

Abstract
Cyclooxygenase-2 (COX-2), a rate-limiting enzyme converting arachidonic acid to prostaglandins and a key player in neuroinflammation, has been implicated in the pathogenesis of neurodegenerative diseases such as multiple sclerosis, Parkinson’s and Alzheimer’s diseases, and in traumatic brain injury- and ischemia-induced neuronal damage, and epileptogenesis. Accumulated information suggests that the contribution of COX-2 to neuropathology is associated with its involvement in synaptic modification. Inhibition or elevation of COX-2 has been shown to suppress or enhance excitatory glutamatergic neurotransmission and long-term potentiation (LTP). These events are mainly mediated via PGE2, the predominant reaction product of COX-2, and the PGE2 subtype 2 receptor (EP2)-protein kinase A pathway. Recent evidence shows that endogenous cannabinoids are substrates for COX-2 and can be oxygenated by COX-2 to form new classes of prostaglandins (prostaglandin glycerol esters and prostaglandin ethanolamides). These COX-2 oxidative metabolites of endocannabinoids, as novel signaling mediators, modulate synaptic transmission and plasticity and cause neurodegeneration. The actions of these COX-2 metabolites are likely mediated by mitogen-activated protein kinase (MAPK) and inositol 1,4,5-trisphosphate (IP3) signal transduction pathways. These discoveries suggest that the contributions of COX-2 to neurotransmission and brain malfunction result not only from its conversion of arachidonic acid to classic prostaglandins but also from its oxidative metabolism of endocannabinoids to novel prostaglandins. Thus, elucidation of COX-2 in synaptic signaling may provide a mechanistic basis for designing new drugs aimed at preventing, treating or alleviating neuroinflammation-associated neurological disorders.
Keywords: Inflammation, Prostaglandin E2, Endocannabinoids, Prostaglandin glycerol esters, Prostaglandin ethanolamides, Synaptic plasticity
Introduction
Neuroinflammation is a biological immune response to various endogenous and exogenous stimuli in the nervous system. However, chronic neuroinflammation may lead to dysfunction in neural circuits and neurological disorders. As a key player in inflammation, the role and mechanisms of cyclooxygenase-2 (COX-2) in neurological disorders are being unraveled. In particular, recent progress on COX-2 as an important mediator participating in excitatory glutamatergic synaptic transmission and long-term synaptic plasticity greatly advances our understanding of mechanisms underlying COX-2-mediated physiological and pathological functions in the brain. As an inducible isoform, the expression and activity of COX-2 are markedly elevated by a wide variety of stimuli ranging from proinflammatory insults and epileptic activity to the activation of NMDA receptors [1–5]. This means that synaptic efficacy will be altered when COX-2 is upregulated in neuroinflammation. Nevertheless, the COX-2-mediated synaptic modification is ultimately attributed to the amount of prostaglandins synthesized and their functional receptors. Thus, in this review, we will discuss the most recent information on COX-2 and its reaction products, prostaglandins, in synaptic signaling.
COX-2 in neural plasticity
It has been well documented that cyclooxygenases (COX) catalyze the first committed step of the conversion of arachidonic acid (AA) into unstable intermediate PGG2, which is rapidly converted to PGH2 by COX. Finally, a series of biologically active prostaglandins (PGD2, PGE2, PGF2α, and PGI2) and thromboxane A2 (TXA2) are formed from PGH2 by various isomerases. Two isozymes of COX, COX-1 and COX-2, have been identified [3, 6–11]. COX-1 and COX-2 are membrane-associated enzymes with a 71 kD molecular weight and a 63% amino acid sequence identity [11, 12]. COX-1 is constitutively expressed in most tissues of the brain, and its reaction products are expected to mainly contribute to normal physiological function. COX-2 is also constitutively expressed in the CNS (neurons, astrocytes, microglia and endothelia), but enriched in the hippocampus and cortex [1]. Importantly, COX-2 as an inducible enzyme is responsible for the ‘pathological’ production of PGs in response to a variety of stimuli ranging from proinflammatory factors (e.g., cytokines, endotoxin), seizure activity, brain injury, growth factors to the activation of NMDA receptors [1–3, 10, 11]. Most commonly used non-steroidal anti-inflammatory drugs (NSAIDs) such as indomethacin and aspirin are nonselective and inhibit both COX-1 and COX-2, whereas selective COX-2 inhibitors (Celecoxib, NS398, Vioxx, Valdecoxib) preferentially inhibit COX-2 over COX-1. A putative third isoform, COX-3, was recently reported to be expressed in the canine cerebral cortex and is targeted by acetaminophen [13]. It is a splice variant of the COX-1 transcript and therefore not a new isoform of COX [14, 15].
Because of a higher level of COX-2 expression in hippocampal and cortical neurons involved in cognitive function, investigators speculated that COX-2 might participate in neural plasticity. Yamagata et al. [1] provided the initial evidence that the basal expression of COX-2 is regulated by NMDA receptor-dependent synaptic activity. Also COX-2 expression is upregulated by high-frequency stimulation (HFS) associated with the induction of long-term potentiation (LTP), a form of synaptic plasticity and cellular model of learning and memory [16]. This information suggests that the expression level and activity of COX-2 may be linked to long-term synaptic plasticity. This speculation was confirmed by the experiments where authors observed that selective COX-2 inhibitors, but not those of COX-1, reduce HFS-induced LTP at hippocampal perforant path-dentate granule cell synapses [17]. This study provided the first direct evidence that COX-2 is involved in hippocampal long-term synaptic plasticity. The involvement of COX-2 in hippocampal LTP has been confirmed by studies performed by others who show that the inhibition of COX-2 suppresses both LTP [18, 19] and long-term depression in the hippocampus [18]. Introducing a small RNA interference (siRNA) technique in conjunction with local electroporation targeting the brain region, Akaneya and Tsumoto [20] reported that the knockdown of COX-2 in the visual cortex significantly decreases the expression levels of COX-2 and almost completely blocks theta-burst stimulation (TBS)-induced LTP, while the gene silencing for COX-1 does not induce significant effects on the induction or maintenance of LTP. This is consistent with the results reported by Chen and Bazan [21], who demonstrated that hippocampal perforant path LTP in COX-1 knockout mice is normal when compared to that in wild type controls. These findings based on the inhibition of COX-2 by selective COX-2 inhibitors or suppression of the COX-2 gene indicate that the constitutively expressed COX-2 participates in long-term synaptic plasticity. To determine whether the inducible form of COX-2 also contributes to synaptic transmission and plasticity, Sang et al. [4] administrated lipopolysaccharide (LPS) or interleukin-1β (IL-1β), proinflammatory factors, in hippocampal neurons in culture to elevate COX-2 expression and observed that the elevation of COX-2 enhances the frequency of miniature excitatory postsynaptic currents (mEPSCs). Recently, this team revealed that in vivo injection of LPS, which elevates COX-2 expression and activity, significantly facilitates basal synaptic transmission and mEPSCs and induces a time-dependent augmentation of LTP at the hippocampal perforant path [5]. Meanwhile, they also showed that hippocampal LTP is potentiated in neuronal COX-2 expression transgenic mice [22]. However, they did not find a significant difference in HFS-induced LTP between COX-2 knockout and wild type control animals [5]. This is probably due to compensatory response by COX-1 in COX-2 null mice [23, 24]. These findings suggest that both constitutive and inducible COX-2 play an important role in refinement of synaptic activity [25]. The participation of COX-2 in synaptic transmission and plasticity is supported by the evidence that COX-2 is localized in neuronal dendritic spines, specialized structures where synaptic signaling occurs [4, 26, 27]. In addition, the involvement of COX-2 in long-term synaptic plasticity and cognition has been supported from the behavioral tests where administration of COX-2 inhibitors impairs passive avoidance task [28, 29], memory acquisition, memory retention [30, 31], and spatial memory consolidation [32, 33]. Since COX-2 plays a key role in neuroinflammation, which is closely associated with brain injury and certain neurologic disorders such as multiple sclerosis, epilepsy, Parkinson’s and Alzheimer’s diseases [2, 15, 34–43], an elucidation of COX-2 in excitatory glutamatergic synaptic transmission and plasticity has greatly advanced our understanding of mechanisms responsible for the occurrence of these neurological disorders. For instance, the elevation of COX-2 expression and activity enhances excitatory glutamatergic neurotransmission [4, 5]. Glutamate is an important neurotransmitter for cell-to-cell communication under physiological conditions. However, an excessive release of glutamate will induce synaptic dysfunction, neuronal injury, or death. This can be used to explain why chronic neuroinflammation leads to neurodegeneration. Therefore, the information gained from the experiments on the contribution of COX-2 to long-term synaptic plasticity and cognitive function may help in the discovery of ways to prevent and treat neurological and psychiatric disorders resulting from excessive expression of COX-2.
