

Abstract
Macrophages are less effective than DC at priming naïve CD4+ T cells, suggesting that DC are unique in initiating T cell-dependent antibody responses. We compared the ability of DC and macrophages, pulsed in vitro with Streptococcus pneumoniae (Pn), to elicit protein- and polysaccharide (PPS)-specific Ig isotype production upon adoptive transfer into naïve mice. Pn-activated DC secreted more pro-and anti-inflammatory cytokines, expressed higher levels of surface MHC-II and CD40, and presented Pn or recombinant pneumococcal surface protein A (PspA) to a PspA-specific T hybridoma more efficiently than macrophages. However, upon adoptive transfer into naïve mice, Pn-pulsed macrophages elicited an IgM and/or IgG anti-PspA and anti-PPS response comparable in serum titers and IgG isotype distribution to that induced by DC. The IgG anti-PspA, in contrast to the IgG anti-PPS, response to Pn-pulsed macrophages was T cell-dependent. Pn-pulsed macrophages that were paraformaldehyde-fixed prior to transfer or lacking expression of MHC-II or CD40 were highly defective in eliciting an anti-PspA response, although the anti-PPS response was largely unaffected. To our knowledge, these data are the first to indicate that macrophages can play an active role in the induction of a T cell-dependent humoral immune response in a naïve host.
Keywords: Rodent, Bacterial, Antibodies, Macrophages, Dendritic cells, Transgenic/Knockout Mice
Introduction
Numerous studies have provided compelling evidence that DC are unique in their capacity to prime naïve T cells (1, 2). The role of other APC, such as macrophages and B cells, is thought instead to be primarily the promotion of various effector functions of T cells previously primed by DC. Thus, early studies demonstrated that DC were at least 100-fold more potent at stimulating a primary MLR than macrophages or B cells (3–5). In addition splenic DC, but not peritoneal macrophages or unfractionated (B cell-rich) spleen cells, pulsed in vitro with a number of different soluble proteins, and injected into the footpads of mice, induced significant priming of CD4+ T cells from the draining LN, as evidenced by DNA synthesis in an in vitro restimulation assay (6). Similarly, in the presence of peptide antigen, splenic DC were significantly more efficient than splenic B cells, or peritoneal, splenic, or BM marrow-derived macrophages in promoting DNA synthesis or IL-2 production in naïve, peptide-specific transgenic CD4+ T cells in vitro (7, 8). However, it has also been demonstrated that macrophages are heterogeneous in their ability to present antigen to naïve CD4+ T cells (9). Thus, ~20% of murine splenic macrophage precursors were found to present native or peptide antigen to, and activate, naïve TCR transgenic CD4+ T cells. This correlated with the ability of presenting macrophages to release IL-12. DC have also been found to be the main APC for stimulating primed T cells in vitro, when cultured ex vivo from mice immunized with different soluble proteins; macrophages from immunized mice were non-stimulatory nor were they able to transfer antigen to DC (10). Purified splenic DC, but not splenic or peritoneal macrophages, are also potent APC for restoring the in vitro T cell-dependent primary anti-SRBC response or TNP-KLH-induced anti-TNP response in mixtures of B and T cells (11, 12).
Consistent with the studies cited above, numerous studies have demonstrated the ability of antigen- or pathogen-pulsed DC to elicit immune responses upon adoptive transfer into naïve mice (13). However, little is known regarding a similar function for adoptively-transferred, antigen-pulsed macrophages. A series of studies by M. Moser and colleagues (14–17) indeed demonstrated the ability of non-elicited peritoneal macrophages, as well as splenic DC, pulsed with soluble protein antigen and adoptively transferred into naïve mice, to both elicit specific antibody responses and T cell priming in vivo. However, it is unclear from these studies whether the antigen-pulsed macrophage itself was playing an active and direct role in initiating immunity or perhaps was serving to transfer antigen to other, endogenous APC. This latter issue, however, was addressed by Pozzi et al who investigated the ability of adoptively-transferred peptide-pulsed BM macrophages and DC to elicit CTL responses in transgenic CD8+ T cells specific for the peptide bound to MHC-I (18). Using the approach of adoptive transfer into BM chimeras, both type of APC were shown to directly induce, without requiring peptide transfer to host APC, CD8+ T cell proliferation, cytokine secretion, differentiate into CTL and memory cells. Although macrophages injected s.c. required pulsing with 10-fold more peptide than DC to elicit a comparable response, this was shown to be a function of lower migration of macrophages to LN, and not APC potency per se. Equivalent migration of APC to the spleen and subsequent CD8+ T cell responses were observed, however, using the i.v. route for APC injection.
Several in vivo studies support the notion that endogenous DC are indeed critical, non-redundant APCs for initiating immunity. Conventional (c)DC (CD11chigh) can be depleted by injection of diphtheria toxin into transgenic mice that express the DT receptor under the control of the CD11c promoter. DC depletion in these mice results in the abrogation of CTL priming in response to cross presentation of cell-associated (19) or exogenous soluble (20) antigen. Further CTL generation in response to Listeria monocytogenes and Plasmodium yoelii was also inhibited following DC depletion (19). Another study demonstrated that DC depletion in mice infected with lymphocytic choriomeningitis virus (LCMV) inhibited priming of LCMV-specific CD8+ T cells, despite the presence of LCMV in all major APC (DC, macrophages and B cells) (21). Priming of CD4+ T cells in response to i.v. injection of soluble or cell-associated protein is also inhibited by splenic cDC depletion, whereas LN plasmacytoid DC can substitute for cDC for CD4+ T cell priming following s.c. immunization (20). Additionally, following DC-depletion, mice immunized i.v. with Mycobaterium tuberculosis, exhibit a substantial delay in specific CD4+ T cell priming (22). Of interest, no studies have been reported to date on the effects of DC depletion on the induction of a T cell-dependent antibody response. Further, although both static and live imaging microscopy has clearly demonstrated the ability of antigen-expressing DC to form stable interactions with, and activate, naïve CD4+ T cells (23), the ability of macrophages to mediate a similar function has not been resolved.
To our knowledge, it remains to be determined whether macrophages can play an active and direct role in initiating a CD4+ T cell-dependent humoral response in vivo. We previously demonstrated that mice immunized with intact Streptococcus pneumoniae, capsular type 14 (Pn14) elicit an IgG response specific for pneumococcal surface protein A (PspA) and type 14 capsular polysaccharide (PPS14) that is dependent on CD4+ T cells and CD40/CD40-ligand interactions (24). In contrast, the IgM anti-PPS14 response is T cell-independent. Further, we reported that BMDC pulsed with Pn14 in vitro and transferred i.v. into naïve, WT mice elicit IgG anti-PPS14 and IgG anti-PPS14 responses that are dependent on viable BMDC prior to transfer, their ability to secrete IL-6, and the presence of endogenous CD4+ T cells in the recipient mice (25). Whereas the IgG anti-PspA response required BMDC expression of MHC-II, CD40, and B7-1/B7-2, the IgG anti-PPS14 did not. Using this approach, we now wished to directly compare Pn14-pulsed BMDC with two distinct populations of macrophage (BM macrophages [BMM] and peritoneal macrophages [PerM]) for their ability to induce anti-Pn humoral immunity upon adoptive transfer, and determine the mechanism by which this might occur. We show for the first time that macrophages pulsed with Pn14 can play an active, direct role in eliciting a specific humoral response in a naïve mouse, comparable to that observed using BMDC.
Materials and Methods
Mice
C57BL/6, BALB/c and athymic nude mice (BALB/c background) were obtained from the National Cancer Institute (Frederick, MD). CD40−/− mice (BALB/c background; strain name: CNCr.129P2-Cd40tm1Kik/J) and MHC class I1−/− mice (C57BL/6 background, strain name: B6.129-H2dlab1-Eα/J) were obtained from the Jackson Laboratory (Bar Harbor, ME). CD40−/− and MHC class II−/− mice were bred and maintained at U.S.U.H.S. in a pathogen-free environment, and were used between 7–12 wk of age.
Reagents
Recombinant pneumococcal surface protein A (PspA) [family 1, seroclade 2] was expressed in Saccharomyces cerevisiae BJ3505 and purified as previously described (26). Purified pneumococcal capsular polysaccharide type 14 (PPS14) was purchased from the American Type Culture Collection (Manassas, VA.). Lipopeptide Pam3Cys-Ser-Lys4 (Pam3Cys) and purified lipopolysaccharide from E. coli K12 strain (LPS) were purchased from InvivoGen (San Diego, CA). Phosphorotriated 30-mer CpG-ODN (27) was synthesized in the Biomedical Instrumentation Center (U.S.U.H.S).
Preparation of S. pneumoniae, capsular type 14 (Pn14)
A frozen stock of S. pneumoniae, capsular type 14 was thawed and sub-cultured on BBL pre-made blood agar plates (VWR International, Bridgeport, NJ). Isolated colonies in blood agar were grown in Todd Hewitt broth (Becton Dickinson, Sparks, MD) to mid-log phase, collected, and heat killed by incubation at 60°C for 1h. Sterility was confirmed by subculture on blood agar plates. After extensive washings, the bacterial suspension was adjusted with PBS to give an absorbance reading at 650 nm of 0.6 which corresponded to 109 CFU/ml. Bacteria were then aliquoted at 1010 CFU/ml and frozen at −80°C until their use as antigen for mouse immunizations.
