

Abstract
The authors evaluate the expression of endothelin-1 (ET-1) and its receptors in the uterus and placenta during maternal nitric oxide synthase (NOS) inhibition. Timed-pregnant rats received L-NAME (2.5 mg/kg/h) or saline from day 14 to 21 of gestation. Uterine and placental tissues collected on day 21 were assayed for preproET-1, ETA, and ETB mRNA expression; localization and expression of ET-1 and receptor proteins; and receptor activity. NOS inhibition did not affect preproET-1 mRNA expression in the placenta or uterus. ETA expression decreased in the uterine free wall, but no other changes in receptor mRNA expression were observed in the uterus or placenta. ET-1 and receptor proteins were unchanged. Placental ETA and ETB receptor binding decreased. Uterine ETA receptor binding decreased in the placental bed. ET-1, a prominent mediator during NOS inhibition, is not of uterine or placental origin. Reduced receptor binding activity is the primary means by which these tissues regulate their response to ET-1 in the setting of NOS inhibition.
Keywords: Endothelin, nitric oxide, placenta, pregnancy, uterus, rat
Endothelin-1 (ET-1) is one of the most potent vasoconstrictors known and is part of a family of peptides that includes 3 isoforms, ET-1, ET-2, and ET-3. The endothelins are produced mainly by endothelial or epithelial cells and act primarily as paracrine mediators. Their predominant effect is vasoconstriction, although they also affect cell proliferation, differentiation, and migration, as well as chemokinesis and apoptosis.1,2 Endothelin mediates its effects via 2 types of receptors in mammalian species. The ETA receptor, which has a high affinity for ET-1, is located on smooth muscle cells and is responsible principally for the vasoconstrictive and proliferative effects of ET-1. The ETB receptor, which has an equal affinity for all 3 isoforms, is located on endothelial cells and is responsible for a vasodilatory effect mediated primarily by nitric oxide.3 It also functions as a clearance receptor to remove excess ET from the circulation.4
Nitric oxide is a potent vasodilator that also has a role in the regulation of production of other vasoactive mediators. In the human fetoplacental circulation, nitric oxide contributes to the maintenance of low vascular tone not only by its direct vasodilatory effects but also by attenuation of ET-1 production.5 Chronic inhibition of nitric oxide synthase (NOS) reduces regional perfusion, which is secondary not only to the lack of nitric oxide production but also to an increase in production of ET-1. In hypertensive rats, aortic and mesenteric arterial ET-1 mRNA expression and plasma ET-1 protein increase in response to NOS inhibition.6 With NOS inhibition during pregnancy, not only do nitric oxide levels decrease but we7 and others8 have also observed significant increases in plasma ET-1 levels. We also demonstrated that administration of an ETA antagonist significantly improves both fetal growth9 and placental perfusion10 in NOS-inhibition–induced fetal growth restriction, suggesting that ET-1 is of primary importance in the pathophysiology of fetal growth restriction in this model. Since ET-1 is primarily locally active, uteroplacental tissue expression of ET-1 and its receptors is of particular interest. The purpose of the present study is to evaluate the effects of chronic maternal NOS inhibition on the expression of endothelin and its receptors in the uterus and placenta of the rat.
MATERIALS AND METHODS
Materials
[125I]ET-1 and [125I]ET-3 (each 2200 Ci/mmol) were obtained from Amersham (Arlington Heights, IL). ET-1 and ET-3 were purchased from American Peptide Company (Sunnyvale, CA). Antibodies to the following proteins were purchased from the sources indicated: ET-1, Phoenix Pharmaceuticals (Burlingame, CA); ETA and ETB, Alomone Laboratories (Jerusalem, Israel); and sodium potassium ATPase, Abcam (Cambridge, MA). Other reagents were purchased from Sigma (St Louis, MO) unless indicated otherwise.
Animals
Female Sprague-Dawley rats were purchased from Harlan Sprague Dawley (Madison, WI), housed in the Evanston Northwestern Healthcare Research Institute (ENHRI) Center for Comparative Medicine, and bred with male rats from the same source. All rats received a standard laboratory rodent diet (PMI Feeds, St Louis, MO) and water ad libitum and were kept on a 12-hour light/12-hour dark cycle. Animal care and the performance of all experiments were in accord with guidelines approved by the ENHRI Institutional Animal Care and Use Committee.
Chronic Inhibition of NOS
Treatment groups of 6 animals received either NG-nitro-L-arginine-methyl ester (L-NAME; 2.5 mg/kg/h) or normal saline by continuous subcutaneous infusion via osmotic pump. The osmotic pump (Alzet model 2ML1, output = 10 μL/h; Durect Corp, Palo Alto, CA) was placed subcutaneously on the back of the rat between the scapulae under anesthesia consisting of a single intraperitoneal injection of xylazine 8 mg/kg, ketamine 40 mg/kg, and acepromazine 1.3 mg/kg in combination. Treatment was begun on gestational day 14 and continued through day 21.
Tissue Collection
On day 21 of gestation, rats were sacrificed and a laparotomy was performed. The uterine horns were opened to expose the fetuses and placentas. Placentas were collected and frozen in liquid nitrogen for storage. The uterine placental beds (placental attachment sites) were dissected free of the remaining uterine wall. The uterine placental bed is the full thickness of the uterine wall at the sites where each placenta was attached. The uterine free wall is the remaining uterine wall where no placental attachment sites were present. The uterine placental beds, the uterine free wall, and placentas were individually frozen and stored at −80°C for further use.