PGE2 is an important signaling mediator in COX-2-mediated synaptic modification
Cyclooxygenases are the enzymes catalyzing AA into PGG2/PGH2, and finally into prostaglandins by cell-specific synthases [3]. Therefore, COX-2-mediated synaptic modification relies on its reaction products, prostaglandins [25]. Both COX-1 and COX-2 are capable of converting AA into five primary prostaglandins (PGD2, PGE2, PGF2α, PGI2, and TXA2), but they exhibit preferentially in synthesizing prostaglandins. It has been demonstrated that PGE2 and PGI2 are mainly derived from the COX-2 pathway [4, 21, 44, 45]. To determine which prostaglandin(s) is (are) the signaling mediator(s) responsible for the COX-2-involved modification of synaptic efficacy, Chen et al. [17] examined the individual effects of PGD2, PGE2, and PGF2α on the COX-2 inhibitor-induced reduction of hippocampal LTP. It appeared that the COX-2 inhibitor-induced reduction of LTP could be reversed by exogenous application of PGE2 but not by PGD2 or PGF2α [17]. This means that PGE2 may be an important signaling molecule in COX-2-mediated modulation of hippocampal synaptic transmission and plasticity [4, 17, 21, 46]. Further studies revealed that endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons [46]. Under the condition of depletion of endogenous PGE2, membrane input resistance and frequency of firing were significantly reduced. Meanwhile, exogenous application of PGE2 reversed this reduction of membrane excitability both in soma and apical dendrites of rat hippocampal CA1 pyramidal neurons. Likewise, exogenous application of PGE2 produced a greater enhancement of EPSPs and temporal summation in slices where endogenous PGE2 had been eliminated with NS938 compared with those in control slices. This is the first demonstration that endogenous PGE2 plays an important role in dynamically maintaining membrane excitability, synaptic transmission, integration, and plasticity in the hippocampus [46]. Akaneya and Tsumoto [20] also provide evidence that PGE2 serves as a messenger in COX-2-mediated synaptic modulation. They showed that the application of PGE2 induces a dose-dependent facilitation of TBS-induced LTP, while PGD2, PGF2α, PGI2, and TXA2 fail to facilitate LTP in the visual cortex. Meanwhile, TBS increases the release of PGE2; this increase is completely blocked by RNAi silencing of the COX-2 gene [20]. COX-2 has been shown to be colocalized with PSD-95, a postsynaptic marker, but has little overlap with synaptophysin, a presynaptic marker, confirming the presence of COX-2 in postsynaptic dendritic spines [4, 26, 27]. This suggests that postsynaptic dendritic spines are an important source of PGE2 synthesis.
PGE2 is produced by prostaglandin E synthase (PGES). Because PGE2 is lipophilic, it diffuses rapidly once synthesized and activates its specific membrane receptors. Three types of PGES (Figure. 1) are responsible for the conversion of PGH2 into PGE2: microsomal PGES-1 (mPGES-1), PGES-2 (mPGES-2), and cytosolic PGES (cPGES). Cytosolic PGES is mainly associated with the COX-1 pathway; mPGES-1, however, is preferentially coupled with COX-2; mPGES-2, a constitutive enzyme initially associated with the Golgi membrane and released into the cytoplasm after N-terminal proteolysis, is coupled with both COX-1 and COX-2 [47–50]. The colocalization of mPGES-1 and mPGES-2 with PSD-95 (a postsynaptic marker) suggests that PGE2 synthases are expressed in postsynaptic dendritic spines [4]. Since COX-2 is also present in postsynaptic dendritic spines, it is likely that PGE2 is catalyzed by COX-2 from the AA pathway in the postsynaptic dendritic spines. Thus, the sequential biosynthetic enzymes are present in the same subcellular compartment (postsynaptic dendritic spines), implying that the PGE2 availability is tightly and efficiently regulated by COX-2. This assumption has been supported by recent studies where the elevation of COX-2 expression or activity induced by LPS or IL-1β enhances expression of mPGES-1 and production of PGE2, which promotes the probability of synaptic glutamate release in cultured hippocampal neurons [4] and in slices [5]. A blockade of the mPGES-1 has also been shown to reduce the production of PGE2 when COX-2 expression is elevated [49]. Therefore, new drugs targeting the mPGES may hold great potential as alternatives for traditional NSAIDs and COX-2 inhibitors for alleviating or treating neuroinflammation-associated neurological disorders.
Figure 1.
