0.039±0.0033 | 5.2±0.68 | 0.0076±0.00035 |
|
| |||
| FAP-1 (−) | 1.0±0.061 | 1.1±0.13 | 0.94±0.057 |
| FAP-1 (+) | 0.85±0.033 | 1.1±0.10 | 0.80±0.045 |
|
| |||
| FAP-1-2364 (−) | 0.20±0.014 | 4.0±0.39 | 0.050±0.0013 |
| FAP-1-2364 (+) | 0.028±0.00082 | 4.5±0.15 | 0.0062±0.00011 |
|
| |||
| PTP-PEST (−) | 0.46±0.00057 | 4.9±0.29 | 0.096±0.0058 |
| PTP-PEST (+) | 0.45±0.038 | 4.7±0.80 | 0.097±0.011 |
|
| |||
| PTP-PEST-204 (−) | 0.077±0.0016 | 4.2±0.10 | 0.018±0.00020 |
| PTP-PEST-204 (+) | 0.014±0.0015 | 5.0±0.92 | 0.0028±0.00020 |
|
| |||
| PTPH1 (−) | 5.3±0.075 | 0.56±0.027 | 9.4±0.36 |
| PTPH1 (+) | 4.8±0.13 | 0.53±0.032 | 9.0±0.30 |
|
| |||
| PTPH1-816 (−) | 5.4±0.20 | 0.84±0.075 | 6.4±0.32 |
| PTPH1-816 (+) | 0.53±0.0074 | 1.0±0.025 | 0.53±0.0064 |
|
| |||
| PTPα (−) | 1.6±0.092 | 8.3±0.81 | 0.19±0.0077 |
| PTPα (+) | 1.6±0.010 | 7.8±0.26 | 0.21±0.0059 |
|
| |||
| PTPα-406 (−) | 2.0±0.20 | 2.6±0.50 | 0.79±0.073 |
| PTPα-406 (+) | 0.10±0.014 | 5.1±1.0 | 0.021±0.0015 |
2.4. Potency of FlAsH-induced inhibition
To determine the potency of FlAsH's inhibitory activity on all of the PTP insertion mutants, we assayed their activities after incubation with varying concentrations of FlAsH. Under the conditions of the assay, none of the wild-type PTPs showed any significant inhibition at any FlAsH concentration (Figure 3). By contrast, the activities of all seven CCPGCC-insertion mutants dropped dramatically in a dose-dependent manner when pre-incubated with FlAsH. From these data we estimated the apparent inhibition constants (KIapp) of FlAsH with the seven sensitized mutants (Table 3). With the exception of FAP-1-2364 (KIapp = 370 nM), all of the insertion mutants demonstrated low-nanomolar sensitivity to FlAsH, with the KIapp values for the more potently inhibited mutants ranging from 17 nM (TCPTP-187) to 89 nM (HePTP-211).
Figure 3.
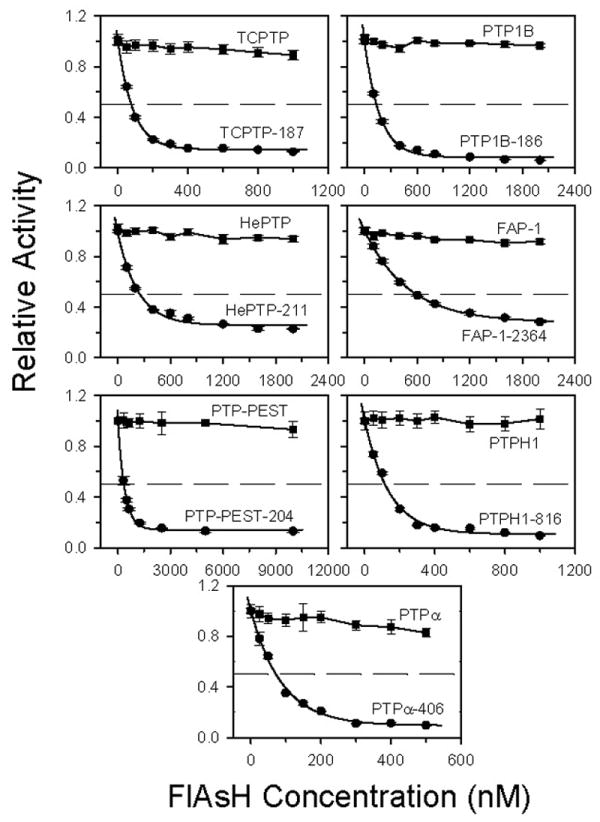
Dose-dependent inhibition of wild-type and CCPGCC-insertion mutant PTPs. PTP activity of each wild-type and mutant was measured at the indicated FlAsH concentrations, and normalized to uninhibited (no-FlAsH) controls. Enzyme concentrations used were as follows: 100 nM for TCPTP and PTPH1; 200 nM for PTP1B, HePTP, and FAP-1; 500 nM for PTP-PEST; and 50 nM for PTPα.