Real-Time Quantitative Reverse Transcription Polymerase Chain Reaction
Tissue samples stored at −80°C were homogenized in RNA STAT-60 (a phenol/guanidinium thiocyanate reagent;TEL-TEST, Friendswood,TX) on ice for extraction of total RNA. Genomic DNA, a possible impurity in the RNA extract, was removed by adding RNase-free DNase (1 U/μg DNA) and incubating at 37°C for 60 minutes. The concentration of RNA was determined by absorbance at 260 nm, and the purity was checked by the 260/280 nm ratio (greater than 1.8). RNA integrity was verified by electrophoresis in a 1% agarose gel. For each sample, 3 μg total RNA was reverse transcribed at 37°C for 1 hour in a total of 20 μL reaction mixture: 50 mM Tris-HCl, 75 mM KCl, 2.0 mM MgCl2, 10 mM dithiothreitol, 1.25 mM of each deoxynucleotide-triphosphate, 7.5 pM random hexamer, 1 U/μL RNaseOUT (RNase inhibitor; Invitrogen, Carlsbad, CA), and 10 U/μL Moloney-murine leukemia virus reverse transcriptase (Gibco-BRL, Grand Island, NY). cDNA was amplified by polymerase chain reaction (PCR) with 0.1 U/μL AmpliTaq DNA polymerase on an Applied Biosystems (Foster City, CA) GeneAmp 5700 real-time quantitative PCR instrument in a total volume of 50 μL consisting of 1.0 to 3.0 μL RT product; 10 mM Tris-HCl; 50 mM KCl; 2 mM MgCl2; 0.1 mM of each dNTP; 0.2 to 1.0 μM of each primer, sense and antisense, respectively; and 0.2 to 1.0 μM TaqMan-MGB fluorescent detection probe (Applied Biosystems). For each PCR reaction, a probe and primer titration was performed to determine optimal concentrations for each. The reaction mixtures were heated at 95°C for 30 seconds and then immediately carried through 60 cycles of PCR using the following schedule: 30 seconds denaturation at 94°C and 2 minutes annealing plus extension at 60°C. Primers and probes specific for rat ET-1, ETA, ETB, and β-actin (internal control gene) were designed by Primer Express (Applied Biosystems). In preliminary experiments, β-actin was shown to be expressed similarly in both vehicle-treated and L-NAME–treated rat placenta and uterus (data not shown). No-template controls were used to exclude the possibility of primer dimer formation. The following represent the primer sense and antisense and TaqMan probe sequences, respectively:
-
Rat preproET-1:
5′-GACCAGCGTCCTTGTTCCAA-3′,
5′-TTGCTACCAGCGGATGCAA-3′,
5′-(6FAM™)-TCCAAGAGAGGTTGAGGTGT-(MGBNFQ)-3′
-
Rat ETA:
5′-CTCAACGCCACGACCAAGTT-3′,
5′-GCAAGCTCCCATTCCTTCTG-3′,
5′-(6FAM)-ATGGAGTTTTACCAAGACGT-(MGBNFQ)-3′
-
Rat ETB:
5′-TGGCCATTTGGAGCTGAGAT-3′,
5′-TCCAAGAAGCAACAGCTCGAT-3′,
5′-(6FAM)-TGCCCTTCATACAGAAGG-(MGBNFQ)-3′
-
Rat β-actin:
5′-AGGCCAACCGTGAAAAGATG-3′,
5′-ACCAGAGGCATACAGGGACAA-3′,
5′-(6FAM)-CCCAGATCATGTTTGAGAC-(MGBNFQ)-3′
Membrane Preparations
Frozen tissues were homogenized on ice in 10 volumes (w/v) of 10 mM Hepes (pH 7.4) containing 0.25 M sucrose, 3 mM EDTA, and protease inhibitors (0.1 mM PMSF and 5 μg/mL Pepstatin A) using a stainless-steel Polytron tissue homogenizer (Brinkmann,Westbury, NY) at 13 500 rpm. The homogenate was centrifuged at 1000g for 15 minutes at 4°C. The supernatant was collected, and the pellet was resuspended in the same buffer, rehomogenized, and recentrifuged. The combined supernatant was centrifuged at 60 000g for 1 hour at 4°C. The precipitate was resuspended in 50 mM Tris (pH 7.4) containing 50 mM EDTA and the previously listed protease inhibitors and then centrifuged at 30 000g for 30 minutes at 4°C. The final pellet was resuspended in the Tris buffer and stored at −80°C. In addition to the placental and uterine tissues, membranes were prepared from an untreated adult rat brain (contains abundant ETA and ETB receptors) for use as control membranes in the Western blot analyses. Protein content was determined by the Bradford assay (BioRad, Hercules, CA).
Western Immunoblots
From each tissue membrane preparation, 20 μg of protein was separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride blotting membranes. Immunoblots were performed using rabbit polyclonal primary antibodies specific for ETA or ETB or mouse monoclonal antibodies to sodium potassium ATPase (a plasma membrane marker protein used as a loading control to ensure equal amounts of protein in all lanes). After blocking nonspecific binding with 5% normal goat serum (NGS) in 1 M trisbuffered saline (TBS; pH 7.5) containing 0.1% Tween-20 (TBST), primary antibodies were incubated on the membranes at 1:250 or 1:200 (ETA and ETB, respectively; dilutions determined in preliminary antibody titration experiments) overnight at 4°C in TBS containing 5% NGS, 0.5% Tween-20, 10% glycerol, and 18% glucose. Following six 10-minute washes in TBST and 1 hour of blocking, the membranes were incubated for 2 hours at 25°C with secondary goat antirabbit IgG antibodies labeled with horseradish peroxidase (1:5000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA). Membranes were washed again as before. Immunoreactive bands were identified using the ECL+Plus Chemifluorescent Detection System (Amersham, Piscataway, NJ) as directed by the manufacturer and then quantified using a Storm Imager (Molecular Dynamics, Sunnyvale, CA). Quantitative comparisons were made between bands run together on the same gel. Band intensity for uterine and placental samples was normalized to the ET receptor–rich control membrane preparation used throughout all Western analyses. Transfer of proteins to the membranes was ascertained by the inclusion of Rainbow molecular weight markers (Amersham, Arlington Heights, IL) on each gel, and specific bands for ETA (48 kDa), ETB (49 kDa), and sodium potassium ATPase (112 kDa) were identified by Cruz molecular weight standards (Santa Cruz Biotechnology).