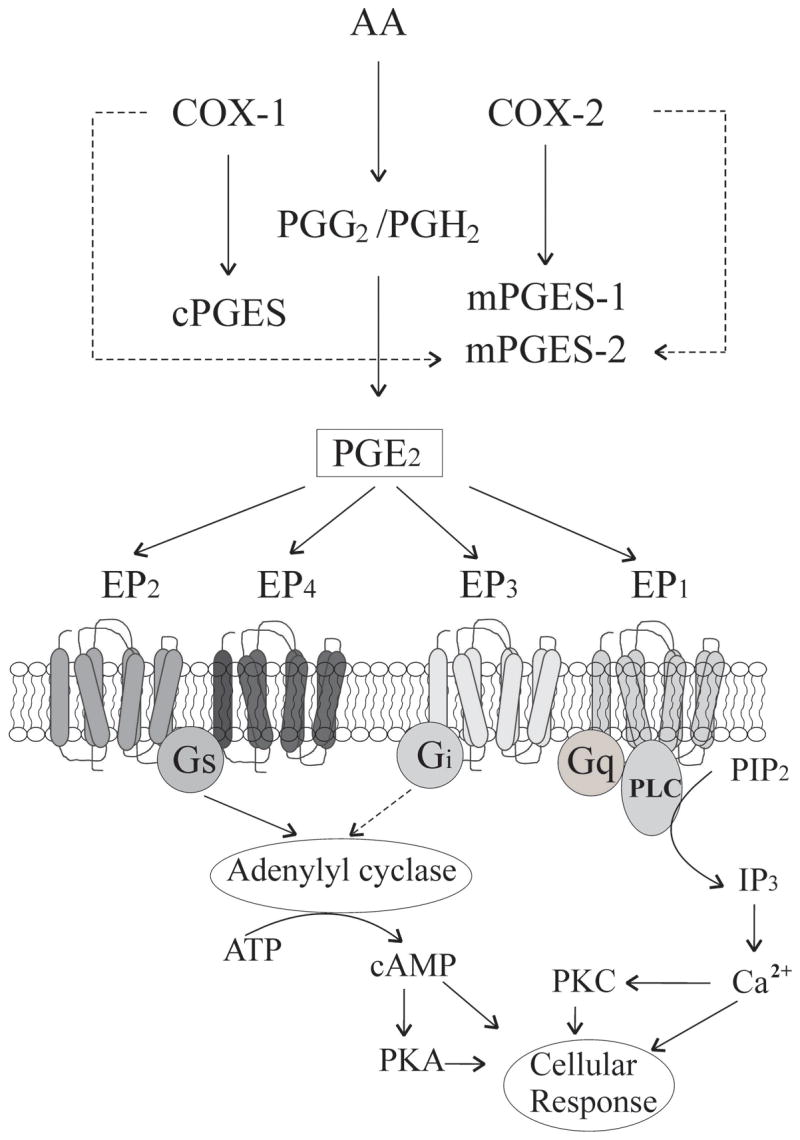
PGE2 formation and its signal transduction pathways. Arachidonic acid (AA) is transformed into unstable intermediate PGG2, which is promptly converted into PGH2 by cyclooxygenases (COXs). PGE2 is generated from PGH2 by cytosolic PGE2 synthase (cPGES) and membrane-bound PGE2 synthase-1 and -2 (mPGEs-1 and -2). Cytosolic PGES is mainly involved in COX-1, mPGES-1, however, is preferentially coupled with COX-2, and mPGES-2 is linked with both COX-1 and COX-2. Once synthesized, PGE2 diffuses immediately and activates its specific membrane receptors (EP1–4), which belong to the family of seven transmembrane-segment G-protein-coupled receptors. EP1 receptors couple with the Gq-phospholipase C(PLC)-inositol trisphosphate (IP3) pathway and its activation results in the release of intracellular Ca2+. EP2 and EP4 receptors couple with the Gs-adenylyl cyclase (AC)-cAMP-protein kinase A (PKA) pathway. EP3 couples with a pertussis toxin–sensitive Gi protein to inhibit AC resulting in a decrease in cAMP.
EP2 mediates the PGE2-induced synaptic modification
Four subtypes of the PGE2 receptors (EPs) have been identified and cloned, which have been designated as EP1, EP2, EP3, EP4, and multiple splicing isoforms of the subtype EP3 [51–55]. EPs belong to the family of seven-transmembrane-domain G protein-coupled receptors (GPCRs).These EP subtypes exhibit a distinctive signal transduction profile and cellular actions (Figure 1). Activation of EP1 receptors increases the levels of intracellular Ca2+, which is coupled with the Gq-PLC-IP3 and protein kinase C (PKC) pathways [56, 57]. EP3 is associated with a pertussis toxin (PTX)-sensitive Gi protein resulting in a decrease of cAMP [51, 58]. On the other hand, EP2 and EP4 receptors couple with the Gs-cAMP-PKA pathway leading to an increase of cAMP [59–61]. Zhu et al. [62] reported that four subtypes of EPs are heterogeneously expressed both in neurons and astroglial cells in the rodent hippocampus and cortex. They observed that EP2 and EP3 are the most abundant EPs expressed in the hippocampus and cortex, while only trace amount of EP1 and EP4 are detectable. Interestingly, EP2 and EP4 are well merged with synaptophysin (a presynaptic marker), but not PSD-95 (a postsynaptic marker), indicating that these two subtypes of EPs are likely present in presynaptic terminals. This is the first evidence that EPs are physically present in synaptic regions in hippocampal neurons, providing important avenues for PGE2 signaling in synaptic activity.
The roles of EPs in synaptic transmission and plasticity are being uncovered. It has been shown that an EP2-like receptor in spinal dorsal neurons may mediate PGE2-induced increase in membrane excitability and inhibition of glycinergic neurotransmission [63, 64]. Activation of EP3 and EP4 inhibits the release of γ-aminobutiric acid (GABA) from the GABAergic terminals innervating supraoptic nucleus neurons by the opening of nonselective cation channels [65]. Sang et al. [4] reported that PGE2 increases excitatory postsynaptic potentials (EPSPs), decreases the paired-pulse ratio (PPR) both at perforant path-granule cell synapses in dentate gyrus and Schaffer-collateral synapses in CA1 area of hippocampal slices, and enhances the frequency of mEPSCs in hippocampal neurons in culture. They further demonstrated that an EP2 agonist mimics the PGE2 effect, whereas EP1 or EP3 agonist fails to enhance the synaptic transmission both in hippocampal slices and primary cultured neurons. As described above, both EP2 and EP4 are associated with the Gs-cAMP-PKA pathway and are expressed in presynaptic terminals [62]. To distinguish the role of EP2 and EP4 in mediating the PGE2-induced facilitation of synaptic transmission, the authors designed an experiment in which EP2 and EP4 genes are individually silenced by the siRNA technique in cultured hippocampal neurons. Transiently silencing the EP2 gene eliminated the PGE2-enhanced miniature synaptic activity, while inhibiting the EP4 failed to prevent the PGE2-induced effect. These results provide the convincing evidence that the efficacy of PGE2 in modulation of synaptic activity is in its activation of presynaptic EP2 [4, 25]. It is likely that PGE2, which is produced at and released from the postsynaptic site, acts as a retrograde messenger in excitatory synaptic transmission via a presynaptic EP2 receptor. A recent study confirmed the role of EP2 in synaptic transmission and plasticity [20]. Suppressing the EP2 gene results in an abolishment of TBS-induced LTP in the visual cortex [20]. However, this study found that EP2 is expressed at postsynaptic sites. This discrepancy in location of EP2 expression between the two studies may be ascribed to the regional specificity of the hippocampus and visual cortex [20]. The function of other EPs in neural plasticity is still not well understood. Akaneya and Tsumoto [20] reported that suppressing the EP3 gene in the visual cortex leads to potentiate LTP. This suggests that EP2 and EP3 may play an opposite effect in long-term synaptic plasticity. Based on the available information, COX-2-derived PGE2 in the facilitation of glutamatergic synaptic transmission and long-term plasticity is mainly mediated via the EP2. The role of other EPs in modulation of synaptic efficacy remains to be determined.