Table 3.
Apparent inhibition constants (KIapp) of FlAsH for CCPGCC-insertion PTPs
| enzyme | KIapp (nM) |
|---|---|
| TCPTP-187 | 17±3.3 |
| PTP1B-186 | 46±5.9 |
| HePTP-211 | 89±12 |
| FAP-1-2364 | 370±140 |
| PTP-PEST-204 | 53±6.8 |
| PTPH1-816 | 38±1.7 |
| PTPα-406 | 39±1.7 |
2.5. Phosphopeptide dephosphorylation by PTPH1-816 and PTPα-406
We selected PTPH1-816 and PTPα-406 as candidates for further characterization. These two mutants are particularly attractive as inhibitor-sensitized enzymes in that they demonstrate kinetic parameters that are essentially indistinguishable (or marginally improved, in the case of PTPα-406) from the corresponding wild-type enzymes in the absence of an allele-specific inhibitor. (Even sensitized protein kinases generally demonstrate substantially decreased kcat/KM values with ATP.28,29) Thus, of the seven sensitized PTPs described above, PTPH1-816 and PTPα-406 are the most likely to meet the critical criterion for cellular signaling studies using engineered proteins: that a targeted mutant protein retains the cellular function of the wild-type in the absence of ligand.
All of the initial characterization of PTPH1-816 and PTPα-406 was carried out with an artificial small-molecule PTP substrate, pNPP, and it is possible that a CCPGCC mutant could demonstrate wild-type-level kinetics with pNPP yet fail to demonstrate robust kinetics with a more physiologically relevant substrate. To further confirm the suitability of PTPH1-816 and PTPα-406 as inhibitor-sensitized enzyme variants, we measured their activities with a phosphopeptide substrate DADEpYLIPQQG, the sequence of which corresponds to the auto-phosphorylation site of the epidermal growth factor receptor (EGFR988-998).30 We found that, in the absence of FlAsH, the kinetic parameters of DADEpYLIPQQG dephosphorylation by PTPH1-816 and PTPα-406 are unchanged with respect to the corresponding wild-type values (Table 4, e.g. compare “PTPH1 (−)” to “PTPH1-816 (−)”). These data show that the catalytic activity of these CCPGGCC mutants is “wild-type-like” regardless of choice of PTP substrate and further support that the proposition that PTPH1-816 and PTPα-406 would function like their parent PTPs in a cellular context.
Table 4.