Immunohistochemistry
Placental and uterine wall tissues were fixed for 2 hours in 10% neutral buffered formalin and either embedded in paraffin (for ET-1) or infiltrated with 10% sucrose in phosphate-buffered saline (PBS) and frozen in OCT compound (Miles Labs, Naperville, IL) for cryosectioning (for ETA and ETB). Sections were deparaffinized (when appropriate) and hydrated in PBS. PBS rinses were interspersed between all steps except following blocking of nonspecific antibody binding. Immunohistochemistry began with 3% hydrogen peroxide for 15 minutes to block endogenous peroxidase activity, followed by 10 mM citrate (pH 6, 30 minutes, 80°C) for antigen retrieval and 1% nonfat milk (5 minutes) to block nonspecific antibody binding. Sections were then incubated with primary antibody for 2 hours at 25°C (all antibodies diluted 1:500 in PBS) in Mach 4 detection reagent (Biocare Medical, Concord, CA) for 45 minutes and then in a diaminobenzidine chromogen (Dako, Cupertino, CA) for 10 minutes. A light methyl green counterstain was applied (1% for 5 minutes). Sections were dehydrated through a graded series of ethyl alcohols and xylene and then mounted in Permount for bright-field microscopy. Controls included elimination of the primary antibody and adsorption of the primary antibody with 10 μM antigen to verify specificity.
Homologous Competitive Endothelin Receptor Binding Assay
Binding assays were performed in triplicate in 96-well microtiter plates pretreated with 0.1% bovine serum albumin. Aliquots of the membrane preparation (2 μg protein/well) were incubated with 12 different concentrations of ET-1 or ET-3 and 0.1 nM [125I]ET-1 or [125I]ET-3, respectively. After a 3-hour incubation at 25°C, unbound ligands were separated from bound ligands by vacuum filtration using glass-fiber filter strips in a PHD cell harvester (Cambridge Technology, Watertown, MA). Filters were washed 3 times with saline (1 mL each), and the radioactivity was measured in a Wallac gamma counter (PerkinElmer Life Sciences, Boston, MA). Nonspecific binding was determined in the presence of 1 μM ET-1 or ET-3. Maximum binding (Bmax, fmoles/μg protein) and affinity (Kd) were computed by nonlinear regression11 using GraphPad Prism (GraphPad Software, San Diego, CA). ET-1 binds to both ETA and ETB with the same potency (IC50 = 0.1 nM), while ET-3 binding has a 2000-fold greater potency for ETB (IC50 = 0.1 nM) than for ETA (IC50 = 200 nM).12 Homologous ET-3 competition determined the Bmax for ETB, homologous ET-1 competition represented binding of both receptors, and the difference determined the Bmax for ETA.
Statistical Analyses
Results are presented as the mean ± SEM. Statistical comparisons among multiple groups were made using an ANOVA followed by a post hoc Newman-Keuls test. When comparisons were made between 2 groups, a t test was used. All statistical tests were 2 tailed, and results were considered statistically significant at P < .05.
RESULTS
Placenta
Neither preproET-1 nor ETA or ETB mRNA expression were influenced by NOS inhibition (Figure 1). The Western blot data showed no difference in protein expression of ETA or ETB between L-NAME–treated rats and vehicle-treated control rats (Figure 2). Similarly, the immunohistochemistry results revealed no difference in stain distribution or intensity between the 2 groups of rats (Figure 3). ET-1 was localized primarily to placental fetal membranes in the labyrinth and to some giant cells and vascular cells in the transitional zone. ETA and ETB staining was observed in the labyrinth and in the small arterial and venous vasculature. Localization of ETA and ETB was not uniform among the vessels in any individual placenta.
Figure 1.
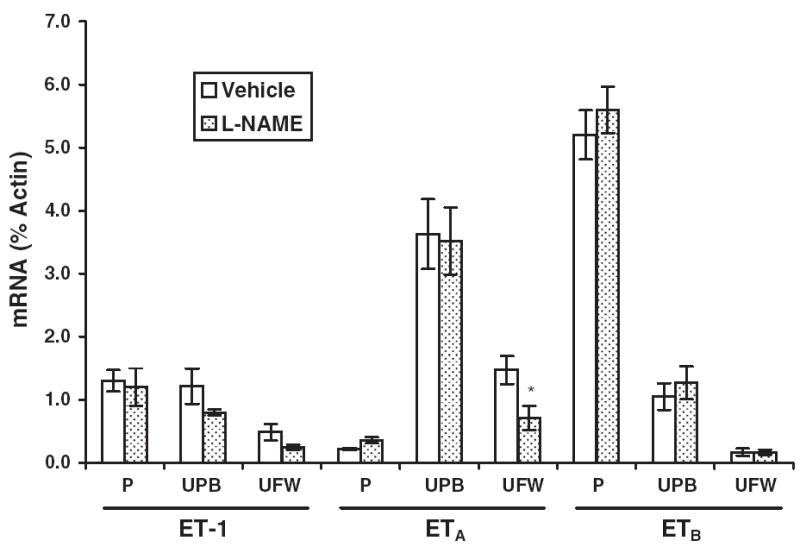
Expression of endothelin-1 (ET-1) as well as ETA and ETB receptor mRNA in rat placentas (P), uterine placental beds (UPB), and the uterine free wall (UFW). Comparative mRNA expression was determined by real-time quantitative reverse transcription polymerase chain reaction in tissues from rats treated with NG-nitro-L-arginine-methyl ester (L-NAME; 2.5 mg/kg/h, gestational days 14-21, n = 6) or with saline vehicle (n = 6). Results are expressed as a percentage of the internal control gene, cytoplasmic β-actin, and each value represents the mean ± SEM. *P < .05 versus vehicle-treated control, by ANOVA.