The intracellular signal transduction mechanisms responsible for the PGE2-mediated synaptic modification are less clear. Chen and Bazan [46] reported that the PGE2-enhanced EPSPs are partially blocked by a PKA inhibitor. However, the effect is completely blocked in the presence of both PKA and PKC inhibitors in rat hippocampal slices. Further evidence from other studies shows that PKA inhibitors attenuate the PGE2-enhanced mEPSCs in cultured hippocampal neurons [4]. Since the PGE2-elevated synaptic activity is mainly mediated by the EP2, which is linked to the Gs-cAMP/PKA pathway, it is believed that the PGE2-potentiate neurotransmission is mediated via the EP2–Gs–cAMP/PKA pathway. Extracellular signal-regulated protein kinase (ERK), p38MAPK, and nuclear factor-κB (NF-κB) have been shown to be involved in the expression COX-2 and production of PGE2. However, little is known about the role of these signal transduction pathways in the PGE2-mediated synaptic modification, which warrants for further studies.
Endocannabinoids as important signaling mediators in neurotransmission
Endocannabinoids (eCBs) are endogenous metabolites of eicosanoid fatty acids and have been demonstrated to be lipid signaling mediators capable of binding to and functionally activating two cannabinoid receptors (CB1 and CB2) that are targeted by Δ9-tetrahydrocannabinol (Δ9-THC), the primary psychoactive ingredient in marijuana [66–73]. Cannabinoid receptors are seven-transmembrane, G-protein-coupled receptors, which are negatively coupled to adenylyl cyclase. The CB1 cannabinoid receptor was molecularly cloned from rat brain in 1990 [74], and is present in various mammalian tissues, mostly in the central nervous system, while the CB2 was identified by sequence homology in 1993 [75], and is most abundant in the immune system. Recent evidence shows that the CB2 is also present in the brain (astroglial cells and brainstem neurons), suggesting that the CB2 may be involved in physiological and pathological functions in the brain [76–82].
The discovery of CBRs prompted the search for eCBs. At least five endogenous ligands, 2-arachidonoyl glycerol (2-AG), arachidonoyl ethanolamide (AEA or anandamide), 2-arachidonoylglycerol ether (2-AGE), N-arachidonoyl-dopamine (NADA) and -arachidonoyl-ethanolamine (AE, virodhamine), have been identified [70–72, 83–89]. While virodhamine, 2-AGE and NADA are a partial or weak agonist for the CB1 or CB2 receptor, and their physiology and pharmacology have not yet been clarified [70, 71, 90], AEA and 2-AG are the two most studied eCBs and have been demonstrated to be involved in a variety of physiological and pathological processes in mammalian tissue via CB1 and CB2 receptors [70–72, 90–95]. The eCB signaling system is composed of eCBs, cannabinoid receptors, and the enzymes that synthesize, degrade, and transport them. The production and degradation of 2-AG and AEA are through different pathways [70–72, 96]. AEA is the first identified cannabinoid agonist, mainly produced from N-arachidonoylphosphatidylethanolamine (NAPE) by phospholipase D and degraded to AA by fatty acid amide hydrolase (FAAH). Several lines of evidence shows that AEA is a partial CB1 and a weak CB2 agonist, and an agonist for the vanilloid receptor [97–100]. 2-AG is the second identified cannabinoid receptor ligand, synthesized from diacylglycerol (DAG) by diacylglycerol lipase and hydrolyzed to AA by monoacylglycerol lipase (MGL). 2-AG has been demonstrated to be the most abundantly endogenous ligand and a full agonist for both CB1 and CB2 receptors [70–72, 90, 94]. 2-AG and AEA are removed from extracellular space through a diffusion-facilitated transporter or uptaken via membrane-associated carrier and simple diffusion [101]. Recent progress reveals that 2-AG and AEA are substrates for COX-2 and can be oxygenated by COX-2 to form new classes of prostaglandins: prostaglandin glycerol esters (PG-Gs) and prostaglandin ethanolamides (PG-EAs) [25, 102–105]. This means that there is another pathway in degrading eCBs in addition to their well known hydrolysis pathways.
Several pieces of information indicate that eCBs mainly induce an inhibitory effect on both GABAergic and glutamatergic neurotransmission and neurotransmitter release. Mechanisms underlying eCB modulation of synaptic function are still not clear. Presumably they are related to the cannabinoid modulation of ion channels, including transient A-type K ulchannels and voltage-gated P/Q and N-type Ca2+channels, and activation of MAPK cascades [106–108]. A recent study shows that eCB-mediated synaptic signaling is involved in a Ca2+-induced Ca2+ release (CICR) by activating the ryanodine receptor Ca2+ release channel [109]. One of the most important breakthroughs in understanding how eCBs modulate synaptic efficacy in the CNS results from the pioneer work by Alger and his colleagues [110, 111] and Wilson and Nicoll [112, 113], who demonstrated the phenomenon of depolarization-induced suppression of inhibition (DSI). DSI refers to eCB-induced suppression of GABAergic synaptic transmission. In DSI, strong depolarization of a postsynaptic neuron induces a release of signal that acts on the presynaptic CB1 receptor and transiently inhibits the release of GABA. Thereafter, a similar phenomenon was demonstrated for glutamatergic synaptic transmission and was designated depolarization-induced suppression of excitation (DSE) [70, 71, 114–116]. Thus, eCBs are proposed to serve as retrograde messengers in modulating both GABAergic and glutamatergic synaptic transmission [11–114, 117]. The latest evidence indicates that 2-AG, but not AEA, is likely a signaling molecule in mediating CB1 dependent DSI or DSE [73, 94; 118–123]. Also enzymes that synthesize 2-AG are present in postsynaptic dendritic spines, providing direct evidence that 2-AG is synthesized in postsynaptic sites and acts on presynaptic CB1 receptors [124, 125]. This information suggests that 2-AG plays an important role as a retrograde messenger in modulating synaptic activity.
COX-2 oxidative metabolism of eCBs in synaptic signaling
Endocannabinoids are substrates for a number of fatty acid oxygenases including the cyclooxygenases, lipoxygenases, and cytochrome P450s [105]. As mentioned above, COX-2 is capable of degrading 2-AG and AEA to form new types of prostaglandins. Interestingly, AEA and 2-AG are poor substrates for COX-1, suggesting that COX-2 oxidative metabolism is an important pathway in degrading or inactivating eCBs and may have functional significance in endocannabinoid signaling in synaptic activity. This has been supported by a recent report where the authors demonstrate that a selective COX-2 inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury [126]. These protective effects appear to result from the reduced COX-2 activity and PGE2, and preserved the levels of 2-AG [126]. The role of COX-2 oxidative metabolism of eCBs has also been supported by studies where the COX-2 inhibition alters eCB signaling in DSI and long-term synaptic plasticity in the hippocampus [19, 25, 118, 127]. Yang et al. [5] demonstrated that the elevation of COX-2 by in vivo injection of LPS or overexpression of neuronal COX-2 by a genetic manipulation abolishes DSI at hippocampal perforant path synapses. Conversely, the inhibition of COX-2 by administration of a selective COX-2 inhibitor or deletion of COX-2 gene augments DSI. These discoveries show an important role of COX-2 oxidative metabolism of eCBs in synaptic signaling. As mentioned above, both COX-2 and the enzymes synthesizing 2-AG are present in postsynaptic dendritic spines of excitatory neurons [4, 26, 124, 125]. Importantly, 2-AG is an endogenous natural substrate for COX-2, and COX-2 preferentially oxygenates 2-AG to prostaglandin glyceryl esters (PG-Gs) as it converts AA to classic prostaglandins, whereas the reaction of COX-2 metabolism of AEA to produce prostaglandin ethanolamides (PG-EAs) is relatively slow [105]. The colocalization of COX-2 and 2-AG in the same subcellular space (postsynaptic dendritic spines) allows COX-2 to rapidly and efficiently metabolize 2-AG when COX-2 expression or activity is elevated. Thus, the inhibition of COX-2 prevents the inactivation of eCBs, raising the eCB levels and promoting the eCB-mediated response, whereas the elevation of COX-2 accelerates the metabolism of eCBs, lowering the eCB levels and attenuating the eCB-mediated response. This means that COX-2 oxidative metabolism of eCBs constitutes an important mechanism contributing to COX-2-mediated synaptic modification, and that increases in COX-2 alter synaptic signaling not only through increased production of PGE2 from AA, but also through the oxidative metabolism of 2-AG to form new types of prostaglandins (Figure 2). Hence, up- or down-regulation of COX-2 expression or activity will significantly influence the COX-2 oxidative metabolism of eCBs and the effects on synaptic activity.