Kinetic constants of wild-Type and CCPGCC-insertion PTPH1 and PTPα in the absence (−) and presence (+) of FlAsH when assayed with DADEpYLIPQQG
| enzyme | kcat (s-1) | KM (μM) | kcat/KM (μM-1s-1) |
|---|---|---|---|
|
| |||
| PTPH1 (−) | 8.0±0.48 | 3.1±0.89 | 2.8±0.76 |
| PTPH1 (+) | 6.9±0.80 | 2.7±0.14 | 2.6±0.20 |
|
| |||
| PTPH1-816 (−) | 9.8±0.65 | 3.5±0.34 | 2.8±0.25 |
| PTPH1-816 (+) | 1.8±0.070 | 7.7±0.62 | 0.23±0.0093 |
|
| |||
| PTPα (−) | 4.2±0.27 | 82±9.4 | 0.052±0.0029 |
| PTPα (+) | 4.2±0.40 | 77±1.8 | 0.055±0.0045 |
|
| |||
| PTPα-406 (−) | 3.4±0.33 | 84±15 | 0.041±0.0043 |
| PTPα-406 (+) | 0.12±0.027 | 100±12 | 0.0012±0.00021 |
Along similar lines, it is important that FlAsH, the target-specific inhibitor of PTPH1-816 and PTPα-406, acts in a substrate-independent manner, as PTPs will encounter a variety of phosphorylated-protein substrates in a cellular context. Indeed, the engineered FlAsH sensitivities of PTPH1-816 and PTPα-406 also proved to be substrate-independent (Figure 4 and Table 4). When incubated with FlAsH, the activities of both mutants dropped dramatically and specifically. The magnitude of these inhibition events is, within error, precisely in line with what was observed when pNPP was used as substrate (PTPH1-816: 12-fold catalytic efficiency drop with pNPP and 12-fold drop with DADEpYLIPQQG; PTPα-406: 38-fold drop with pNPP and 34-fold drop with DADEpYLIPQQG). As with pNPP, the FlAsH-induced inhibition of DADEpYLIPQQG dephosphorylation is potent and specific to the engineered CCPGCC mutants; moreover, FlAsH appears to operate by a conserved mechanism that is independent of the particular PTP and substrate being investigated.
Figure 4.
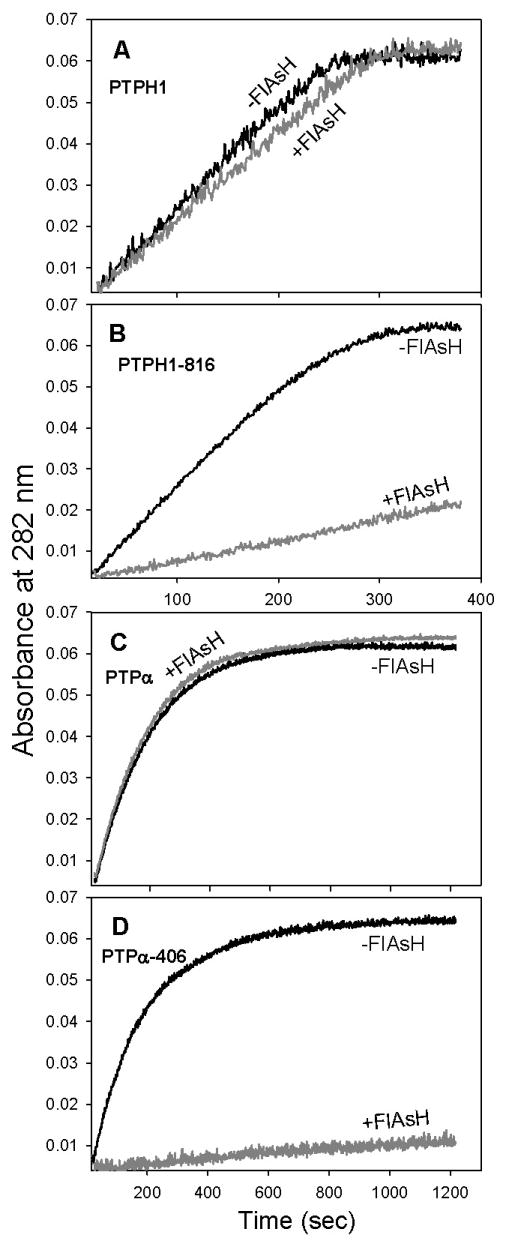
Specific inhibition of PTPH1-816 and PTPα-406 when assayed with the phosphopeptide substrate DADEpYLIPQQG. PTP enzymes (2.5 μM) were incubated in the absence (black lines) or presence (gray lines) of FlAsH (10 μM), diluted to the final enzyme concentration noted for each panel, and assayed at pH 7.0. (A) Wild-type PTPH1 (36 nM). (B) PTPH-816 (36 nM). (C) Wild-type PTPα (140 nM). (D) PTPα-406 (180 nM).