Figure 2.
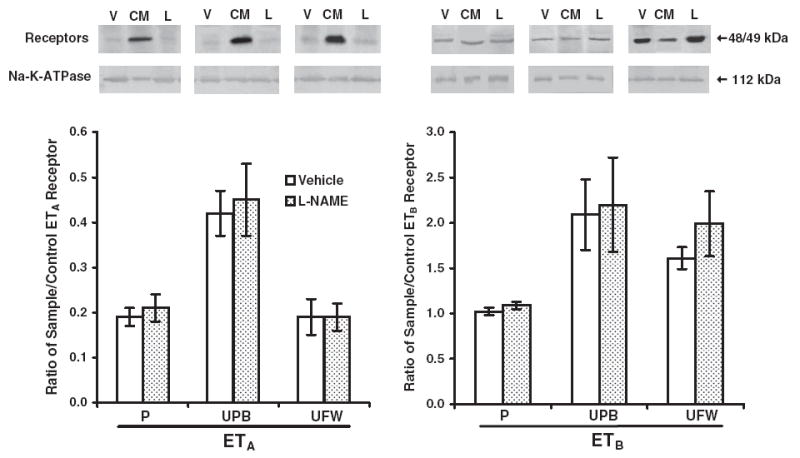
Protein expression of ETA (48 kDa, left) and ETB (49 kDa, right) receptors in pregnant rats. Western blots were performed on membranes isolated from placental (P), uterine placental bed (UPB), and uterine free wall (UFW) homogenates from NG-nitro-L-arginine-methyl ester (L-NAME)–treated rats (L, gestational days 14-21, n = 6) and vehicle-treated controls (V, n = 6) pregnant rats. All gels included control membranes (CM) from a rat brain, and the densitometric results are expressed as the mean ± SEM of the ratio of reproductive tissue and control receptor band densities. Na+/K+ transporting ATPase (112 kDa) was used as a control membrane protein to verify loading of equal amounts of protein into each well.
Figure 3.
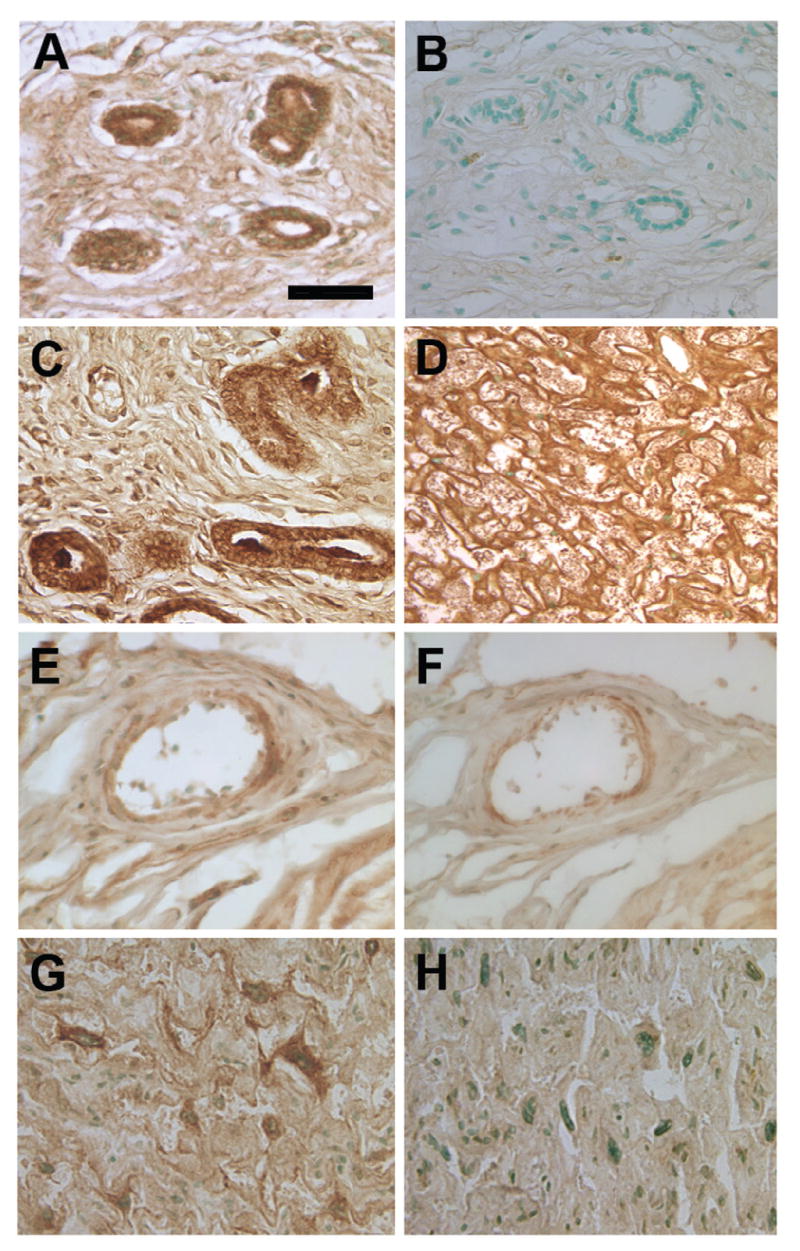
Immunolocalization of endothelin-1 (ET-1), ETA, and ETB in uterine vessels and placental labyrinth from pregnant rats treated with saline vehicle or with 2.5 mg/kg/h NG-nitro-L-arginine-methyl ester (L-NAME) from gestational day 14 to 21. Immunostaining patterns and intensities were similar between the 2 treatment groups. ET-1 localized to the uterine vascular walls in both the vehicle-treated group (A) and the L-NAME–treated group (C). A negative control (omission of primary antibody) from the vehicle-treated group indicates no binding of the secondary antibody-chromogen complex in the absence of primary antibody (B). Placental ET-1 localized primarily to the membranes of the labyrinth in both groups; a representative photomicrograph from the vehicle-treated group is shown (D). ETA and ETB receptors in the uterus (E and F, respectively) were localized in the vascular wall in both vehicle and L-NAME–treated groups. In the placenta, these receptors were distributed along the labyrinthine membranes, with ETA appearing in patches (G) and ETB having a more uniform distribution (H). Light methyl green counterstain. Bar = 100 μm (A-C and E-H) and 200 μm (D).