Pneumococcal strain R6-14 (Pn-R6-14)
The S. pneumoniae strain R6J (R6cps::Janus), a variant of the strain R6 in which the capsule locus is substituted by a Janus cassette, was kindly provided by Drs Kryzysztof Trzcinski and Marc Lipsitch (Harvard School of Public Health, Boston MA). An IgG1κ mouse mAb specific for the capsular polysaccharide of S. pneumoniae type 14 (44.1) was kindly provided by Dr Alex Lucas (Children’s Hospital Oakland Research Institute, Oakland, CA). Two IgG2a mouse mAbs specific for the family 1, seroclade 2 of PspA (DC10-IA5 and CF9IIB7), were kindly provided by Dr. Rick Schuman (Biosynexus, Inc., Gaithersburg, MD). An isogenic variant of S. pneumoniae strain R6 expressing capsular polysaccharide type 14 (R6-14), was constructed by transformation with chromosomal DNA, essentially as described (28). The expression of serologically intact Cps14 on the selected smooth colonies was confirmed by colony-blot using the mouse mAb 44.1 specific for Cps14 as detection antibody. DNA from one isolate was purified and used to retransform R6J into a Cps14-expressing strain again. This process of backcross transformation was repeated three times in order to minimize uncontrolled recombinational replacements in loci other than cps. R6-14 expresses quantitatively equivalent levels of Cps14 relative to the donor strain (Pn14) as determined by quantitative sandwich ELISA. The PspA is serologically identical to the PspA of the recipient R6 strain.
PspA-specific T hybridomas
CD4+ T cell hybridomas specific for PspA peptide in association with MHC-IId were produced and screened according to a previously published protocol (29). Briefly, BALB/c mice were injected in the footpad with 50 μg of recombinant PspA emulsified in complete Freund’s adjuvant (Sigma, St. Louis, MO). On d7, popliteal LN cells were cultured with PspA at 5 μg/ml in the presence of IL-2. Cells were then harvested on d6 and fused, using polyethylene glycol (Boehringer Mannheim, Mannheim, Germany), with the fusion partner BW1100. Clones that grew in hypoxanthine/aminopterin/thymidine (HAT) (Sigma) selection media were screened for PspA-specificity and MHC restriction. One CD4+CD8− T hybridoma, BALD4, was selected for use in these studies.
Preparation of BMDC
BMDC were prepared as previously described (25). Briefly, BM cells were cultured at 1.25 × 106 cells/ml (24-well plates) in RPMI 1640 + 5% FBS, 10,000 IU penicillin & 10μg/ml streptomycin, 1 mM sodium pyruvate, 2 mM L-glutamine, 0.1 mM nonessential amino acids, and 25 mM HEPES (“culture medium”), supplemented with 10 ng/ml of murine rGM-CSF (Sigma). After 7d of culture, non-adherent cells were harvested.
Preparation of BMM
BMM were obtained using a similar approach to that for BMDCs, with slight modifications. BM cells were cultured at 1 × 106 cells/ml in cell culture medium supplemented with 10 ng/ml of murine M-CSF (Sigma). Cells were plated in 6 well plates in a volume of 4–5 ml/well. On d3 and 5, 3/4 culture media were removed and fresh culture media were added. On d7, BMM were harvested by washing plates with sterile PBS to remove nonadherent cells. Cells were detached from the plate by adding 2 ml of detachment buffer (4 mg/ml lidocaine, 5 mM EDTA, PBS) for 3–5 minutes with pipetting.
Isolation of peritoneal macrophages (PerM)
Culture medium (10 ml) was injected into the peritoneal cavity using a 23-gauge needle. After 3 minutes, lavage fluid containing the peritoneal cells was withdrawn using an 18-gauge needle. Cells were pelleted by centrifugation at 400 × g for 10 min. Red blood cells were removed by lysis with ACK lysing buffer. Cells were resuspended in culture medium, and cultured in 6 well plates (5 ml/plate) at 1 × 106 cells/ml overnight, followed by washing of plates with sterile PBS to remove nonadherent cells. Adherent cells were then detached by adding 2 ml of detachment buffer (4 mg/ml lidocaine, 5 mM EDTA, PBS) for 3–5 minutes with pipetting.
In vitro culture of APC
APC were plated in culture medium without added GM-CSF or M-CSF, at 1 × 106 cells/ml in 24-well cell culture plates (Costar, Corning, NY). After 30 min, to allow cells to settle and to completely reverse the transient metabolic effects of the lidocaine on the macrophage population, varying concentrations of Pn14 or Toll-like receptor (TLR) ligands were added to the cultures for 24h.
APC stimulation of IL-2 production by PspA-specific T-cell hybridoma
APC were plated at 2×104 APC/well in 96-well flat bottom tissue-culture plates (Costar), containing serial dilutions of PspA or bacteria. After 2h of pulse with the antigen at 37°C, the T cell hybridoma was added to the cultures at 1 × 105 cells/well and co-cultured with the APCs for 24h in the presence of free antigen. APCs derived from C57BL/6 BM cells were used in every experiment as a control of MHC-restricted antigen presentation. Negative controls included cultures of the T-cell hybridoma and antigen in the absence of APCs, and APCs cultured with antigen in the absence of T-cell hybridoma.
Adoptive transfer of Pn14-pulsed APCs
APC were cultured at 1 × 106 cells/ml for 4 h in the presence of 2 × 109 CFU of Pn14 in vitro. Free bacteria were then removed from the cell cultures by washing ~6X in cold PBS, each time followed by centrifugation for 10 min at 350 × g at 4°C. A control tube containing a mixture of thymocytes and CM-DiI fluorescent-labeled Pn14 at the same initial bacterial density was used to monitor the progress of the washings. If >100 free bacteria were found in pelleted cells, using an inverted fluorescent microscope, the washings were continued. Pn14-pulsed cells were then resuspended in fresh medium at 1 × 106 cells/200 μl, and 200 μl were injected i.v. into each mouse.
Measurement of cytokine concentrations in culture SN by ELISA
IL-6, IL-10, IL-12, and TNF-α concentrations released into the culture medium by activated APC or T hybridoma cells, were measured by ELISA according to manufacturer’s instructions (BD Pharmingen, San Diego, CA). TGF-β was measured by ELISA (“TGF-β1 mouse/rat/prcine/canine Quantikine ELISA kit”, Cat#MB100B) [R&D Systems, Inc., Minneapolis, MN]. The ELISA measured only active TGF-β. To measure total TGF-β (active + latent) the TGF-β in culture SN was first purposefully activated using HCl followed by neutralization using NaOH/HEPES according to manufacturer’s instructions, then assayed immediately by ELISA. Standards were included in every plate, and the samples were tested in duplicate.
Flow cytometric analysis
All steps were performed on ice. FcγR receptors were \blocked with 10 μg/ml of rat IgG2b,κ anti-mouse FcγRI/II/III (clone 2.4G2). Cells were stained for 30 min with either PE-Armenian hamster IgG1,λ2 anti-mouse CD11c (clone HL3), FITC-mouse IgG2b,κ anti-mouse MHC-IId (clone AMS-32.1), FITC-rat IgG2a,κ anti-mouse CD40 (clone 3/23), PE-rat IgG2a,κ anti-mouse CD86 (clone GL1), Armenian hamster IgG2a,κ anti-mouse CD80 (clone 16-10A1), or PE-rat IgG1,κ anti-mouse CD14 (clone rmc5-3). All mAbs were purchased from BD Pharmingen. FITC rat anti-mouse F4/80 (clone C1:A3-1) was purchased from Accurate Chemicals (Westbury, NY). Irrelevant isotype- and species-matched mAbs were used as staining controls. Cells were analyzed on an EPICS XL-MCL flow cytometer (Beckman Coulter, Miami, FL). Dead cells and debris were eliminated from analysis by excluding cells positive for propidium iodide and gating on the appropriate forward and side scatter profile.
Measurement of serum Ig isotype titers
Serum titers of PspA- and PPS14-specific Ig isotypes were measured by ELISA as previously described (30).
Fluorescent labeling of Pn14 with CM-DiI
Pn14 (1 × 109 CFU) were incubated at 37°C for 10 min, in 5 mM CM-DiI (chloromethylbenzamido derivative DiI) [Molecular Probes, Eugene, OR] in PBS, followed by additional 30 min incubation at 4°C, and washed before use.
Phagocytosis assay
APC were pulsed with CM-DiI-labeled Pn14. At different times, APC were washed in PBS, and viable cells were analyzed by flow cytometry with gating according to size, to exclude free bacteria. To control for cell surface binding of Pn14, cytochalasin D (CytD) [5 μg/ml] (Calbiochem, La Jolla, CA) was added to APC cultures (31). The treatment markedly inhibited the internalization of CM-DiI-labeled bacteria (<4% of the MFI uptake by untreated cells).
Fixation of BMM
BMM were fixed by incubation for 30 min in 1% paraformaldehyde in PBS at RT and washed 3× in PBS before use.
Statistics
Data were expressed as geometric mean ± S.E.M. of the individual results. Student’s t-test was used to determine statistical significance between groups. P<0.05 was considered statistically significant.