2.6. Time dependence of PTPH1-816 and PTPα-406 inhibition by FlAsH
One of the advantages of chemical, as opposed to genetic, approaches for studying protein function is that, generally, chemical inhibitors act rapidly (seconds to minutes) while DNA- or RNA-based approaches take much longer (hours to days) to reduce levels of a target protein's activity. To ensure that FlAsH acts on a timescale that is consistent with its potential use in chemical-genetic experiments, we measured the time course of FlAsH-induced inhibition for PTPH1-816 and PTPα-406. In these experiments, the FlAsH-responsive mutants were assayed after incubation with FlAsH for various amounts of time. As shown in Figure 5, the activities of both PTPH1-816 and PTPα-406 drop immediately upon FlAsH addition, with maximum inhibition being achieved in approximately 30 minutes. These experiments show that the majority of FlAsH-sensitive PTP inhibition occurs within minutes and suggest that FlAsH may be able to rapidly “knockout” PTP activity in a cell or lysate that expresses a sensitized PTP.
Figure 5.
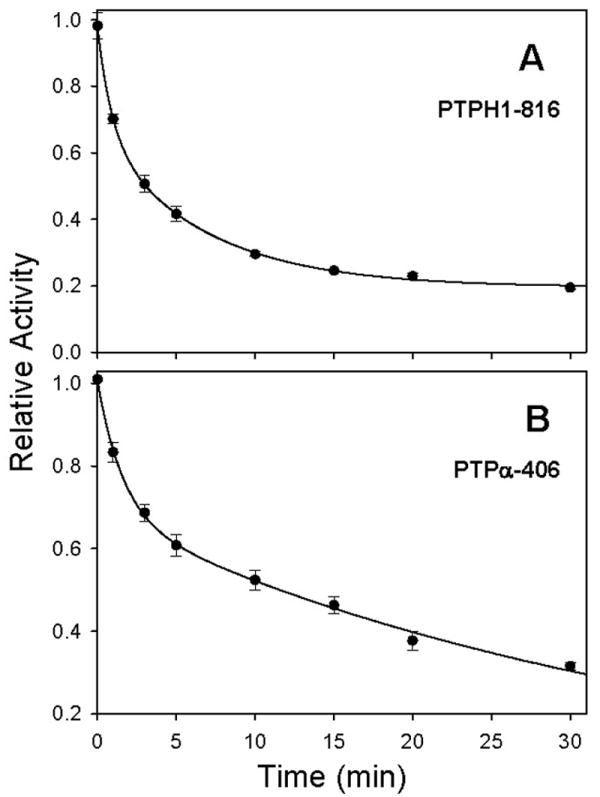
Time-dependent inhibition of PTPH1-816 and PTPα-406. PTP enzymes (250 nM) were incubated with FlAsH (1 μM). At the indicated time points after FlAsH addition, pNPP was added to initiate PTP activity. Percentage activity at each time point was normalized to uninhibited (no-FlAsH) controls. Data were fitted according to two-phase exponential decay using SigmaPlot 10.0. (A) PTPH1-816. (B) PTPα-406.
Collectively, the data above show that FlAsH can rapidly, potently, and specifically target engineered WPD loops via a conserved mechanism that is independent of the particular PTP that has been sensitized to inhibition. There is no correlation between PTP subfamily and FlAsH potency. For example, PTPH1 and PTPα, which belong to different subfamilies (and neither of which belongs to the same subfamily as the model phosphatase, TCPTP) are among the most potently inhibited in our panel of sensitized PTPs. Moreover, the PTPs whose activities drop most dramatically at a fixed FlAsH concentration include both non-transmembrane (PTP1B) and receptor-like (PTPα) phosphatases.