In homologous competition studies, unlabeled ET-1 or ET-3 at various concentrations was used to compete against the respective radiolabeled ligand. The Kd values were 3.12 nM for ET-1 and 0.87 nM for ET-3 in vehicle-treated (control) placenta. NOS inhibition decreased the Kd values to 13% and 2% of control values, respectively (0.42 nM for ET-1 and 0.02 nM for ET-3), indicating a larger increase in affinity for ETB than for ETA binding. ETA receptor Bmax was significantly higher than ETB Bmax with both vehicle and L-NAME treatments (2.4-fold and 4-fold, P < .01 and P < .001, vehicle and L-NAME, respectively), indicating greater abundance of ETA than ETB available to bind ligand on cells in these tissues (Figure 4). Bmax for both receptors was downregulated with NOS inhibition (43% and 66%, P < .01 for ETA and ETB, respectively), indicating a significant reduction in the number of receptors available for ligand binding in the placenta, despite the lack of change in ETA receptor mRNA or protein expression for both receptors.
Figure 4.
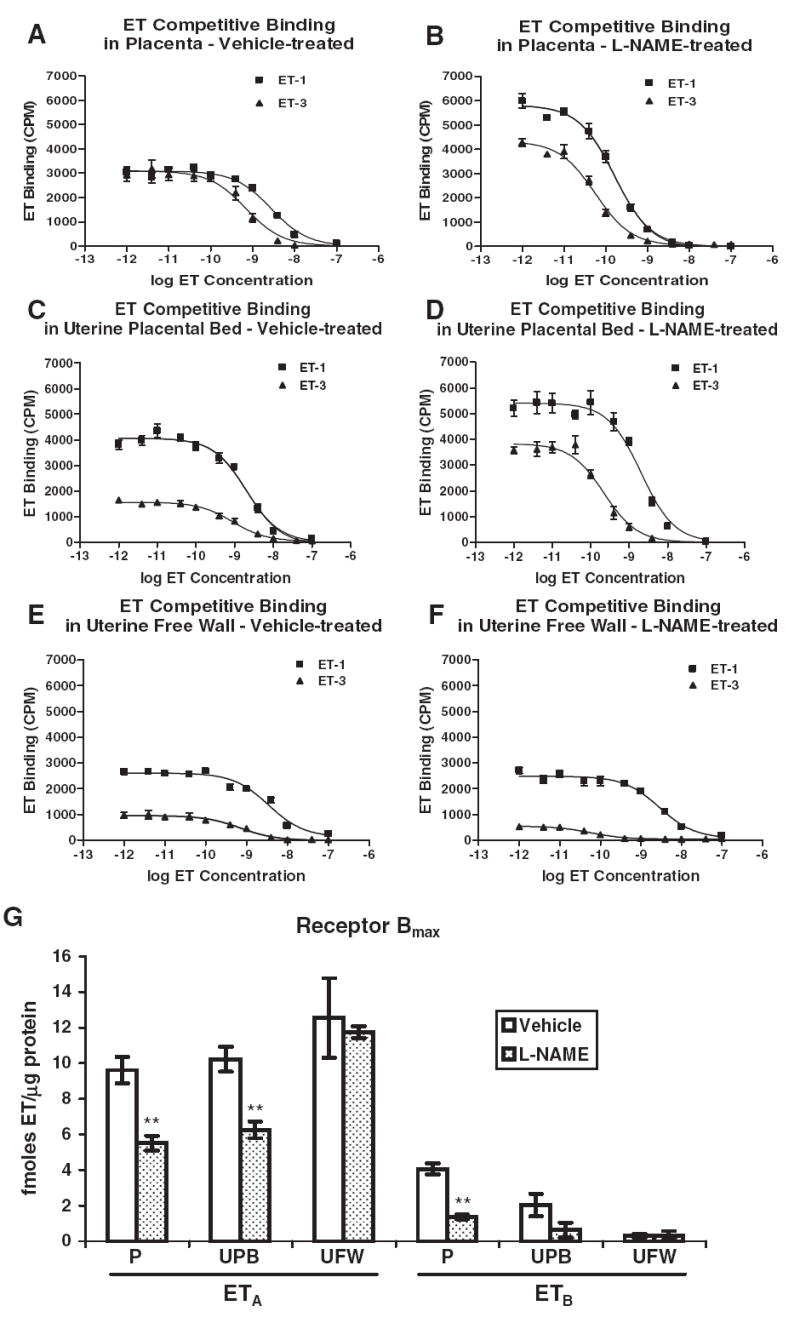
Homologous competitive binding of endothelin-1 (ET-1) and ET-3 by ETA and ETB receptors in rat placenta (P), uterine placental bed (UPB), and uterine free wall (UFW). Placental membranes were prepared from vehicle-treated (n = 6) and L-NAME-treated (2.5 mg/kg/h, gestational days 14-21, n = 6) pregnant rats. (A, C, E) Representative binding curves for P, UPB, and UFW membrane preparations, respectively, from vehicle-treated rats. (B, D, F) Representative binding curves for P, UPB, and UFW membrane preparations, respectively, from L-NAME-treated rats. Each experiment was performed in triplicate. (G) The maximum binding coefficient, Bmax, for both receptors is expressed as fmoles endothelin/μg protein, and each value represents the mean ± SEM. ET in captions and axis labels refers to both ET-1 and ET-3. **P < .01 versus vehicle-treated control by ANOVA.