Results
Phenotypic characterization of unstimulated and Pn14-activated BMDC, BMM, and PerM populations in vitro
We previously demonstrated that BMDC pulsed with intact heat-killed Pn14 elicited, via distinct mechanisms, both protein- and PS-specific Ig isotype responses upon adoptive transfer into naïve mice (25). Using this approach, we now wished to compare the ability of Pn14-pulsed macrophages and DC to elicit antigen-specific Ig isotype responses in vivo. Heat-killed Pn14 was used in all experiments. Two distinct macrophage populations were utilized: freshly-harvested peritoneal macrophages (PerM) obtained in the absence of thioglycolate elicitation, and macrophages obtained after culturing BM cells for 6d in M-CSF (BMM). A similar approach, in which BM cells were instead cultured in GM-CSF, was used to produce BMDC. BMDC were only lightly adherent to the tissue culture plastic, and were easily removed by pipetting (32). In contrast, BMM and PerM were firmly adherent. We effected detachment by using lidocaine as previously described (33), instead of the more traditional method of cell scraping, since we found the latter caused significant cell death (data not shown). Although reports have shown that lidocaine can inhibit macrophage O2 consumption, digestion of immune complexes, or metabolic activity, all of these inhibitory effects could be completely reversed within 30 min following washing of the macrophages in medium (34–36). In this regard, we cultured lidocaine-detached macrophages in medium alone for at least 30 min before adding bacteria or other activators.
The 3 cell populations cultured in medium alone each exhibited distinct patterns of expression of a number of key cell surface phenotypic markers (Figure 1). As expected (37, 38), BMM and PerM both expressed, in a comparable fashion, significantly higher levels of F4/80, CD11b and CD14 than BMDC. Whereas all 3 populations expressed CD11c, BMM exhibited the highest expression. Of interest, whereas BMDC broadly expressed a high mean level of MHC-II, compared to the uniformly low expression by BMM, a major sub-population of PerM constitutively exhibited high levels of MHC-II. In contrast, all 3 populations expressed low levels of CD40, although PerM exhibited somewhat higher levels than either BMDC or BMM. CD86 expression on unstimulated BMDC was low, whereas BMM and PerM exhibited moderately higher and comparable levels. In contrast, significant CD80 expression was observed on both BMDC and BMM, but found at much lower levels on PerM. These data confirm two distinct populations of macrophages that each contrast with BMDC and suggest <3% cross-contamination between the macrophage and DC populations.
Figure 1. Cell surface phenotype of unstimulated and Pn14-activated BMDC, BMM, and PerM.
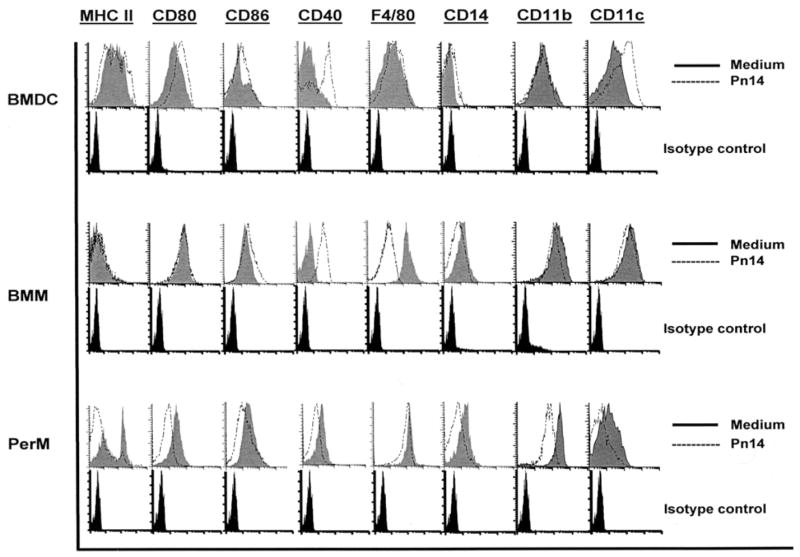
APC were stained with fluorochrome-labelled mAbs specific for the indicated cell surface proteins after a 24h culture period (1 × 106 cells/ml) in medium alone or in the presence of Pn14 (2 × 109 CFU/ml), and analyzed by flow cytometry. Staining with species/isotype control mAbs are below each set of tracings. Results representative of 2 independent experiments.
Following 24h of culture with intact Pn14, expression of MHC-II, CD80, CD86, CD40, CD14, CD11b, and CD11c each decreased significantly on viable PerM relative to cells cultured in medium alone (Figure 1). In contrast, CD40 expression was markedly upregulated on both BMDC and BMM following Pn14-mediated activation, whereas each moderately increased CD86. No significant modulation of either MHC-II or CD80 was observed in BMM, whereas BMDC exhibited a moderate increase in CD80, although no significant change in mean MHC-II expression. BMDC also upregulated CD11c. Collectively, the phenotypic profiles of both unstimulated PerM and Pn14-activated BMDC suggest that these cells may, during some time interval, be potentially effective APC for CD4+ T cells. However, low expression of MHC-II on BMM, in the absence or presence of Pn14, despite relatively high expression of costimulatory molecules following activation, suggest that these cells are likely to function poorly as APCs.
BMDC produce higher levels of pro- and anti-inflammatory cytokines than BMM or PerM in response to Pn14
Secretion of cytokines by activated APC can significantly impact on CD4+ T cell activation, and differentiation into distinct subsets, with subsequent differing effects on humoral immunity (39–41). In particular we previously demonstrated that endogenous pro-inflammatory cytokines stimulate, and the anti-inflammatory cytokine IL-10, inhibits humoral immunity in response to Pn14 (40). In this regard, we wished to compare the ability of BMDC, BMM, and PerM to secrete key pro- (IL-6, TNF-α, and IL-12), and anti- (IL-10) inflammatory cytokines in vitro in response to graded doses of Pn14. As illustrated in Figure 2A, BMDC secreted significantly (p<0.05) higher levels of all 4 cytokines than BMM or PerM, at the two highest doses of Pn14, and higher levels of IL-6 at all doses. BMM made significantly higher levels of IL-12 than PerM at all doses of Pn14, whereas the latter was more sensitive to low dose Pn14 for TNF-α secretion. We previously demonstrated that Pn14 elicits cytokine production by splenic macrophages and DC through the combined action of TLR2, TLR4, and TLR9 (42). In this regard, a similar pattern of IL-12 secretion as that observed for intact Pn14, was also observed using optimal amounts of individual TLR ligands (i.e. LPS [TLR4], CpG-ODN [TLR9], and Pam3Cys [TLR2]) (43) (Figure 2B). Likewise, BMDC secreted significantly higher amounts of TNF-α than BMM or PerM in response to all 3 TLR ligands, and PerM made significantly more TNF-α than BMM at least in response to LPS. In a separate experiment to measure release of TGF-β, an anti-inflammatory cytokine, BMDC, BMM, and perM were cultured for 24h in the presence of either medium alone or Pn14. No detectable (< 1 pg/ml) active TGF-β was observed in either medium alone or in the presence of Pn14 from any of the 3 cell types (Fig. 2C). Detectable amounts of total (active + latent) TGF-β were released constitutively by all three cell populations with only modest, though significant differences between the cell types (perM>BMDC>BMM). In the presence of Pn14, BMDC showed a only modest, though significant, increase in total TGF-β release whereas BMM and perM showed modest, though significant, decreases (Fig. 2C). Collectively, although these data indicate that BMDC elicit a more robust in vitro, pro-inflammatory cytokine response to Pn14 than either BMM or PerM, it is not clear whether the higher levels of IL-10 secreted by BMDC would counteract the potential in vivo Ig-stimulatory effects of the enhanced IL-6, IL-12, and TNF-α response (40). Alternatively, as we reported previously, autocrine IL-10 might enhance APC-mediated induction of Ig secretion by delaying APC apoptosis (41).
Figure 2. Cytokine secretion by BMDC, BMM, or PerM in the absence or presence of Pn14 or TLR ligands.

APC were cultured for 24 h in medium alone or in the presence of (A) varying concentrations of Pn14 or (B) LPS (2 μg/ml), CpG-ODN (2 μg/ml), or Pam3CSK4 (150 ng/ml) or (C) 2 × 109 CFU/ml of Pn14. Concentrations of cytokines in the culture SN were measured by ELISA. Values represent the geometric mean ± SEM. Significance (referred to in text, p<0.05). Representative of 2 independent experiments.
BMM>BMDC>PerM for phagocytosis of Pn14
Although both macrophages and DC can phagocytose intact bacteria, the rate by which this occurs could impact on their ability to act as APC. In particular, we were interested in establishing conditions to obtain roughly comparable bacterial uptake by BMDC, BMM, and PerM in vitro, in order to better compare their ability to induce humoral responses upon adoptive transfer into naïve mice. In a first set of experiments, Pn14 was labeled with the fluorescent dye CM-Dil and added to cultures of APC at a ratio of 800 CFU Pn14/APC for varying periods of time. The mean fluorescence intensity of the 3 APC populations was measured to compare the relative uptake of bacteria at each time point. Since fluorescence could reflect cell surface binding rather than actual uptake, we included control groups consisting of the various APC cultured with Pn14 in the presence of cytochalasin D, an inhibitor of microfilament assembly, and hence phagocytosis, for 300 min (the latest time point). We previously demonstrated that cytochalasin D blocks Pn14 uptake into BMDC (31). As illustrated in Figure 3A the efficiency of uptake of Pn14 was BMM>BMDC>PerM. With the addition of concentrations of Pn14 >800 CFU/APC these differences could be somewhat narrowed over time (Figure 3B). In a separate experiment we determined that BMDC, BMM, and PerM internalized 27, 58, and 14 CFU of Pn14 per cell, respectively following 4h of culture with 2,000 CFU Pn14/APC. We used these conditions for our subsequent adoptive transfer experiments.