The key advantage of using protein engineering for targeting large protein families is that, by introducing chemical diversity into the target protein (through mutagenesis), novel and specific ligand/receptor pairs can be designed efficiently. Our current data support this assertion: due to the conserved structure and mechanism of the WPD loop, FlAsH sensitization was systematically applied to seven divergent classical PTPs for which “traditional” inhibitors have not been identified. Importantly, WPD-loop sensitization requires no detailed structural information for a target PTP. Of the seven PTPs investigated to date, only one (PTP1B) has been crystallized in both its open and closed WPD loop structures (Figure 1B). Moreover, no crystal structure has been reported for the catalytic domain of PTP-PEST, which, like other members of our PTP panel, is readily sensitized through primary-sequence-based CCPGCC insertion. These findings suggest a large fraction of the classical PTPs will be similarly susceptible to FlAsH sensitization, including other PTPs for which no three-dimensional structures are known.
3. Conclusion
Few systematic means for chemically targeting members of large protein families have been described. In the present work we show that protein tyrosine phosphatases, a critical family of cell-signaling enzymes, can be readily sensitized to inhibition by FlAsH, a small molecule that has no significant effect on the activity of wild-type PTPs. We have identified a site in the conserved WPD loop of PTP domains that can be modified to display a FlAsH-binding peptide, CCPGCC; and we show that CCPGCC insertion systematically confers strong FlAsH sensitivity to enzymes from six different PTP subfamilies. Since essentially all known classical PTPs utilize the WPD-loop mechanism targeted in this study, it is likely that other PTPs can be sensitized through an analogous approach, and, therefore, that FlAsH can be widely used for chemically targeting PTPs in cell-signaling studies.
4. Experimental
4.1. Materials, equipment, and general procedures
FlAsH was synthesized as described14-16 and dissolved in DMSO. The phosphopeptide substrate DADEpYLIPQQG was purchased from EMD. All DNA sequencing was carried out at the Cornell Biotechnology Resource Center. All PTP assays were performed in triplicate, and all error bars and “±” values represent the standard deviations of at least three independent experiments. Sequences of all cloning and mutagenic primers are provided as supplementary material.
4.2. Cloning of PTP catalytic domains
PTP1B and HePTP: The plasmids encoding glutathione-S-transferase (GST)-tagged PTP1B31 and six-histidine (His6)-tagged HePTP32 were generous gifts from Professor Zhong-Yin Zhang (Indiana University School of Medicine) and Professor Rebecca Page (Brown University), respectively. TCPTP and PTPH1: The plasmids encoding His6-tagged TCPTP (pHEH042) and PTPH1 (pERB047) have been previously described.10,11 FAP-1: A fragment encoding FAP-1's PTP domain (residues 2160–2481) was amplified (template: Open Biosystems Clone ID 6718945) with Pfu DNA polymerase (Stratagene). The PCR product and pET21b vector DNA were cut with Hind III and gel purified, and then re-cut with Bam H1 and gel purified again. Ligation reactions (1 μL T4 DNA ligase, 2 μL 10 × ligase buffer (New England Biolabs), ∼10 ng of insert and ∼10 ng of vector in a 20 μL reaction) were performed overnight at 15 °C. Ligation products were transformed into competent DH5α E. coli and plated on LB/Agar containing 100 μg/mL ampicillin. A single colony was isolated, and the presence of the insert in the resulting plasmid (pAML001) was confirmed by restriction digests and by DNA sequencing. PTP-PEST: A fragment encoding the catalytic domain of PTP-PEST (residues 1–307) was amplified (template: Open Biosystems Clone ID 5268732) and cloned into pET21b essentially as described above (resulting plasmid: pERB041). Restriction sites used were Eco RI and Xho I. PTPα: A fragment encoding the catalytic domain of PTPα (residues 212–503) was amplified (template: Open Biosystems Clone ID 3920094) and cloned into pET21b as above (resulting plasmid: pERB043). Restriction sites used were Sac I and Hind III.