Uterine Placental Bed
PreproET-1 mRNA expression did not change significantly in response to NOS inhibition (Figure 1). Placental bed ETA and ETB mRNA expression was not significantly altered by L-NAME compared with control (Figure 1). L-NAME did not affect the ratio of ETA to ETB mRNA expression. ETA and ETB protein expression in the L-NAME–treated uterine placental bed also was not significantly different from vehicle-treated controls (Figure 2).
Competition studies in the vehicle-treated placental bed resulted in Kd values of 3.33 nM for ET-1 and 0.50 nM for ET-3 similar to those in placenta. NOS inhibition decreased these values to 1.25 nM for ET-1 and 0.03 nM for ET-3, with the greater change for ET-3 indicating a larger increase in ETB binding affinity than in ETA binding affinity. Placental bed ETA receptor Bmax was significantly higher than ETB Bmax with both vehicle and L-NAME treatments (5-fold and 10-fold, P < .001, vehicle and L-NAME, respectively), indicating a greater availability of ETA than ETB receptor to bind ligand in this tissue (Figure 4). Bmax for ETA was downregulated 39% with NOS inhibition (P < .01), indicating a decrease in the number of ETA receptors available for binding in the uterine placental bed when NOS is inhibited.
Uterine Free Wall
PreproET-1 mRNA expression was not changed significantly in response to NOS inhibition (Figure 1) in the uterine free wall. ETA, but not ETB, receptor mRNA expression decreased 50% (P < .05, Figure 1). Western blots showed no difference in either ETA or ETB protein expression in response to NOS inhibition (Figure 2).
Endothelin-1, ETA, and ETB were each localized by immunohistochemistry to the vascular lining in small arteries and veins in the uterine wall. The staining patterns were the same as reported by many others for ET-1 and its receptors (Figure 3). Labeling for all these antigens was present on only some of the vessels in any particular section, and no difference in distribution or intensity was observed between groups.
Competition studies in the vehicle-treated uterine free wall resulted in Kd values of 5.50 nM for ET-1 and 0.23 nM for ET-3. L-NAME decreased these values to 1.93 nM for ET-1 and 0.05 nM for ET-3. ETA was responsible for most of the binding of ET-1 in the uterine free wall (Figure 4), with ETA Bmax being 40-fold and 37-fold higher than ETB Bmax (P < .01, vehicle and L-NAME, respectively). There was no change in ETA or ETB binding in response to NOS inhibition in the uterine free wall.
DISCUSSION
ET-1 expression in either the uterus or placenta is not increased in response to chronic maternal NOS inhibition. This is in contrast to maternal hypoxia, another model of fetal growth restriction, in which the placenta appears to be the primary source of increased ET-1 production.13 PreproET-1 expression was also measured in the lung and kidney, common prominent sources of ET-1, and no increase in ET-1 was observed in these organs (data not shown). Therefore, in the NOS-inhibition model of fetal growth restriction, the source of the observed increased circulating ET-1 is unknown. However, a generalized endothelial response to NOS inhibition is a likely source, as has been demonstrated in the hypertensive rat.6
The reaction of the placenta and uterus to the excess circulating ET-1 is revealed in part by differences in receptor expression. The predominant receptor in the placenta is ETB. Stimulation of the ETB receptor results in release of nitric oxide as well as clearance of ET-1, both serving to preserve perfusion. Thus, the persistence of ETB expression may be a cellular response that ordinarily could maintain perfusion in the placenta via increased nitric oxide and decreased circulating ET-1. In contrast, stimulation of ETA, the predominant receptor in the uterus, results in vasoconstriction and decreased perfusion. The decreased expression of ETA mRNA in the uterine free wall may again be a cellular reaction that normally should reduce responsiveness to ET-1 and maintain perfusion in uterine tissues.
Although receptor expression varied by tissue, it was minimally affected by NOS inhibition in either the uterus or placenta. Receptor activity, however, differed significantly in both the placenta and the uterine placental bed. Receptor binding capacity (Bmax) decreased for both receptors in the placenta and for ETA in the uterine placental bed, accompanied in each case by increased receptor affinity (Kd) with NOS inhibition. Decreased binding capacity and increased affinity can be consequences of posttranslational modifications such as receptor phosphorylation, for which both receptors have multiple potential sites.14 Phosphorylation produces desensitization and decreased activity of both ETA and ETB.15 Reduced Bmax of both ETA and ETB can also be the result of deglycosylation, which decreases low-affinity sites while leaving high-affinity sites unaffected, thereby increasing the net receptor affinity.16 Similarly, it has been shown that arachidonate decreases adenosine A1 receptor Bmax while increasing receptor affinity to both agonists and antagonists.17
Alternatively, sequestration of receptors intracellularly in recycling endosomes is another well-established mechanism for cellular regulation of G protein–coupled receptor activity. Receptors in recycling endosomes are unavailable for ligand binding, resulting in reduced cell surface receptors, presumably leading to a reduced cellular response to the ligand. These mechanisms for changes in endothelin receptor binding capacity have not been studied or described in the setting of NOS inhibition per se. However, the in vivo interaction of the nitric oxide and endothelin systems is well established.18 Thus, decreased endothelin receptor binding capacity through 1 or more of these mechanisms would not be an unexpected response to the increased vascular tone caused by decreased nitric oxide production. We have previously shown that receptor binding is consistently downregulated in hypoxia-induced fetal growth restriction in the uterus and placenta.13 Taken together with the current results, this suggests that receptor modification or sequestration/recycling may be consistent methods by which these tissues respond to ET-1.