Figure 3. Uptake of Pn14 by BMDC, BMM, and PerM.

APC were cultured in the presence of CM-Dil-labeled Pn14. (A). APC were cultured for the indicated times with 8 × 108 CFU of labeled Pn14 (800 CFU/APC). (B). APC were cultured for 240 min with the indicated concentrations of labeled Pn14. Cytochalasin D (“cyto”) was used to control for surface binding of labeled Pn14. APC were analyzed by flow cytometry. Results are representative of 2 independent experiments.
Pn14-pulsed BMM and PerM elicit humoral immune responses comparable to BMDC upon adoptive transfer into naïve mice
BMDC, BMM, and PerM were pulsed with Pn14 in vitro under the conditions cited above. After thorough washing to remove free bacteria, 1 × 106 Pn14-pulsed APC were injected i.v. per mouse. As controls, free Pn14 and unpulsed APC were also injected i.v. into separate groups of mice. The percentages of Pn14-pulsed BMDC and BMM found in the spleen, 24h after i.v. injection were 0.01% and 0.03% of the total spleen cell population, respectively (significant, p<0.05, one representative of two independent experiments). Fourteen days post-immunization with either Pn14-pulsed APC or free Pn14, all groups were boosted i.p. with free Pn14 to asses the potential generation of memory during the primary immunization. Sera were collected on d0 (pre-bleed), 7, 14 (following primary immunization with Pn14-pulsed APC or free Pn14), and 21 (following secondary immunization with free Pn14) for determination of titers of IgG specific for pneumococcal surface protein A (PspA), and IgM and IgG specific for the type 14 capsular polysaccharide (PPS14). As illustrated in Figure 4A, Pn14-pulsed BMDC, BMM, and PerM each effectively primed naïve mice, in a comparable manner, for a secondary IgG anti-PspA response following boosting, on d14, with free Pn14. Thus, d21 serum titers of IgG anti-PspA were significantly (p<0.05) higher in all groups primed with Pn14-pulsed APC relative to mice primed with unpulsed APC, and relative to naïve mice, 7d after immunization with free Pn14. Additionally, a small, but significant, primary IgG anti-PspA response was also elicited by all Pn14-pulsed APC, in contrast to undetectable titers in mice primed with unpulsed APC (Figure 4A). In contrast, none of the Pn14-pulsed APC populations primed mice for a secondary IgM or IgG anti-PPS14 response following boosting with free Pn14 (Figure 4A). However, all Pn14-pulsed APC induced substantial primary IgM and IgG anti-PPS14 responses, in contrast to the undetectable titers observed in mice injected with unpulsed APC. Additionally, no significant differences in d21 serum titers of PspA-specific IgG3, IgG1, IgG2b, or IgG2a were observed between any of the groups primed with Pn14-pulsed APC and boosted with free Pn14 (Figure 4B). With the exception of significantly higher d14 serum tiers of PPS14-specific IgG3 and IgG1 in mice injected with Pn14-pulsed BMM relative to BMDC, PPS14-specific IgG isotypes were otherwise comparable among the 3 groups (Figure 4B).
Figure 4. Adoptive transfer of Pn14-pulsed BMDC, BMM, and PerM into naïve, WT mice.

APC were cultured in medium alone or pulsed with Pn14, and injected i.v. into naïve BALB/c mice (6 mice per group). Mice were boosted i.p. with free Pn14 (2 × 108 CFU/mouse) 14d following injection of APC. Another group of BALB/c mice (n=6) were immunized and boosted on d14 with free Pn14 alone. Sera were collected on the indicated days for (A) measurement of serum Ig titers. (B) Titers of secondary (d21) PspA- and primary (day 14) PPS14-specific IgG isotypes were measured from sera analyzed in “A”. The data show the geometric mean ± SEM of the individual titers. Significance (p<0.05) is indicated in the text. Representative of 2 independent experiments.
Pn14-pulsed BMM play an active role as APC for initiating the PspA-specific IgG response
The ability of Pn14-pulsed macrophages to initiate a humoral immune response upon adoptive transfer into naïve mice could reflect an active process mediated by the transferred macrophages or, alternatively, the passive transfer by macrophages of intact Pn14 and/or Pn14-derived antigens to other immune cell types. Further, Pn14-pulsed macrophages could be playing a direct and critical role in presenting Pn14-derived proteins to CD4+ T cells in vivo or, alternatively, might be actively transferring Pn14 proteins to endogenous APC. In the following series of studies to test these possibilities, we focused solely upon BMM, as opposed to PerM, because of our ability to obtain substantially larger numbers of BMM for our experiments. BMM were pulsed with Pn14 as described above for adoptive transfer (see Figure 4), then either fixed with paraformaldehyde or left untreated, immediately prior to i.v. injection. The percentages of Pn14-pulsed viable and fixed BMM found in the spleen, 24h after i.v. injection were 0.02% and 0.06% of the total spleen cell population, respectively (significant, p<0.05, one representative of two independent experiments). As illustrated in Figure 5, viable, but not fixed, Pn14-pulsed BMM elicited a primary PspA-specific IgG response, and primed mice for a secondary IgG response following boosting with free Pn14, similar to that observed in Figure 4. In distinct contrast, fixed Pn14-pulsed BMM elicited an IgG anti-PPS14 response that was not significantly different than that for viable BMM, whereas the IgM anti-PPS14 was only modestly, though, significantly reduced (Figure 5). In a separate experiment, serving as a control, BALB/c mice (7 per group) were immunized and boosted with free heat-killed Pn14 with or without further paraformaldehyde fixation. No significant differences in the serum titers of IgG anti-PspA, as well as IgM or IgG anti-PPS14 were observed (data not shown) strongly suggesting that paraformaldehyde did not destroy key antigenic epitopes in the bacteria. Thus, viable BMM are required in vivo for induction of the PspA-, though not PPS14-specific, Ig response.
Figure 5. Adoptive transfer of viable and paraformaldehyde-fixed Pn14-pulsed BMM.

BMM obtained from BALB/c mice were pulsed in vitro with Pn14. BMM were then either left untreated or fixed in paraformaldehyde, and immediately injected i.v. into naïve BALB/c mice (5 mice per group). Mice were immunized i.p. 14d later with free Pn14 (2 × 108 CFU/mouse). A separate group of 5 mice were immunized and boosted i.p. on d14 with free Pn14 alone (2 × 108 CFU/mouse). Sera were collected on the indicated days for measurement of serum Ig titers. The data show the geometric mean +/− SEM of the individual titers. *Significance (p<0.05) in comparing viable vs. fixed BMM. Representative of 2 independent experiments.
To assess whether transferred Pn14-pulsed BMM were playing an active role as APC in vivo for induction of the PspA-specific IgG response, we first wished to determine whether this response required endogenous T cells. We previously demonstrated that both the IgG anti-PspA and IgG anti-PPS14 response to free Pn14 was dependent on CD4+, but not CD8+, T cells, whereas the IgM anti-PPS14 response was T cell-independent (24, 44). Pn14-pulsed BMM or free Pn14 were injected into either naïve WT or athymic nude mice, the latter being markedly deficient in T cells. As illustrated in Figure 6, both Pn14-pulsed BMM and free Pn14 significantly (p<0.05) primed WT mice, but not athymic nude mice, for a subsequent secondary IgG anti-PspA response following boosting with free Pn14. In distinct contrast Pn14-pulsed BMM elicited an equivalent primary IgG anti-PPS14 response in WT and athymic nude mice (Figure 6). Boosting of WT mice, initially injected with Pn14-pulsed BMM, with free Pn14 increased the IgG anti-PPS14 response only to those levels seen in WT mice immunized with free Pn14 alone, confirming that there was no significant priming by Pn14-pulsed BMM for induction of PPS14-specific IgG. These data thus demonstrate that the IgG anti-PspA response to Pn14-pulsed BMM is dependent on endogenous CD4+ T cells.
Figure 6. Adoptive transfer of Pn14-pulsed BMM into athymic nude mice.

BMM obtained from BALB/c mice were pulsed in vitro with Pn14. BMM (i.v.) or free Pn14 (2 × 108 CFU/mouse, i.p.) were then injected into either naïve BALB/c or athymic nude (BALB/c background) mice (5 mice per group). All groups of mice were immunized i.p. 14d later with free Pn14 (2 × 108 CFU/mouse). Sera were collected on the indicated days for measurement of serum Ig titers. The data show the geometric mean +/− SEM of the individual titers. Significance (p<0.05) is indicated in the text. Representative of 2 independent experiments.