Figure 2.
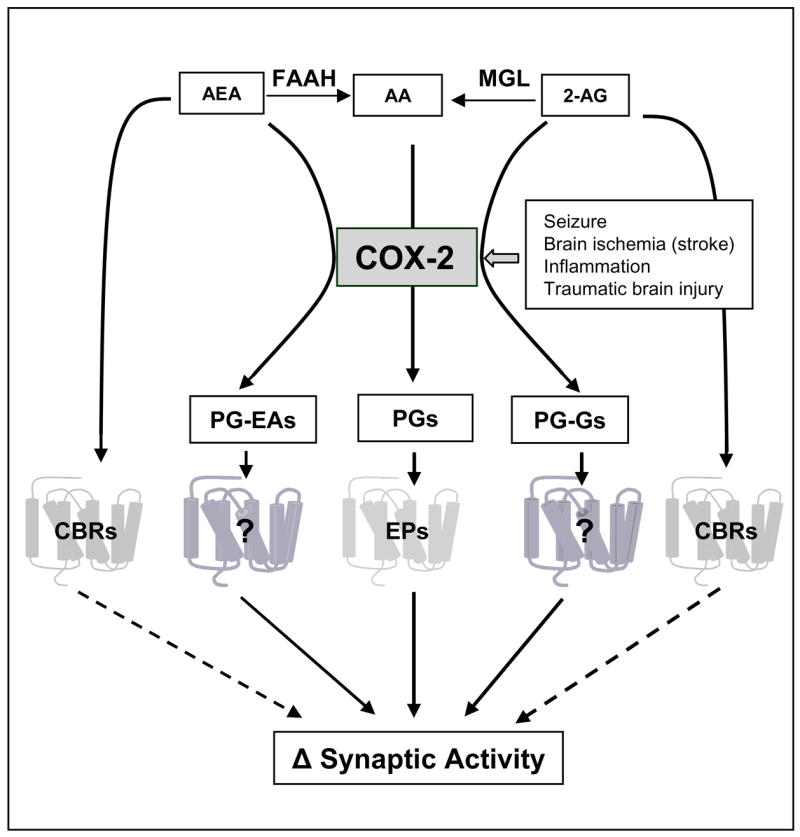
Scheme illustrating COX-2 oxidative metabolism of arachidonic acid and endocannabinoids in synaptic signaling. Neuroinflammation, brain ischemia, epileptic activity and traumatic brain injury elevate COX-2 expression and activity, which oxygenates AA, 2-AG and AEA to produce prostaglandins (PGs), and PG-Gs and PG-EAs. PGs (mainly PGE2) facilitate excitatory glutamatergic synaptic transmission via acting on the PGE2 receptors (EPs, mainly EP2). Endocannabinoids (2-AG and AEA) activate the presynaptic CB1 receptors and inhibit the release of neurotransmitters (GABA or glutamate depending upon the types of synapses). COX-2 oxidative metabolites of 2-AG or AEA (PG-Gs or PG-EAs) may exert their role in modulating synaptic events via unidentified receptors (novel receptors?). 2-AG and AEA are also hydrolyzed by MGL or FAAH to AA. 2-AG: 2-arachidonoylglycerol; AEA: arachidonoyl ethanolamide; MGL: monoacylglycerol lipase; FAAH: fatty acid amide hydrolase; AA: arachidonic acid; PGs: prostaglandins; PG-Gs: prostaglandin glycerol esters; PG-EAs: prostaglandin ethanolamides; EPs: prostaglandin receptors; CBRs: cannabinoid receptors
Novel prostaglandins derived from endocannabinoids in synaptic plasticity
Novel prostaglandins derived from 2-AG include PG-Gs (PGE2-G, PGI2-G, PGD2-G, PGF2α-G, and TXA2-G); those derived from AEA include PG-EAs (PGE2-EA, PGI2-EA, PGD2-EA, PGF2α -EA, and TXA2-EA) [25, 102–106]. Both 2-AG- and AEA-derived novel prostaglandins are significantly more stable metabolically than AA-derived classic prostaglandins, implying that COX-2 metabolism of eCBs may provide oxygenated lipids with sufficiently long half-lives to act as signaling mediators in physiological and pathological functions [105]. Although functional roles of endocannabinoid-derived PG-Gs and PG-EAs in the brain have not been well understood, they may represent unique signal mediators with potent activities distinct from their precursors and corresponding AA-derived prostaglandins [14, 25, 105]. Nirodi et al. [128] showed that the PGE2-G, a principal COX-2 oxidative metabolite of 2-AG, mobilizes intracellular calcium, triggers IP3 synthesis, and activates PKC in RAW264.7 macrophage cells, indicating PGE2-G may be a signaling mediator. Sang et al. [129] provided the first evidence that COX-2 metabolites of eCBs modulate GABAergic receptor-mediated inhibitory synaptic transmission in primary cultured hippocampal neurons. They found that PGE2-G, a principal COX-2 oxidative metabolite of 2-AG, significantly increases the frequency of miniature inhibitory postsynaptic currents (mIPSCs). The increase is not blocked by SR141716, a selective CB1 antagonist, suggesting the PGE2-G-mediated effect is not via the CB1. PGD2-G, PGF2α-G and PGD2-EA also increased the frequency of mIPSCs in a similar way to PGE2-G, but PGE2-EA and PGF2α-EA did not. These findings indicate that COX-2 oxidative metabolites of eCBs serve as signal modulators in modulating inhibitory synaptic transmission. In a consecutive study, Sang et al. [130] shows that PGE2-G enhances excitatory glutamatergic synaptic transmission which is CB1-independent in hippocampal neurons in culture. In particular, they demonstrated that this COX-2 oxidative metabolite of 2-AG induces neuronal injury and apoptosis and potentiates the NMDA-induced neuronal death in a dose-dependent manner. The PGE2-G-induced neurotoxicity is blocked by APV, an NMDA receptor antagonist or kynurenic acid, a broad spectrum glutamate receptor antagonist, suggesting that the PGE2-G-induced neurodegeneration results from the elevated release of glutamate. This is the first evidence showing that the COX-2 oxidative metabolite of 2-AG contributes to neurodegeneration. Endocannabinoids have been demonstrated to exert neuroprotection against β-amyloid-, NMDA-, and kainic acid-induced neurotoxicity and traumatic brain injury- and inflammation-induced neuronal damage [82, 93, 99, 131–140]. This means that eCBs as precursors protect neurons from harmful insults while their COX-2 oxidative metabolites may induce neurotoxicity. It is likely that the levels of eCBs are lowered and the levels of COX-2 metabolites of eCBs are elevated when COX-2 is abnormally elevated or activated in neuroinflammation [25]. Thus, some therapeutic benefits of selective COX-2 inhibitors in relieving inflammation-associated disorders may be derived from the preserved eCBs and reduced endocannabinoid oxidative metabolites. This is new evidence for the role of COX-2 in neuroinflammation-associated neurological disorders.