4.3. Insertional mutagenesis
PTP1B-186, HePTP-211, FAP1-2364, PTP-PEST-204, and PTPH1-816: Insertion of CCPGCC-encoding DNA into the relevant templates (see above) was carried out using Quikchange mutagenesis essentially as previously described for the plasmid encoding TCPTP-187.14 The resulting CCPGCC-insertion-encoding plasmids are: pXYZ092 (PTP1B-186), pACB148 (HePTP-211), pACB146 (FAP1-2364), pACB149 (PTP-PEST-204), and pMSR001 (PTPH1-816). PTPα-406: For reasons that are not clear, the 18-base insertion could not be achieved with the PTPα-encoding template using Quikchange-based approaches, and “inverse” PCR13 was used instead: plasmid DNA (pERB043, ∼50 ng in 1 μL), cloned Pfu 10× reaction buffer (5 μL), primers (5 μL of each at a concentration of 10 μM), cloned Pfu DNA polymerase (1 μL, 2.5 U, Stratagene), 2 mM dNTP mix (5 μL), and water (28 μL) were combined and placed in a temperature cycler. The reaction mixture was subjected to one cycle of 95 °C for 2 minutes, then 25 cycles of 95 °C for 1 minute, 55 °C for 1 minute, and 68 °C for 16 minutes. At the end of temperature cycling, 10 U (1 μL) of Dpn I restriction enzyme were added, and the reaction mixture was incubated at 37 °C for 1 hour. Following Dpn I digestion of parental DNA, the reaction mixture was EtOH precipitated and re-suspended in 8 μL of water, 1 μL of T4 DNA ligase buffer, and 1 μL of polynucleotide kinase (ABgene), and the reaction mixture was incubated at 37 °C for 1 hour. T4 DNA ligase (1 μL) was added and the mixture was incubated at 15 °C overnight. The ligation mixture was used to transform DH5α competent cells, and plasmids from ampicillin-resistant colonies were purified. Upon DNA sequencing it was revealed that all of the mutated plasmids contained an extra base in the inserted section. One further round of mutagenesis was carried out to delete the extra base using a standard Quikchange protocol. The resulting PTPα-406-encoding plasmid is pXYZ290.
4.4. Protein expression and purification
PTPs that were expressed from pET21-based plasmids (PTPH1, TCPTP, PTP-PEST, PTPα) contained His6-tags and were purified using SwellGel Nickel Chelated Discs (Pierce) according to the manufacturer's instructions and as described,12 with one change: IPTG inductions were carried out at 26°C for 16 hours. GST-tagged PTP1B was purified as described.31
4.5. PTP activity assays
All PTP activity assays with para-nitrophenyl phosphate (pNPP) were carried out at 22 °C in a total reaction volume of 200 μL containing pNPP (0.25-20 mM) and the appropriate phosphatase (10-500 nM) in 1×PTP buffer (50 mM 3,3-dimethylglutarate at pH 7.0, 1 mM EDTA, and 50 mM NaCl). Reactions were quenched by the addition of 40 μL of 5 M NaOH. The reaction mixtures (200 μL) were loaded onto a 96-well plate, and the absorbances at 405 nm were measured. Kinetic constants were determined by fitting the data to the Michaelis-Menten equation. To measure the effect of FlAsH on PTP activity, PTP solutions (2.5 μM) in 1×PTP buffer were incubated in the absence (DMSO vehicle only) or presence of FlAsH (10 μM). After 2.5 hours at room temperature, PTP activity in the presence and absence of FlAsH was assayed as described above. For inhibition-constant measurements, PTP assays were carried out as described above, but with varying concentrations of FlAsH at a pNPP concentration equal to the KM for the enzyme. The percentage activity was obtained by dividing the absorbance in the presence of FlAsH by the absorbance of the no-FlAsH control. Inhibition constants were estimated from these data using non-linear regression analysis as previously described.27 For time-course inhibition experiments, PTPH1-816 or PTPα-406 (250 nM) and FlAsH (1 μM) were mixed and incubated for 1, 3, 5, 10, 15, 20, and 30 minutes. At the noted time points, pNPP (concentration equal to KM for the PTP being assayed) was added to an aliquot of the PTP/FlAsH mixture to initiate PTP activity. Reactions were quenched after 30-60 sec, and percentage activity at each time point was obtained as described above.