We next wished to address whether the IgG anti-PspA response to Pn14-pulsed BMM required cognate interactions between the transferred BMM and the endogenous CD4+ T cells. Thus, BMM were obtained from mice genetically deficient in MHC-II. WT and MHC-II−/− BMM were pulsed with Pn14 in vitro and injected i.v. into naïve WT mice followed by boosting i.p. with free Pn14 on d14. A separate group of WT mice were immunized and boosted with free Pn14 alone as a control. In contrast to WT Pn14-pulsed BMM, MHC-II−/− BMM exhibited a complete failure to prime for a secondary IgG anti-PspA response (Figure 7A). In contrast, both WT and MHC-II−/− Pn14-pulsed BMM elicited equivalent IgM and IgG anti-PPS14 responses. In a second, analogous set of studies, we compared WT and CD40−/− Pn14-pulsed BMM for their ability to elicit Ig responses upon adoptive transfer into WT mice. During cognate interactions, CD40-ligand on activated T cells binds CD40 expressed by APC, resulting in delivery of a potent costimulus for cytokine secretion and phenotypic maturation of the APC (45). In this regard, we observed that the primary IgG anti-PspA response elicited by Pn14-pulsed CD40−/− BMM was markedly reduced relative to WT BMM, whereas priming by CD40−/− BMM for the secondary response was partially, though significantly, reduced as well (Figure 7B). In contrast, CD40−/− and WT BMM elicited essentially equivalent IgM and IgG anti-PPS14 responses (Figure 7B)
Figure 7. Adoptive transfer of Pn14-pulsed BMM from WT versus MHC-II−/− or CD40−/− mice into naïve WT mice.

BMM obtained from (A) C57BL/6 [WT] or MHC-II−/− mice (C567BL/6 background) or (B) BALB/c [WT] or CD40−/− mice (BALB/c background) were pulsed in vitro with Pn14. BMM (i.v.) or free Pn14 (2 × 108 CFU/mouse i.p.) were then injected into naïve C57BL/6 [A] or BALB/c [B] mice (5 mice per group). All groups of mice were immunized i.p. 14d later with free Pn14 (2 × 108 CFU/mouse). Sera were collected on the indicated days for measurement of serum Ig titers. The data show the geometric mean +/− SEM of the individual titers. *Significance (p<0.05) in comparing BMM obtained from WT vs knockout mice. Both “A” and “B” are each representative of 2 independent experiments.
BMDC pulsed with either recombinant PspA or intact Pn14 are more potent stimulators of IL-2 secretion by a PspA-specific T hybridoma, than similarly treated BMM
Collectively, the data so far strongly suggest that Pn14-pulsed BMM, in addition to BMDC, can play an active role as APC for CD4+ T cells when adoptively transferred into naïve mice. In a final set of experiments we therefore wished to assess the relative ability of BMDC and BMM to act as APC for CD4+ T cells upon uptake of Pn14. We thus created an MHC-IId-restricted CD4+ T hybridoma (BALD4) specific for the Pn14 cell wall protein, pneumococcal surface protein A (PspA) [see Methods]. In preliminary studies we observed that BMDC cultured with Pn14 failed to stimulate BALD4 for IL-2 secretion (data not shown). This was in distinct contrast with either recombinant PspA, intact Pn, capsular type 2 (strain D39) or its unencapsulated isogenic mutant (strain R36A). In light of previous reports that different strains of Pn can express antigenically-distinct families/seroclades of PspA, we concluded that BALD4 recognized an epitope of PspA not expressed by our Pn14 strain. We thus genetically engineered a new strain of capsular type 14 Pn (R6-14) that produced the same family 1/seroclade 2 of PspA as strain R36A (both recognized by the mAbs DC10-IA5 and CF9IIB7, and able to stimulate BALD4), but expressed the same serotype 14 capsular PS as Pn14 (both recognized by the mAb 44.1) [see “Methods”].
BMDC and BMM were co-cultured with BALD4 for 24h in the presence of varying concentrations of PspA (0.001–25 μg/ml) or R6-Pn14 or its unencapsulated isogenic mutant (R6) [0.03–67 × 106 CFU/ml]. At the end of the culture period, SN was collected for determination of IL-2 concentrations by ELISA. In 3 independent experiments BMDC were found to be 12 +/− 2 (arithmetic mean +/− SEM)-fold more efficient at presenting PspA than BMM (significance p<0.05) (Figure 8). This efficiency reflects the amount of additional PspA required for BMM to give an IL-2 response equivalent to that induced by BMDC. The difference in efficiency between BMDC and BMM co-cultured with either R6-14 or R6, for induction of IL-2 by BALD4 was even more marked than that observed for PspA: 443 +/− 34 for R6-14 (2 independent experiments) and 418 +/− 117 for R6 (3 independent experiments) (Figure 8). Of interest, although it has been suggested that the capsular PS might interfere with, or otherwise modulate, the presentation of bacterial protein to T cells (46–49), the IL-2 induction profiles using R6-14 and R6 were essentially identical. The graphs in Figure 8 were aligned so that the amount of PspA, calculated by an inhibition ELISA assay, contained in a given dose of R6-14 or R6 is directly (vertically) beneath the concentration given for PspA in the uppermost graph. This indicates that BMDC, though not BMM, present PspA ~100-fold more efficiently to BALD4 when expressed by the intact bacteria as opposed to being present in an isolated, soluble form (Figure 8). Collectively, these data strongly suggest that Pn14-pulsed BMM can play an active role as APC for eliciting a robust TD humoral immune response in vivo, despite a markedly lower APC efficiency in vitro relative to BMDC.
Figure 8. APC capacity of BMDC versus BMM pulsed with either free PspA or unencapsulated or encapsulated Pn for a PspA-specific T hybridoma.
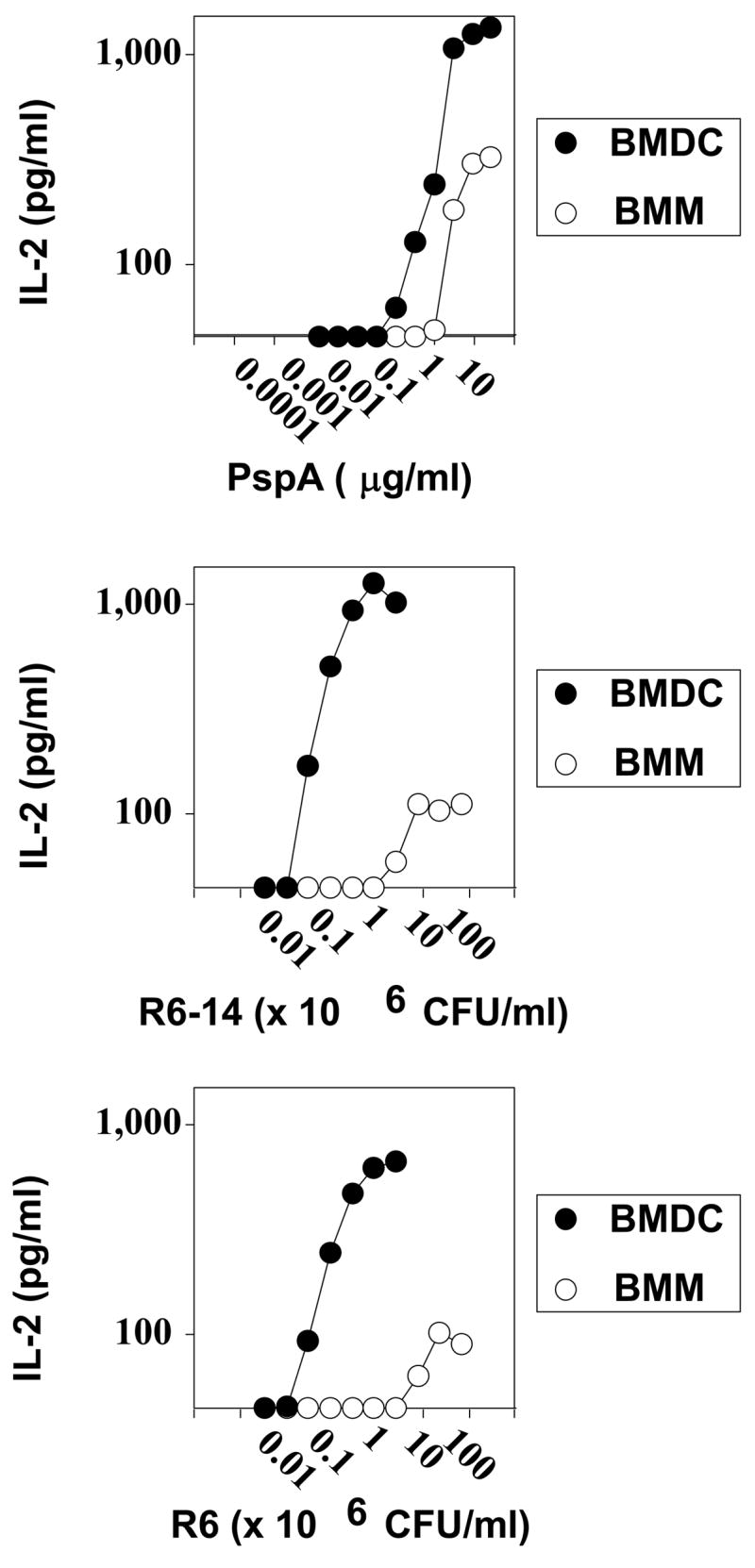
BMDC and BMM obtained from BALB/c mice were cultured in the presence of the indicated concentrations of (A) PspA, (B) R6-14, or (C) R6. BALD4 (PspA-specific T hybridoma) cells were then added to these cultures, in the continued presence of the added antigen/bacteria, and cultured for 24h. Concentrations of IL-2 released by BALD4 cells in the culture SN were measured by ELISA. Representative of 2 independent experiments.