The inhibition of COX-2 in excitatory synaptic response and long-term synaptic plasticity is CB1-dependent by most recent studies [19, 127]. Yang and his colleagues recently provided direct evidence showing that COX-2 oxidative metabolites of eCBs alter hippocampal long-term synaptic plasticity [5]. They observed that PGE2-G, PGE2-EA, and PGF2α-EA, COX-2 oxidative metabolites derived from eCBs, elevate hippocampal LTP, the effect opposite to that of their parent molecules 2-AG and AEA [5].
The receptors for these novel prostaglandins have not been cloned and remain to be identified. Sang et al. [129] reported that PG-G- and PG-EA-induced effects on inhibitory synaptic transmission in hippocampal neurons are different from those induced by corresponding AA-derived prostaglandins, suggesting that the endocannabinoid oxidative metabolite-induced effects are not mediated via known prostaglandin receptors, instead, mediated by novel receptors (Figure 2). Hence, identification of the receptors for these novel prostaglandins will further our understanding of the actions of these endocannabinoid oxidative metabolites in synaptic function. Also the signal transduction pathways mediating the endocannabinoid-derived COX-2 metabolites in modulation of synaptic transmission and long-term synaptic plasticity are largely unknown. Recent work [129, 130] shows that PGE2-G-enhanced hippocampal GABAergic and glutamatergic synaptic transmissions are not mediated via PKA and PKC pathways, but appear to be mediated through ERK, p38MAPK, IP3, and NF-κB signal transduction pathways. This finding has been confirmed by more recent work of Yang et al. [5]. They demonstrated that the PGE2-G-induced increase in hippocampal LTP is attenuated by an IP3 inhibitor, indicating the involvement of the IP3-mediated mobilization of intracellular Ca2+ in PGE2-G-induced increase in LTP. In addition, ERK and p38MAPK inhibitors block the PGE2-G-potenitated LTP. The involvement of ERK and p38MAPK pathways is further supported from the molecular evidence. PGE2-G induces a time-dependent phosphorylation of ERK and p38MAPK, and this phosphorylation is attenuated by ERK and p38MAPK inhibitors [5]. Although the role of COX-2 oxidative metabolism of eCBs and their metabolites in physiological and pathological functions and the signal transduction mechanisms in the CNS has not been well characterized, this pioneer work has begun to unravel the functional role of novel prostaglandins derived from eCBs in synaptic transmission and plasticity.
Perspective
In the past decade, significant progress has been made in the elucidation of COX-2 regulation of PGE2 signaling in synaptic transmission and plasticity. This greatly advances our understanding of basic mechanisms underlying neuroinflammation-associated neurological and psychiatric disorders. The strategy for new drug design has been focused on targeting the downstream of the COX-2 pathways (e.g., mPGES, EPs) after reports of the occurrence of a series of cardiovascular complications in patients receiving COX-2 inhibitors. Recent work on COX-2 oxidative metabolism and their metabolites in modulating neurotransmission and synaptic plasticity may provide new clues to explore novel therapeutic approaches aimed at preventing, ameliorating or treating neuroinflammation-associated neurological disorders resulting from excessive activation of COX-2.
Acknowledgments
We would like to thank Jian Zhang for her excellent technical assistance. The authors’ work was supported by National Institutes of Health grants R01NS054886 and P20RR16816, and the Alzheimer’s Association grant IIRG-05-13580.
References
- 1.Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]
- 2.Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004;63:901–910. doi: 10.1093/jnen/63.9.901. [DOI] [PubMed] [Google Scholar]
- 3.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 4.Sang N, Zhang J, Marcheselli V, Bazan NG, Chen C. Postsynaptically synthesized prostaglandin E2 (PGE2) modulates hippocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J Neurosci. 2005;25:9858–9870. doi: 10.1523/JNEUROSCI.2392-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang H, Zhang J, Andreasson K, Chen C. COX-2 oxidative metabolism of endocannabinoids augments hippocampal synaptic plasticity. Mol Cell Neurosci. 2008 doi: 10.1016/j.mcn.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 7.Kujubu DA, Fletcher BS, Varnum BC, Lim RW, Herschman HR. TIS 10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/Cyclooxygenase homologue. J Biol Chem. 1991;266:12866–12872. [PubMed] [Google Scholar]
- 8.Hla T, Neilson K. Human cyclooxygenases-2 cDNA. Proc Natl Acad Sci U S A. 1992;89:7384–7388. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Banion MK, Winn V, Young DA. CDNA cloning and functional activity of a glucocorticoid-regulated inflammatory cyclooxygenase. Proc Natl Acad Sci U S A. 1992;89:4888–4892. doi: 10.1073/pnas.89.11.4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenase)-1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 11.Vane JR, Bakhle YS, Botting RM. Cyclooxygenase 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 12.Hawkey CJ. COX-2 inhibitors. Lancet. 1999;353:307–314. doi: 10.1016/s0140-6736(98)12154-2. [DOI] [PubMed] [Google Scholar]
- 13.Chandrasekharan NV, Dai H, Roos KLT, Evanson NK, Tomsik J, Elton TS, et al. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci USA. 2002;99:13926–13931. doi: 10.1073/pnas.162468699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patrignani P, Tacconelli S, Sciulli MG, Capone ML. New insights into COX-2 biology and inhibition. Brain Res Rev. 2005;48:352–359. doi: 10.1016/j.brainresrev.2004.12.024. [DOI] [PubMed] [Google Scholar]
- 15.Hewett SJ, Bell SC, Hewett JA. Contributions of cyclooxygenase-2 to neuroplasticity and neuropathology of the central nervous system. Pharmacol Ther. 2006;112:335–357. doi: 10.1016/j.pharmthera.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 16.Bliss VTP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 17.Chen C, Magee JC, Bazan NG. Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol. 2002;87:2851–2857. doi: 10.1152/jn.2002.87.6.2851. [DOI] [PubMed] [Google Scholar]
- 18.Murray HJ, O’Connor JJ. A role for COX-2 and p38 mitogen activated protein kinase in long-term depression in the rat dentate gyrus in vitro. Neuropharmacol. 2003;44:374–380. doi: 10.1016/s0028-3908(02)00375-1. [DOI] [PubMed] [Google Scholar]