Phosphopeptide-dephosphorylation assays using DADEpYLIPQQG were carried out by measuring increasing absorbance at 282 nm essentially as described.33 Assays were performed at 22 °C in a total volume of 140 μL and contained the following: 50 mM 3,3-dimethylglutarate pH 7.0, 125 mM NaCl, 1 mM EDTA, 70 μM DADEpYLIPQQG, and the appropriate phosphatase pre-incubated with FlAsH or DMSO vehicle. Pre-incubation mixtures contained 2.5 μM enzyme and 10 μM FlAsH. Final enzyme concentrations for the experiments shown in Figure 4 were 36 nM for PTPH1 and PTPH-816, 140 nM for PTPα, and 180 nM for PTPα-406. Due to the strong inhibition of PTPH-816 and PTPα-406 in the presence of FlAsH, higher enzyme concentrations were used to extract the relevant kinetic constants shown in Table 4: 180 nM for PTPH1-816 (+) and 1820 nM for PTPα-406 (+). Kinetic constants were obtained by non-linear regression to the integrated Michaelis-Menten equation using SigmaPlot 10.0.
Supplementary Material
Sequences of all cloning and mutagenic primers.
Acknowledgments
This research was supported by the National Institutes of Health (1 R15 GM071388-01A1) and Research Corporation (CC6372).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walsh DP, Chang YT. Chem Rev. 2006;106:2476. doi: 10.1021/cr0404141. [DOI] [PubMed] [Google Scholar]
- 2.Bishop A, Buzko O, Heyeck-Dumas S, Jung I, Kraybill B, Liu Y, Shah K, Ulrich S, Witucki L, Yang F, Zhang C, Shokat KM. Annu Rev Biophys Biomol Struct. 2000;29:577. doi: 10.1146/annurev.biophys.29.1.577. [DOI] [PubMed] [Google Scholar]
- 3.Bishop AC, Buzko O, Shokat KM. Trends Cell Biol. 2001;11:167. doi: 10.1016/s0962-8924(01)01928-6. [DOI] [PubMed] [Google Scholar]
- 4.Elphick LM, Lee SE, Gouverneur V, Mann DJ. ACS Chem Biol. 2007;2:299. doi: 10.1021/cb700027u. [DOI] [PubMed] [Google Scholar]
- 5.Lin Q, Jiang F, Schultz PG, Gray NS. J Am Chem Soc. 2001;123:11608. doi: 10.1021/ja011423j. [DOI] [PubMed] [Google Scholar]
- 6.Alaimo P, Knight ZA, Shokat KM. Bioorg Med Chem. 2005;13:2825. doi: 10.1016/j.bmc.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 7.Tonks NK. Nat Rev Mol Cell Biol. 2006;7:833. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 8.Bialy L, Waldmann H. Angew Chem Int Ed. 2005;44:3814. doi: 10.1002/anie.200461517. [DOI] [PubMed] [Google Scholar]
- 9.Bishop AC, Blair ER. Bioorg Med Chem Lett. 2006;16:4002. doi: 10.1016/j.bmcl.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 10.Blair ER, Hoffman HE, Bishop AC. Bioorg Med Chem. 2006;14:464. doi: 10.1016/j.bmc.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 11.Hoffman HE, Blair ER, Johndrow JE, Bishop AC. J Am Chem Soc. 2005;127:2824. doi: 10.1021/ja043378w. [DOI] [PubMed] [Google Scholar]
- 12.Bishop AC, Zhang XY, Lone AM. Methods. 2007;42:278. doi: 10.1016/j.ymeth.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erster O, Eisenstein M, Liscovitch M. Nat Methods. 2007;4:393. doi: 10.1038/nmeth1046. [DOI] [PubMed] [Google Scholar]
- 14.Zhang XY, Bishop AC. J Am Chem Soc. 2007;129:3812. doi: 10.1021/ja069098t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffin BA, Adams SR, Tsien RY. Science. 1998;281:269. doi: 10.1126/science.281.5374.269. [DOI] [PubMed] [Google Scholar]