The expression of ET-1 and its receptors in the uterus and placenta of normal pregnant rats has previously been examined. In the rat placenta, ET-1 is expressed in cytotrophoblasts and trophoblastic giant cells of the basal zone, as well as in endothelial cells.19 In the rat uterus, endometrial glandular and myometrial cells also produce ET-1.20 Receptors for endothelin are well represented in normal rat reproductive tissues. ETA is most abundant in the decidual tissue and vascular wall, while ETB has been localized to the basal zone cytotrophoblasts and trophoblastic giant cells.19 Both receptors are equally represented in the labyrinth. ETA is responsible for the primary ET-1 binding activity in rat myometrium,21 which is in agreement with our receptor binding results in both the uterine placental bed and the uterine free wall. The expression and distribution of ET-1 and its receptors suggest that ET-1 may play an important autocrine or paracrine role in the uteroplacental tissues in the rat.22
ET-1 and its receptors are expressed in the human uterus and placenta as well. ETA is located primarily on the uterine endometrial stromal cells and ETB on glandular epithelium.23 While both ETA and ETB are expressed in the human placenta, ETB predominates throughout gestation,24,25 in contrast to the greater abundance of ETA in the rat. In the human, ETA increases near term and is the primary ET-1 receptor in stem villi vessels, whereas ETB expression is more prominent in peripheral vessels and in the decidua and does not change during gestation.24-26 Both ET-1 and ET-3 are produced in placental stem villi vessels, the primary site of placental vascular resistance.27 ET-1 acting via ETA receptors, then, may cause vasoconstriction leading to placental ischemia in the human, corresponding with what we have reported in NOS inhibition–induced fetal growth restriction as well as in hypoxia-induced fetal growth restriction in the rat.10,13 These findings suggest that ET-1 may play an important autocrine or paracrine role in human uteroplacental tissues, as it does in the rat.
We have previously demonstrated that ET-1 plays an important role in the pathophysiology of NOS inhibition-induced fetal growth restriction.9 In this model, in contrast to hypoxia-induced fetal growth restriction,13 the increased circulating ET-1 does not appear to be of uterine or placental origin. The effects of ET-1 are mediated by regulation of receptor activity in both the uterus and the placenta. Taken in the context of our prior study results demonstrating that ETA antagonism significantly improves both fetal growth9 and placental perfusion10 in the setting of chronic NOS inhibition, these findings describe the interaction of ET-1 and its receptors in the pathophysiology of this model of fetal growth restriction. The extent to which ET-1 plays a similar role in human fetal growth restriction remains to be evaluated.
Acknowledgments
This work was supported by National Institutes of Health grants HD34777 and HD01484 (to LGT). We thank Ms Karyn Brasky, Mr Dan Hutchins, Ms LynAriane Lucas, and Ms Erica Schwartz at Evanston Northwestern Healthcare for their technical assistance. We also thank Drs Jinshyun R. Wu-Wong and William Chiou at Abbott Laboratories for their expertise and assistance with the binding experiments.
References
- 1.Cervar M, Puerstner P, Kainer F, Desoye G. Endothelin-1 stimulates the proliferation and invasion of first trimester trophoblastic cells in vitro—a possible role in the etiology of pre-eclampsia? J Investig Med. 1996;44:447–453. [PubMed] [Google Scholar]
- 2.Shichiri M, Kato H, Marumo F, Hirata Y. Endothelin-1 as an autocrine/paracrine apoptosis survival factor for endothelial cells. Hypertension. 1997;30:1198–1203. doi: 10.1161/01.hyp.30.5.1198. [DOI] [PubMed] [Google Scholar]
- 3.De Nucci G, Thomas R, D’Orleans-Juste P, et al. Pressor effects of circulating endothelin are limited by its removal in the pulmonary circulation and by the release of prostacyclin and endothelium-derived relaxing factor. Proc Natl Acad Sci U S A. 1988;85:9797–9800. doi: 10.1073/pnas.85.24.9797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukuroda T, Fujikawa T, Ozaki S, Ishikawa K, Yano M, Nishikibe M. Clearance of circulating endothelin-1 by ETB receptors in rats. Biochem Biophys Res Commun. 1994;199:1461–1465. doi: 10.1006/bbrc.1994.1395. [DOI] [PubMed] [Google Scholar]
- 5.Myatt L, Brewer AS, Langdon G, Brockman DE. Attenuation of the vasoconstrictor effects of thromboxane and endothelin by nitric oxide in the human fetal-placental circulation. Am J Obstet Gynecol. 1992;166:224–230. doi: 10.1016/0002-9378(92)91863-6. [DOI] [PubMed] [Google Scholar]
- 6.Sventek P, Li J-S, Grove K, Deschepper CF, Schiffrin EL. Vascular structure and expression of endothelin-1 gene in L-NAME-treated spontaneously hypertensive rats. Hypertension. 1996;27:49–55. doi: 10.1161/01.hyp.27.1.49. [DOI] [PubMed] [Google Scholar]
- 7.Neerhof MG, Thaete LG. Chronic NOS-inhibition-induced IUGR is associated with increased circulating endothelin-1 levels in the rat. Hypertens Pregnancy. 1997;16:319. [Google Scholar]