- 19.Slanina KA, Roberto M, Schweitzer P. Endocannabinoids restrict hippocampal long-term potentiation via CB1. Neuropharmacol. 2005;49:660–668. doi: 10.1016/j.neuropharm.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 20.Akaneya Y, Tsumoto T. Bidirectional trafficking of prostaglandin E2 receptors involved in long-term potentiation in visual cortex. J Neurosci. 2006;26:10209–10221. doi: 10.1523/JNEUROSCI.3028-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C, Bazan NG. Lipid signaling: sleep, synaptic plasticity, and neuroprotection. Prostaglandins Other Lipid Mediators. 2005;77:65–76. doi: 10.1016/j.prostaglandins.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Andreasson KI, Savoneko A, Vidensky S, Goellner JJ, Yang Y, Shaffer A, et al. Age-dependent cognitive deficits and neuronal apoptosis in cyclooxygenase-2 transgenic mice. J Neurosci. 2001;21:8198–8209. doi: 10.1523/JNEUROSCI.21-20-08198.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ballou LR, Botting RM, Goorha S, Zhang J, Vane JR. Nociception in cyclooxygenase isozyme-deficient mice. Proc Natl Acad Sci USA. 2000;97:10272–10276. doi: 10.1073/pnas.180319297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosetti F, Langenbach R, Weerasinghe GR. Prostaglandin E2 and microsomal prostaglandin E synthase-2 expression are decreased in the cyclooxygenase-2-deficient mouse brain despite compensatory induction of cyclooxygenase-1 and Ca2+-dependent phospholipase A2. J Neurochem. 2004;91:1389–1397. doi: 10.1111/j.1471-4159.2004.02829.x. [DOI] [PubMed] [Google Scholar]
- 25.Sang N, Chen C. Lipid signaling and synaptic plasticity. Neuroscientist. 2006;2:425–34. doi: 10.1177/1073858406290794. [DOI] [PubMed] [Google Scholar]
- 26.Kaufmann WE, Worley PF, Pegg J, Bremer M, Isakson P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc Natl Acad Sci U S A. 1996;93:2317–2321. doi: 10.1073/pnas.93.6.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang X, Wu L, Wang Q, Hand T, Bilak M, McCullough L, Andreasson K. Function of COX-2 and Prostaglandins in Neurological Disease. J Mol Neurosci. 2007;33:94–99. doi: 10.1007/s12031-007-0058-8. [DOI] [PubMed] [Google Scholar]
- 28.Holscher C. Inhibitors of cyclooxygenases produce amnesia for a passive avoidance task in the chick. Eur J Neurosci. 1995;7:1360–1365. doi: 10.1111/j.1460-9568.1995.tb01127.x. [DOI] [PubMed] [Google Scholar]
- 29.Sato T, Ishida T, Irifune M, Tanaka K, Hirate K, Nakamura N, et al. Effect of NC-1900, an active fragment analog of arginine vasopressin, and inhibitors of arachidonic acid metabolism on performance of a passive avoidance task in mice. Eur J Pharmacol. 2007;560:36–41. doi: 10.1016/j.ejphar.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 30.Rall JM, Mach SA, Dash PK. Intrahippocampal infusion of a cyclooxygenase-2 inhibitor attenuates memory acquisition in rats. Brain Res. 2003;968:273–276. doi: 10.1016/s0006-8993(03)02248-0. [DOI] [PubMed] [Google Scholar]
- 31.Teather LA, Packard MG, Bazan NG. Post-training cyclooxygenase-2 (COX-2) inhibition impairs memory consolidation. Learn Mem. 2002;9:41–47. doi: 10.1101/lm.43602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharifzadeh M, Tavasoli M, Soodi M, Mohammadi-Eraghi S, Ghahremani MH, Roghani A. A time course analysis of cyclooxygenase-2 suggests a role in spatial memory retrieval in rats. Neurosci Res. 2006;54:171–179. doi: 10.1016/j.neures.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 33.Shaw KN, Commins S, O’Mara SM. Deficits in spatial learning and synaptic plasticity induced by the rapid and competitive broadspectrum cyclooxygenase inhibitor ibuprofen are reversed by increasing endogenous brain-derived neurotrophic factor. Eur J Neurosci. 2003;17:2438–2446. doi: 10.1046/j.1460-9568.2003.02643.x. [DOI] [PubMed] [Google Scholar]
- 34.Bazan NG. COX-2 as a multifunctional neuronal modulator. Nat Med. 2001;7:414–415. doi: 10.1038/86477. [DOI] [PubMed] [Google Scholar]
- 35.Bazan NG. Synaptic lipid signaling: significance of polyunsaturated fatty acids and platelet-activating factor. J Lipid Res. 2003;44:2221–2233. doi: 10.1194/jlr.R300013-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Hewett S, Uliasz TF, Vidwans AS. Hewett JACyclooxygenase-2 contributes to N-methyl-D-aspartate-mediated neuronal cell death in primary cortical cell culture. J Pharmacol Exp Ther. 2000;293:417–425. [PubMed] [Google Scholar]
- 37.Ho L, Pieroni C, Winger D, Purohit DP, Aisen PS, Pasinetti GM. Regional distribution of cyclooxygenase-2 in the hippocampal formation in Alzheimer’s disease. J Neurosci Res. 1999;57:295–303. doi: 10.1002/(SICI)1097-4547(19990801)57:3<295::AID-JNR1>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 38.Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, et al. Reduced susceptibility to ischemic brain injury and NMDA mediated neurotoxicity in cyclooxygenase-2 deficient mice. Proc Natl Acad Sci USA. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGeer PL, McGeer EG. The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative disease. Brain Res Rev. 1995;21:195–218. doi: 10.1016/0165-0173(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 40.McGeer PL, McGeer EG. Inflammation and the degenerative disease of aging. Ann N Y Acad Sci. 2004;1035:104–116. doi: 10.1196/annals.1332.007. [DOI] [PubMed] [Google Scholar]
- 41.Miettinen S, Fusco FR, Yrjänheikki J, Keinänen R, Hirvonen T, Roivainen R, et al. Spreading depression and focal brain ischemia induce cyclooxygenase-2 in cortical neurons through N-methyl-D-aspartic acid-receptors and phospholipase A2. Proc Natl Acad Sci U S A. 1997;94:6500–6505. doi: 10.1073/pnas.94.12.6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Minghetti L. Role of COX-2 in inflammatory and degenerative brain diseases. Subcell Biochem. 2007;42:127–141. doi: 10.1007/1-4020-5688-5_5. [DOI] [PubMed] [Google Scholar]
- 43.Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, et al. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci U S A. 1998;95:10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brock TG, McNish RW, Peters-Golden M. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J Biol Chem. 1999;274:11660–11666. doi: 10.1074/jbc.274.17.11660. [DOI] [PubMed] [Google Scholar]
- 45.Vidensky S, Zhang Y, Hand T, Goellner J, Shaffer A, Isakson P, et al. Neuronal overexpression of COX-2 results in dominant production of PGE2 and altered fever response. Neuromol Med. 2003;3:15–28. doi: 10.1385/NMM:3:1:15. [DOI] [PubMed] [Google Scholar]