- 16.Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. J Am Chem Soc. 2002;124:6063. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 17.Jia Z, Barford D, Flint AJ, Tonks NK. Science. 1995;268:1754. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 18.Stuckey JA, Schubert HL, Fauman EB, Zhang ZY, Dixon JE, Saper MA. Nature. 1994;370:571. doi: 10.1038/370571a0. [DOI] [PubMed] [Google Scholar]
- 19.Tabernero L, Aricescu AR, Jones EY, Szedlacsek SE. FEBS J. 2008;275:867. doi: 10.1111/j.1742-4658.2008.06251.x. [DOI] [PubMed] [Google Scholar]
- 20.Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Mol Cell Biol. 2001;21:7117. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang S, Zhang ZY. Drug Discov Today. 2007;12:373. doi: 10.1016/j.drudis.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 22.Hallé M, Liu YC, Hardy S, Théberge JF, Blanchetot C, Bourdeau A, Meng TC, Tremblay ML. Mol Cell Biol. 2007;27:1172. doi: 10.1128/MCB.02462-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Côté JF, Chung PL, Théberge JF, Hallé M, Spencer S, Lasky LA, Tremblay ML. J Biol Chem. 2002;277:2973. doi: 10.1074/jbc.M106428200. [DOI] [PubMed] [Google Scholar]
- 24.Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, Parmigiani G, Yan H, Wang TL, Riggins G, Powell SM, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. Science. 2004;304:1164. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 25.Pallen CJ. Curr Top Med Chem. 2003;3:821. doi: 10.2174/1568026033452320. [DOI] [PubMed] [Google Scholar]
- 26.Zanke B, Squire J, Griesser H, Henry M, Suzuki H, Patterson B, Minden M, Mak TW. Leukemia. 1994;8:236. [PubMed] [Google Scholar]
- 27.Zhang XY, Bishop AC. Biochemistry. 2008;47:4491. doi: 10.1021/bi800014c. [DOI] [PubMed] [Google Scholar]
- 28.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. Nature. 2000;407:395. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 29.Blethrow J, Zhang C, Shokat KM, Weiss EL. Curr Protoc Mol Biol. 2004;18:18.11.1. doi: 10.1002/0471142727.mb1811s66. [DOI] [PubMed] [Google Scholar]
- 30.Zhang ZY, Thieme-Sefler AM, Maclean D, McNamara DJ, Dobrusin EM, Sawyer TK, Dixon JE. Proc Natl Acad Sci USA. 1993;90:4446. doi: 10.1073/pnas.90.10.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen K, Keng YF, Wu L, Guo XL, Lawrence DS, Zhang ZY. J Biol Chem. 2001;276:47311. doi: 10.1074/jbc.M106568200. [DOI] [PubMed] [Google Scholar]
- 32.Mustelin T, Tautz L, Page R. J Mol Biol. 2005;354:150. doi: 10.1016/j.jmb.2005.09.049. [DOI] [PubMed] [Google Scholar]
- 33.Zhang ZY, Maclean D, Thieme-Sefler AM, Roeske RW, Dixon JE. Anal Biochem. 1993;211:7. doi: 10.1006/abio.1993.1224. [DOI] [PubMed] [Google Scholar]
- 34.Barford D, Flint AJ, Tonks NK. Science. 1994;263:1397. [PubMed] [Google Scholar]
- 35.Andersen HS, Iversen LF, Jeppesen CB, Branner S, Norris K, Rasmussen HB, Moller KB, Moller NP. J Biol Chem. 2000;275:7101. doi: 10.1074/jbc.275.10.7101. [DOI] [PubMed] [Google Scholar]
- 36.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Cell. 2004;117:699. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of all cloning and mutagenic primers.