- 8.Edwards DL, Arora CP, Bui DT, Castro LC. Long-term nitric oxide blockade in the pregnant rat: effects on blood pressure and plasma levels of endothelin-1. Am J Obstet Gynecol. 1996;175:484–488. doi: 10.1016/s0002-9378(96)70166-7. [DOI] [PubMed] [Google Scholar]
- 9.Thaete LG, Neerhof MG, Silver RK. Differential effects of endothelin A and B receptor antagonism on fetal growth in normal and nitric oxide-deficient rats. J Soc Gynecol Investig. 2001;8:18–23. [PubMed] [Google Scholar]
- 10.Thaete LG, Kushner DM, Dewey ER, Neerhof MG. Endothelin and the regulation of uteroplacental perfusion in nitric oxide synthase inhibition-induced fetal growth restriction. Placenta. 2005;26:242–250. doi: 10.1016/j.placenta.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Motulsky H, Christopoulos A. Fitting Models to Biological Data Using Linear and Nonlinear Regression. San Diego, CA: GraphPad Software; 2003. pp. 222–232. [Google Scholar]
- 12.Opgenorth TJ. Endothelin receptor antagonism. Adv Pharmacol. 1995;33:1–65. doi: 10.1016/s1054-3589(08)60665-1. [DOI] [PubMed] [Google Scholar]
- 13.Thaete LG, Jilling T, Synowiec S, Khan S, Neerhof MG. Expression of endothelin 1 and its receptors in the hypoxic pregnant rat. Biol Reprod. 2007;77:526–532. doi: 10.1095/biolreprod.107.061820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stannard C, Lehenkari P, Godovac-Zimmermann J. Functional diversity of endothelin pathways in human lung fibroblasts may be based on structural diversity of the endothelin receptors. Biochemistry. 2003;42:13909–13918. doi: 10.1021/bi0354132. [DOI] [PubMed] [Google Scholar]
- 15.Freedman NJ, Ament AS, Oppermann M, Stoffel RH, Exum ST, Lefkowitz RJ. Phosphorylation and desensitization of human endothelin A and B receptors: evidence for G protein-coupled receptor kinase specificity. J Biol Chem. 1997;272:17734–17743. doi: 10.1074/jbc.272.28.17734. [DOI] [PubMed] [Google Scholar]
- 16.Shraga-Levine Z, Sokolovsky M. Functional role for glycosylated subtypes of rat endothelin receptors. Biochem Biophys Res Commun. 1998;246:495–500. doi: 10.1006/bbrc.1998.8646. [DOI] [PubMed] [Google Scholar]
- 17.Cunha RA, Constantino MD, Fonseca E, Ribeiro JA. Age-dependent decrease in adenosine A1 receptor binding sites in the rat brain: effect of cis unsaturated free fatty acids. Eur J Biochem. 2001;268:2939–2947. doi: 10.1046/j.1432-1327.2001.02183.x. [DOI] [PubMed] [Google Scholar]
- 18.Hocher B, Schwarz A, Slowinski T, et al. In-vivo interaction of nitric oxide and endothelin. J Hypertens. 2004;22:111–119. doi: 10.1097/00004872-200401000-00020. [DOI] [PubMed] [Google Scholar]
- 19.Shigematsu K, Nakatani A, Kawai K, et al. Two subtypes of endothelin receptors and endothelin peptides are expressed in differential cell types of the rat placenta: in vitro receptor autoradiographic and in situ hybridization studies. Endocrinology. 1996;137:738–748. doi: 10.1210/endo.137.2.8593825. [DOI] [PubMed] [Google Scholar]
- 20.Kajihara T, Tomioka Y, Hata T, Ghazizadeh M, Asano G. Synthesis of endothelin-1 in rat uterus during pregnancy. J Histochem Cytochem. 1996;44:953–957. doi: 10.1177/44.9.8773560. [DOI] [PubMed] [Google Scholar]
- 21.Sakamoto S, Obayashi S, Aso T, Sato J, Hamasaki H, Azuma H. The mechanism of myometrial contractions induced by endothelin-1 in rat. Mol Human Reprod. 1997;3:1029–1035. doi: 10.1093/molehr/3.12.1029. [DOI] [PubMed] [Google Scholar]
- 22.Horwitz MJ, Clarke MR, Kanbour-Shakir A, Amico JA. Developmental expression and anatomical localization of endothelin-1 messenger ribonucleic acid and immunoreactivity in the rat placenta: a Northern analysis and immunohistochemistry study. J Lab Clin Med. 1995;125:713–718. [PubMed] [Google Scholar]
- 23.Kohnen G, Campbell S, Irvine GA, et al. Endothelin receptor expression in human decidua. Mol Human Reprod. 1998;4:185–193. doi: 10.1093/molehr/4.2.185. [DOI] [PubMed] [Google Scholar]
- 24.Mondon F, Anouar A, Ferr F. Endothelin receptor subtypes in the microvillous trophoblastic membrane of early gestation and term human placentas. Eur J Endocrinol. 1998;139:231–237. doi: 10.1530/eje.0.1390231. [DOI] [PubMed] [Google Scholar]
- 25.Cervar M, Huppertz B, Barth S, et al. Endothelin A and B receptors change their expression levels during development of human placental villi. Placenta. 2000;21:536–546. doi: 10.1053/plac.2000.0542. [DOI] [PubMed] [Google Scholar]
- 26.Kohnen G, Mackenzie F, Collett GP, et al. Differential distribution of endothelin receptor subtypes in placentae from normal and growth-restricted pregnancies. Placenta. 1997;18:173–180. doi: 10.1016/s0143-4004(97)90090-4. [DOI] [PubMed] [Google Scholar]
- 27.Bourgeois C, Robert B, Rebourcet R, et al. Endothelin-1 and ETA receptor expression in vascular smooth muscle cells from human placenta: a new ETA receptor messenger ribonucleic acid is generated by alternative splicing of exon 3. J Clin Endocrinol Metab. 1997;82:3116–3123. doi: 10.1210/jcem.82.9.4209. [DOI] [PubMed] [Google Scholar]