Discussion
Although many studies have documented the ability of macrophages to internalize pathogen-associated antigens, and present them to preprimed T cells (50–54), it is widely assumed that the induction of CD4+ T cell-dependent antibody responses in naïve mice is uniquely dependent upon initial antigen presentation by DC (1, 2). Following this event, antigen-specific B cells engage in cognate interactions with DC-primed CD4+ T cells to elicit effector functions critical for induction of Ig secretion and class switching (55). Although a role for macrophages as APC in this latter process is largely obscure, macrophages may regulate humoral immunity through other mechanisms, including transfer of intact antigen to B cells (56, 57), secretion of BAFF, a B cell survival factor and inducer of Ig class switching (58) and/or local release of C3 (59). Although the indirect evidence in support of a central role for DC as APC in naïve mice is extensive, there is to our knowledge, no published data directly demonstrating that endogenous DC are indeed indispensable for eliciting in vivo humoral immune responses. Thus, although several studies, using CD11c-DTR Tg mice, treated with DT to deplete DC, demonstrated a critical role for endogenous DC in eliciting a CD8+ CTL response (19–21) or CD4+ T cell priming (22), in the model systems studied, no comparable data on humoral immunity have as yet been reported. Further, although a number of imaging studies have documented early DC-CD4+ T cell interactions in situ, following immunization with protein antigen (23), there is little indication that potential macrophage-CD4+ T cells were extensively investigated, especially in the spleen, leaving open their possible role as initiating APC for naïve T cells.
Two groups studied the ability of antigen-pulsed macrophages to elicit immunity upon adoptive transfer into naïve mice, and surprisingly both reported that they were indeed capable of acting in a manner comparable to that of antigen-pulsed DC. In the series of studies by Moser and colleagues (15–17, 60) macrophages pulsed in vitro with protein antigen were shown to elicit specific antibody production upon injection into naïve mice. However, the possibility that the injected macrophages were acting largely via passive transfer of antigen to endogenous DC was not ruled out. However, a study by Pozzi et al, demonstrated the ability of peptide-pulsed macrophages to elicit CD8+ CTL responses upon injection into naïve mice, in which a role for endogenous APC in the recipients was clearly ruled out (18).
In our current study we demonstrate that Pn14-pulsed macrophages play an active role in eliciting the IgG anti-PspA response, upon their transfer into naïve mice. This response was abrogated in T cell-deficient recipients, consistent with our earlier studies using free Pn14, in which an absolute dependence on CD4+ T cells was demonstrated (24). We further show that the induction of the IgG anti-PspA response by Pn14-pulsed macrophages was not due exclusively to passive transfer of PspA to endogenous APC. Thus, macrophages from mice genetically deficient in MHC-II or CD40, or WT macrophages made non-viable prior to transfer, were defective in eliciting the IgG anti-PspA response in WT mice, following in vitro pulsing with intact Pn14. This requirement for macrophage expression of MHC-II and CD40 further supports the notion that these cells were acting as APC for CD4+ T cells. We believe that it is highly unlikely that the observed Ig-inducing effects of the Pn14-pulsed macrophage populations used in this study were due to contaminating DC. Firstly, we utilized cells that were firmly adherent to the tissue culture plastic following 7d of culture in the presence of M-CSF-1, a feature not traditionally observed for DC. Secondly, phenotypic analysis of the macrophage populations revealed an essentially unimodal population of cells with a relatively high expression of F4/80, CD14, and CD11b, characteristic of macrophages (37, 61). Finally, the ability of the Pn-pulsed BMM population to present the bacterial PspA to a PspA-specific T hybridoma was ~400-fold less efficient than Pn-pulsed BMDC.
In contrast to the in vivo IgG anti-PspA response induced by Pn14-pulsed BMM, the IgG anti-PPS14 response was not dependent on BMM expression of MHC-II or CD40, was largely independent of BMM viability immediately prior to transfer, and was T cell-independent. However, as previously described (24), the IgG anti-PPS14 response to free Pn14 was T cell-dependent. The IgG anti-PPS14 response elicited by Pn14-pulsed BMM also differed in several respects with what we previously observed using Pn14-pulsed BMDC (25). Using BMDC, the IgG anti-PPS14 response did dependent on BMDC viability and was T cell-dependent, although with the exception of the IgG1 isotype, not dependent on BMDC expression of MHC-II or CD40. The mechanism by which Pn14-pulsed BMM elicited the anti-PPS14 response is not clear, although macrophages have been previously implicated in stimulating T cell-independent antibody responses. Thus, it was demonstrated that macrophages can directly enhance proliferation of B cells in vitro (58). Further, in vitro T cell-independent B cell responses to TNP-Ficoll, TNP-LPS and TNP-Brucella abortus are dependent on the presence of macrophages (62–65). B cells that are stimulated by TNP-Ficoll-pulsed macrophages appear to be a distinct B cell subset that is uniquely responsive to macrophage signals (66). Hence, direct activation of B cells by macrophages, even perhaps macrophages in a non-viable state, might account for their inductive effect.
We demonstrate that Pn14-pulsed macrophages can stimulate a PspA-specific T hybridoma in an MHC-II-restricted manner following uptake of intact Pn14 or free PspA, although with much lower efficiency than DC. Others, using naïve or primed CD4+ T cells have also demonstrated a similar difference in APC efficiency (1, 2). We directly compared presentation of a protein expressed by an intact extracellular bacterium with the same protein, produced in an isolated, soluble form. In this regard, we show that DC exhibit a much greater APC efficiency than macrophages when using intact Pn, as opposed to soluble PspA, where the differences are less marked. The reason for this difference is unclear, although the requirements for APC presentation of soluble and particulate antigens are distinct (67–69). Our data appear to contrast with earlier work reporting that macrophages, but not DC, are capable of presenting particulate OVA to primed T cells (70). The poor efficiency of macrophages as APC, relative to DC, may relate both to their lower surface expression of MHC-II and their tendency to be highly degradative (71). Of note, although it has been suggested that polysaccharides might interfere with, or otherwise regulate, APC presentation of associated proteins (46, 48, 49, 72), we find no difference in APC efficiency when using an encapsulated Pn vs. its unencapsulated, isogenic variant.
Our data notwithstanding, studies documenting the ability of DC to act as superior APC for naïve CD4+ T cells, would appear to argue against a role for endogenous macrophages as initiators of humoral immunity. However, this could be an oversimplification, especially in regard to Ig responses induced by intact bacteria. Thus, the differences observed in APC efficiency between macrophages and DC are relative, not absolute. The threshold level for initial priming of naïve CD4+ T cells required to initiate an in vivo humoral immune response is not clear. Further, macrophages are uniquely positioned in secondary lymphoid organs, and functionally specialized, to efficiently internalize incoming bacteria (38, 73). In this regard, we observed that BMM were more efficient than BMDC in uptake of Pn14. Large numbers of macrophages may also be recruited into secondary lymphoid organs such as the spleen, especially in response to a systemic bacterial infection. Macrophages present in an inflammatory milieu will likely upregulate cell surface costimulatory molecules and release cytokines that may enhance naïve CD4+ T cell priming. We demonstrate that in response to Pn14, which contain multiple TLR ligands, macrophages upregulate CD40 and CD86, and release pro-inflammatory cytokines, including the Th1-inducing cytokine, IL-12 (74). Of interest, it was previously demonstrated that macrophages pulsed in vitro with soluble protein in the absence of a TLR ligand, elicit a predominant Th2 antibody response upon adoptive transfer (15, 60), whereas additional stimulation with a TLR ligand resulted in the induction of both Th1 and Th2 IgG isotypes, similar to DC pulsed with antigen in the absence of TLR stimulation (16). In this regard, we demonstrate that Pn14-pulsed DC and macrophages also elicited a similar PspA-specific IgG isotype profile upon transfer in vivo, and subsequent boosting with free Pn14. Macrophages are also functionally heterogeneous (73, 75, 76), even in regard to their ability to act as APC. In particular, one study demonstrated that 20% of splenic macrophage precursors could process and present a soluble protein antigen to naïve CD4+ T cells, that was positively correlated with their ability to secrete IL-12 (9). Thus, a particular macrophage subset, used in studies of APC function, may not accurately reflect the overall APC potential of the mixed macrophage population in vivo, especially within an inflammatory milieu.
Acknowledgments
This study was supported by N.I.H. grant 2R01 AI49192 and the U.S.U.H.S. Dean’s Research and Education Endowment Fund.
Opinions and assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of Defense or the Uniformed Services University of the Health Sciences.
We thank Dr. Alex Lucas (Children’s Hospital Oakland Research Institute, Oakland, CA) for the IgG1κ mouse mAb specific for the capsular polysaccharide of S. pneumoniae type 14 (44.1) and Dr. Rick Schuman (Biosynexus, Inc., Gaithersburg, MD) for the two IgG2a mouse mAbs specific for PspA (DC10-IA5 and CF9IIB7). We also thank Drs Kryzysztof Trzcinski and Marc Lipsitch (Harvard School of Public Health, Boston, MA) for the R6J strain.
Abbreviations used in this paper
- Pn14
intact Streptococcus pneumoniae, capsular type 14
- PPS14
capsular polysaccharide, serotype 14
- BMM
bone marrow-derived macrophage
- PerM
peritoneal macrophage
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- 3.Steinman RM, Nogueira N, Witmer MD, Tydings JD, Mellman IS. Lymphokine enhances the expression and synthesis of Ia antigens on cultured mouse peritoneal macrophages. J Exp Med. 1980;152:1248–1261. doi: 10.1084/jem.152.5.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinman RM, Gutchinov B, Witmer MD, Nussenzweig MC. Dendritic cells are the principal stimulators of the primary mixed leukocyte reaction in mice. J Exp Med. 1983;157:613–627. doi: 10.1084/jem.157.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinman RM, Witmer MD. Lymphoid dendritic cells are potent stimulators of the primary mixed leukocyte reaction in mice. Proc Natl Acad Sci U S A. 1978;75:5132–5136. doi: 10.1073/pnas.75.10.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inaba K, Metlay JP, Crowley MT, Steinman RM. Dendritic cells pulsed with protein antigens in vitro can prime antigen-specific, MHC-restricted T cells in situ. J Exp Med. 1990;172:631–640. doi: 10.1084/jem.172.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levin D, Constant S, Pasqualini T, Flavell R, Bottomly K. Role of dendritic cells in the priming of CD4+ T lymphocytes to peptide antigen in vivo. J Immunol. 1993;151:6742–6750. [PubMed] [Google Scholar]
- 8.Croft M, Duncan DD, Swain SL. Response of naive antigen-specific CD4+ T cells in vitro: characteristics and antigen-presenting cell requirements. J Exp Med. 1992;176:1431–1437. doi: 10.1084/jem.176.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Askew D, Gatewood J, Olivas E, Havenith K, Walker WS. A subset of splenic macrophages process and present native antigen to naive antigen-specific CD4+ T-cells from mice transgenic for an alpha beta T-cell receptor. Cell Immunol. 1995;166:62–70. doi: 10.1006/cimm.1995.0008. [DOI] [PubMed] [Google Scholar]
- 10.Crowley M, Inaba K, Steinman RM. Dendritic cells are the principal cells in mouse spleen bearing immunogenic fragments of foreign proteins. J Exp Med. 1990;172:383–386. doi: 10.1084/jem.172.1.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inaba K, Steinman RM, Van Voorhis WC, Muramatsu S. Dendritic cells are critical accessory cells for thymus-dependent antibody responses in mouse and in man. Proc Natl Acad Sci U S A. 1983;80:6041–6045. doi: 10.1073/pnas.80.19.6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inaba K, Steinman RM. Protein-specific helper T-lymphocyte formation initiated by dendritic cells. Science. 1985;229:475–479. doi: 10.1126/science.3160115. [DOI] [PubMed] [Google Scholar]
- 13.Colino J, Snapper CM. Dendritic cells, new tools for vaccination. Microbes Infect. 2003;5:311–319. doi: 10.1016/s1286-4579(03)00033-9. [DOI] [PubMed] [Google Scholar]
- 14.De Becker G, Sornasse T, Nabavi N, Bazin H, Tielemans F, Urbain J, Leo O, Moser M. Immunoglobulin isotype regulation by antigen-presenting cells in vivo. Eur J Immunol. 1994;24:1523–1528. doi: 10.1002/eji.1830240710. [DOI] [PubMed] [Google Scholar]
- 15.De Becker G, Moulin V, Tielemans F, De Mattia F, Urbain J, Leo O, Moser M. Regulation of T helper cell differentiation in vivo by soluble and membrane proteins provided by antigen-presenting cells. Eur J Immunol. 1998;28:3161–3171. doi: 10.1002/(SICI)1521-4141(199810)28:10<3161::AID-IMMU3161>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 16.De Becker G, Moulin V, Pajak B, Bruck C, Francotte M, Thiriart C, Urbain J, Moser M. The adjuvant monophosphoryl lipid A increases the function of antigen-presenting cells. Int Immunol. 2000;12:807–815. doi: 10.1093/intimm/12.6.807. [DOI] [PubMed] [Google Scholar]
- 17.Moser M. Regulation of Th1/Th2 development by antigen-presenting cells in vivo. Immunobiology. 2001;204:551–557. doi: 10.1078/0171-2985-00092. [DOI] [PubMed] [Google Scholar]
- 18.Pozzi LA, Maciaszek JW, Rock KL. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J Immunol. 2005;175:2071–2081. doi: 10.4049/jimmunol.175.4.2071. [DOI] [PubMed] [Google Scholar]
- 19.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sapoznikov A, Fischer JA, Zaft T, Krauthgamer R, Dzionek A, Jung S. Organ-dependent in vivo priming of naive CD4+,but not CD8+, T cells by plasmacytoid dendritic cells. J Exp Med. 2007;204:1923–1933. doi: 10.1084/jem.20062373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Probst HC, van den Broek M. Priming of CTLs by lymphocytic choriomeningitis virus depends on dendritic cells. J Immunol. 2005;174:3920–3924. doi: 10.4049/jimmunol.174.7.3920. [DOI] [PubMed] [Google Scholar]
- 22.Tian T, Woodworth J, Skold M, Behar SM. In Vivo Depletion of CD11c+ Cells Delays the CD4+ T Cell Response to Mycobacterium tuberculosis and Exacerbates the Outcome of Infection. J Immunol. 2005;175:3268–3272. doi: 10.4049/jimmunol.175.5.3268. [DOI] [PubMed] [Google Scholar]
- 23.Cahalan MD. Choreography of cell motility and interaction dynamics imaged by two-photon microscopy in lymphoid organs. Annu Rev Immunol. 2008;26:585–626. doi: 10.1146/annurev.immunol.24.021605.090620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan AQ, Lees A, Snapper CM. Differential regulation of IgG anti-capsular polysaccharide and antiprotein responses to intact Streptococcus pneumoniae in the presence of cognate CD4+ T cell help. J Immunol. 2004;172:532–539. doi: 10.4049/jimmunol.172.1.532. [DOI] [PubMed] [Google Scholar]
- 25.Colino J, Shen Y, Snapper CM. Dendritic cells pulsed with intact Streptococcus pneumoniae elicit both protein- and polysaccharide-specific immunoglobulin isotype responses in vivo through distinct mechanisms. J Exp Med. 2002;195:1–13. doi: 10.1084/jem.20011432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Q, Sen G, Snapper CM. Endogenous IL-1R1 signaling is critical for cognate CD4+ T cell help for induction of in vivo type 1 and type 2 antipolysaccharide and antiprotein Ig isotype responses to intact Streptococcus pneumoniae, but not to a soluble pneumococcal conjugate vaccine. J Immunol. 2006;177:6044–6051. doi: 10.4049/jimmunol.177.9.6044. [DOI] [PubMed] [Google Scholar]
- 27.Sen G, Chen Q, Snapper CM. Immunization of aged mice with a pneumococcal conjugate vaccine combined with an unmethylated CpG-containing oligodeoxynucleotide restores defective immunoglobulin G antipolysaccharide responses and specific CD4+-T-cell priming to young adult levels. Infect Immun. 2006;74:2177–2186. doi: 10.1128/IAI.74.4.2177-2186.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trzcinski K, Thompson CM, Lipsitch M. Construction of otherwise isogenic serotype 6B, 7F, 14, and 19F capsular variants of Streptococcus pneumoniae strain TIGR4. Appl Environ Microbiol. 2003;69:7364–7370. doi: 10.1128/AEM.69.12.7364-7370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canaday DH, Gehring A, Leonard EG, Eilertson B, Schreiber JR, Harding CV, Boom WH. T-cell hybridomas from HLA-transgenic mice as tools for analysis of human antigen processing. J Immunol Methods. 2003;281:129–142. doi: 10.1016/j.jim.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Chattopadhyay G, Khan AQ, Sen G, Colino J, Dubois W, Rubtsov A, Torres RM, Potter M, Snapper CM. Transgenic Expression of Bcl-xL or Bcl-2 by Murine B Cells Enhances the In Vivo Antipolysaccharide, but Not Antiprotein, Response to Intact Streptococcus pneumoniae. J Immunol. 2007;179:7523–7534. doi: 10.4049/jimmunol.179.11.7523. [DOI] [PubMed] [Google Scholar]
- 31.Colino J, Snapper CM. Two distinct mechanisms for induction of dendritic cell apoptosis in response to intact Streptococcus pneumoniae. J Immunol. 2003;171:2354–2365. doi: 10.4049/jimmunol.171.5.2354. [DOI] [PubMed] [Google Scholar]
- 32.Steinman RM, Kaplan G, Witmer MD, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. V. Purification of spleen dendritic cells, new surface markers, and maintenance in vitro. J Exp Med. 1979;149:1–16. doi: 10.1084/jem.149.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies JQ, Gordon S. Isolation and culture of murine macrophages. Methods Mol Biol. 2005;290:91–103. doi: 10.1385/1-59259-838-2:091. [DOI] [PubMed] [Google Scholar]
- 34.Duddridge M, Kelly CA, Ward C, Hendrick DJ, Walters EH. The reversible effect of lignocaine on the stimulated metabolic activity of bronchoalveolar lavage cells. Eur Respir J. 1990;3:1166–1172. [PubMed] [Google Scholar]
- 35.Hijlstra A, Van Dorp W, Daha MR, Leslie GQ. The effect of lidocaine on the processing of soluble immune aggregates and immune complexes by peritoneal macrophages. Immunology. 1980;41:237–244. [PMC free article] [PubMed] [Google Scholar]
- 36.Hoidal JR, White JG, Repine JE. Influence of cationic local anesthetics on the metabolism and ultrastructure of human alveolar macrophages. J Lab Clin Med. 1979;93:857–866. [PubMed] [Google Scholar]
- 37.Hume DA. The mononuclear phagocyte system. Curr Opin Immunol. 2006;18:49–53. doi: 10.1016/j.coi.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 38.Gordon S. Macrophages and the immune response. In: Paul William E., editor. Fundamental, Immunology. 4. 1999. pp. 533–545. [Google Scholar]
- 39.Reinhardt RL, Kang SJ, Liang HE, Locksley RM. T helper cell effector fates--who, how and where? Curr Opin Immunol. 2006;18:271–277. doi: 10.1016/j.coi.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 40.Khan AQ, Shen Y, Wu ZQ, Wynn TA, Snapper CM. Endogenous pro- and anti-inflammatory cytokines differentially regulate an in vivo humoral response to Streptococcus pneumoniae. Infect Immun. 2002;70:749–761. doi: 10.1128/iai.70.2.749-761.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colino J, Snapper CM. Opposing signals from pathogen-associated molecular patterns and IL-10 are critical for optimal dendritic cell induction of in vivo humoral immunity to Streptococcus pneumoniae. J Immunol. 2003;171:3508–3519. doi: 10.4049/jimmunol.171.7.3508. [DOI] [PubMed] [Google Scholar]
- 42.Lee KS, Scanga CA, Bachelder EM, Chen Q, Snapper CM. TLR2 synergizes with both TLR4 and TLR9 for induction of the MyD88-dependent splenic cytokine and chemokine response to Streptococcus pneumoniae. Cell Immunol. 2007;245:103–110. doi: 10.1016/j.cellimm.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–389. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 44.Wu ZQ, Shen Y, Khan AQ, Chu CL, Riese R, Chapman HA, Kanagawa O, Snapper CM. The mechanism underlying T cell help for induction of an antigen-specific in vivo humoral immune response to intact Streptococcus pneumoniae is dependent on the type of antigen. J Immunol. 2002;168:5551–5557. doi: 10.4049/jimmunol.168.11.5551. [DOI] [PubMed] [Google Scholar]
- 45.Clark EA, Craxton A. A CD40 bridge between innate and adaptive immunity. Immunity. 2003;18:724–725. doi: 10.1016/s1074-7613(03)00146-8. [DOI] [PubMed] [Google Scholar]
- 46.Leonard EG, Canaday DH, Harding CV, Schreiber JR. Antigen processing of the heptavalent pneumococcal conjugate vaccine carrier protein CRM(197) differs depending on the serotype of the attached polysaccharide. Infect Immun. 2003;71:4186–4189. doi: 10.1128/IAI.71.7.4186-4189.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leyva-Cobian F, Unanue ER. Intracellular interference with antigen presentation. J Immunol. 1988;141:1445–1450. [PubMed] [Google Scholar]
- 48.Ishioka GY, Lamont AG, Thomson D, Bulbow N, Gaeta FC, Sette A, Grey HM. MHC interaction and T cell recognition of carbohydrates and glycopeptides. J Immunol. 1992;148:2446–2451. [PubMed] [Google Scholar]
- 49.Mouritsen S, Meldal M, Christiansen-Brams I, Elsner H, Werdelin O. Attachment of oligosaccharides to peptide antigen profoundly affects binding to major histocompatibility complex class II molecules and peptide immunogenicity. Eur J Immunol. 1994;24:1066–1072. doi: 10.1002/eji.1830240509. [DOI] [PubMed] [Google Scholar]
- 50.Hsieh CS, Macatonia SE, O’Garra A, Murphy KM. Pathogen-induced Th1 phenotype development in CD4+ αβ-TCR transgenic T cells is macrophage dependent. Int Immunol. 1993;5:371–382. doi: 10.1093/intimm/5.4.371. [DOI] [PubMed] [Google Scholar]
- 51.Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- 52.Hamilton-Easton A, Eichelberger M. Virus-specific antigen presentation by different subsets of cells from lung and mediastinal lymph node tissues of influenza virus-infected mice. J Virol. 1995;69:6359–6366. doi: 10.1128/jvi.69.10.6359-6366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Usherwood EJ, Hogg TL, Woodland DL. Enumeration of antigen-presenting cells in mice infected with Sendai virus. J Immunol. 1999;162:3350–3355. [PubMed] [Google Scholar]
- 54.Ziegler HK. The processing and presentation of Listeria monocytogenes antigens by macrophages. Clin Invest Med. 1984;7:269–272. [PubMed] [Google Scholar]
- 55.Allen CD, Okada T, Cyster JG. Germinal-center organization and cellular dynamics. Immunity. 2007;27:190–202. doi: 10.1016/j.immuni.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Phan TG, Grigorova I, Okada T, Cyster JG. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol. 2007;8:992–1000. doi: 10.1038/ni1494. [DOI] [PubMed] [Google Scholar]
- 57.Carrasco YR, Batista FD. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity. 2007;27:160–171. doi: 10.1016/j.immuni.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 58.Craxton A, Magaletti D, Ryan EJ, Clark EA. Macrophage- and dendritic cell--dependent regulation of human B-cell proliferation requires the TNF family ligand BAFF. Blood. 2003;101:4464–4471. doi: 10.1182/blood-2002-10-3123. [DOI] [PubMed] [Google Scholar]
- 59.Kopf M, Herren S, Wiles MV, Pepys MB, Kosco-Vilbois MH. Interleukin 6 influences germinal center development and antibody production via a contribution of C3 complement component. J Exp Med. 1998;188:1895–1906. doi: 10.1084/jem.188.10.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Becker G, Sornasse T, Nabavi N, Bazin H, Thielemans F, Urbain J, Leo O, Moser M. Immunoglobulin isotype regulation by antigen-presenting cells in vivo. Eur J Immunol. 1994;24:1523–1528. doi: 10.1002/eji.1830240710. [DOI] [PubMed] [Google Scholar]
- 61.Gordan S. Macrophages and the immune response. In: Paul William E., editor. Fundamental Immunology. 4. 1999. pp. 533–545. [Google Scholar]
- 62.Boswell HS, Sharrow SO, Singer A. Role of accessory cells in B cell activation. I. Macrophage presentation of TNP-Ficoll: evidence for macrophage-B cell interaction. J Immunol. 1980;124:989–996. [PubMed] [Google Scholar]
- 63.Chused TM, Kassan SS, Mosier DE. Macrophage requirement for the in vitro response to TNP Ficoll: a thymic independent antigen. J Immunol. 1976;116:1579–1581. [PubMed] [Google Scholar]
- 64.Corbel C, Melchers F. Requirement for macrophages or for macrophage- or T cell-derived factors in the mitogenic stimulation of murine B lymphocytes by lipopolysaccharides. Eur J Immunol. 1983;13:528–533. doi: 10.1002/eji.1830130703. [DOI] [PubMed] [Google Scholar]
- 65.Sinha AA, Guidos C, Lee KC, Diener E. Functions of accessory cells in B cell responses to thymus-independent antigens. J Immunol. 1987;138:4143–4149. [PubMed] [Google Scholar]
- 66.Boswell HS, Ahmed A, Scher I, Singer A. Role of accessory cells in B cell activation. II. The interaction of B cells with accessory cells results in the exclusive activation of an Lyb5+ B cell subpopulation. J Immunol. 1980;125:1340–1348. [PubMed] [Google Scholar]
- 67.Kovacsovics-Bankowski M, Clark K, Benacerraf B, Rock KL. Efficient major histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc Natl Acad Sci U S A. 1993;90:4942–4946. doi: 10.1073/pnas.90.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 69.Ziegler HK, Orlin CA, Cluff CW. Differential requirements for the processing and presentation of soluble and particulate bacterial antigens by macrophages. Eur J Immunol. 1987;17:1287–1296. doi: 10.1002/eji.1830170911. [DOI] [PubMed] [Google Scholar]
- 70.Kapsenberg ML, Teunissen MB, Stiekema FE, Keizer HG. Antigen-presenting cell function of dendritic cells and macrophages in proliferative T cell responses to soluble and particulate antigens. Eur J Immunol. 1986;16:345–350. doi: 10.1002/eji.1830160405. [DOI] [PubMed] [Google Scholar]
- 71.Savina A, Amigorena S. Phagocytosis and antigen presentation in dendritic cells. Immunol Rev. 2007;219:143–156. doi: 10.1111/j.1600-065X.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 72.Leyva-Cobian F, Unanue ER. Intracellular interference with antigen presentation. J Immunol. 1988;141:1445–1450. [PubMed] [Google Scholar]
- 73.Gordon S, Fraser I, Nath D, Hughes D, Clarke S. Macrophages in tissues and in vitro. Curr Opin Immunol. 1992;4:25–32. doi: 10.1016/0952-7915(92)90119-y. [DOI] [PubMed] [Google Scholar]
- 74.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 75.Rutherford MS, Witsell A, Schook LB. Mechanisms generating functionally heterogeneous macrophages: chaos revisited. J Leukoc Biol. 1993;53:602–618. doi: 10.1002/jlb.53.5.602. [DOI] [PubMed] [Google Scholar]
- 76.Celada A, Nathan C. Macrophage activation revisited. Immunol Today. 1994;15:100–102. doi: 10.1016/0167-5699(94)90150-3. [DOI] [PubMed] [Google Scholar]