- 46.Chen C, Bazan NG. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;93:929–941. doi: 10.1152/jn.00696.2004. [DOI] [PubMed] [Google Scholar]
- 47.Claveau D, Sirinyan M, Guay J, Gordon R, Chan CC, Bureau Y, et al. Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenases-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J Immunol. 2003;170:4738–4744. doi: 10.4049/jimmunol.170.9.4738. [DOI] [PubMed] [Google Scholar]
- 48.Murakami M, Nakashima K, Kamei D, Masuda S, Ishikawa Y, Ishii T, et al. Cellular prostaglandin E2 production by membranebound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem. 2003;278:37937–37947. doi: 10.1074/jbc.M305108200. [DOI] [PubMed] [Google Scholar]
- 49.Murakami M, Kudo I. Recent advance in molecular biology and physiology of the prostaglandin E2-biosynthetic pathway. Prog Lipid Res. 2004;43:3–35. doi: 10.1016/s0163-7827(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 50.Murakami M, Kudo I. Prostaglandin E synthase: a novel drug target for inflammation and cancer. Curr Pharm Des. 2006;12:943–954. doi: 10.2174/138161206776055912. [DOI] [PubMed] [Google Scholar]
- 51.Breyer RM, Bagdassarian CK, Myers SA. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 52.Boie Y, Stocco R, Sawyer N, Slipetz DM, Ungrin MD, Neuschäfer-Rube F, et al. Molecular cloning and characterization of the four rat prostaglandin E2 prostanoid receptor subtypes. Eur J Pharmacol. 1997;340:227–241. doi: 10.1016/s0014-2999(97)01383-6. [DOI] [PubMed] [Google Scholar]
- 53.Namba T, Sugimoto Y, Negishi M, Irie A, Ushikubi F, Kakizuka A, et al. Alternative splicing of C-terminal tail of prostaglandin E receptor subtype EP3 determines G-protein specificity. Nature. 1993;365:166–170. doi: 10.1038/365166a0. [DOI] [PubMed] [Google Scholar]
- 54.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structure, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 55.Sugimoto Y, Narumiya S. Prostaglandin Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 56.Shibuya I, Tanaka K, Uezono Y, Ueta Y, Toyohira Y, Yanagihara N, et al. Prostaglandin E2 induces Ca2ulrelease from ryanodine/caffeine-sensitive stores in bovine adrenal medullary cells via EP1-like receptors. J Neurochem. 1999;73:2167–2174. [PubMed] [Google Scholar]
- 57.Zonta M, Sebelin A, Gobbo S, Fellin T, Pozzan T, Carmignoto G. Glutamate-mediated cytosolic calcium oscillations regulate a pulsatile prostaglandin release from cultured rat astrocytes. J Physiol. 2003;553:407–414. doi: 10.1113/jphysiol.2003.046706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Breyer RM, Emeson RB, Tarng JL, Breyer MD, Davis LS, Abromson RM, et al. Alternative splicing generates multiple isoforms of a rabbit prostaglandin E2 receptor. J Biol Chem. 1994;269:6163–6169. [PubMed] [Google Scholar]
- 59.Bastien L, Sawyer N, Grygorczyk R, Metters KM, Adam M. Cloning, functional expression, and characterization of the human prostaglandin E2 receptor subtype. J Biol Chem. 1994;269:11873–11877. [PubMed] [Google Scholar]
- 60.Honda A, Sugimoto Y, Namba T, Watabe A, Irie A, Negishi M, et al. Cloning and expression of a cDNA for mouse prostaglandin E2 receptor to induce wakefulness in rats. J Neurosci. 1993;23:5975–5983. [Google Scholar]
- 61.Nishigaki N, Negishi M, Honda A, Sugimoto Y, Namba T, Narumiya S, et al. Identification of prostaglandin E receptor “EP2” cloned from mastocytoma cells EP4 subtype. FEBS Lett. 1995;364:339–341. doi: 10.1016/0014-5793(95)00421-5. [DOI] [PubMed] [Google Scholar]
- 62.Zhu PM, Genc Ai, Zhang X, Zhang J, Bazan NG, Chen C. Heterogeneous expression and regulation of hippocampal prostaglandin E2 receptors. J Neurosci Res. 2005;81:817–826. doi: 10.1002/jnr.20597. [DOI] [PubMed] [Google Scholar]
- 63.Baba H, Kohno T, Moore KA, Woolf CJ. Direct activation of rat spinal dorsal neurons by prostaglandin E2. J Neurosci. 2001;1:1750–1756. doi: 10.1523/JNEUROSCI.21-05-01750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahmadi S, Lippross S, Neuhuber WL, Zeilhofer HU. PGE2 selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat Neurosci. 2002;5:34–40. doi: 10.1038/nn778. [DOI] [PubMed] [Google Scholar]
- 65.Shibuya I, Setiadji SV, Ibrahim N, Harayama N, Maruyama T, Ueta Y, et al. Involvement of postsynaptic EP4 and presynaptic EP3 receptors in actions of prostaglandin E2 in rat supraoptic neurons. J Neuroendocrinol. 2002;14:64–72. doi: 10.1046/j.1365-2826.2002.00741.x. [DOI] [PubMed] [Google Scholar]
- 66.Gaoni Y, Mechoulam R. The isolation and structure of delta-1-tetrahydrocannabinol and other neutral cannabinoids from hashish. J Am Chem Soc. 1971;93:217–224. doi: 10.1021/ja00730a036. [DOI] [PubMed] [Google Scholar]
- 67.Felder CC, Glass M. Cannabinoid receptors and their endogenous agonists. Annu Rev Pharmacol Toxicol. 1998;38:179–200. doi: 10.1146/annurev.pharmtox.38.1.179. [DOI] [PubMed] [Google Scholar]
- 68.Sullivan JM. Cellular and molecular mechanisms underlying learning and memory impairments produced by cannabinoids. Learn Mem. 2000;7:132–139. doi: 10.1101/lm.7.3.132. [DOI] [PubMed] [Google Scholar]
- 69.McAllister SD, Glass M. CB(1) and CB(2) receptor-mediated signalling: a focus on endocannabinoids. Prostaglandins Leukot Essent Fatty Acids. 2002;66:161–171. doi: 10.1054/plef.2001.0344. [DOI] [PubMed] [Google Scholar]
- 70.Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–84. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- 71.Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–66. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- 72.Sugiura T, Kishimoto S, Oka S, Gokoh M, Wake K. Metabolism and physiological significance of anadamide and 2-arachidonoylglycerol, endogenous cannabinoid receptor ligands. In: Fonteh AN, Wykle RL, editors. Arachidonate remodeling and inflammation. Basel, Switzerland: Birkhäuser Verlag; 2004. pp. 211–37. [Google Scholar]
- 73.Mackie K. Cannabinoid receptors as therapeutic targets. Annu Rev Pharmacol Toxicol. 2006;46:101–22. doi: 10.1146/annurev.pharmtox.46.120604.141254. [DOI] [PubMed] [Google Scholar]
- 74.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–4. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 75.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 76.Skaper SD, Buriani A, Dal Toso R, Petrelli L, Romanello S, Facci L, et al. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc Natl Acad Sci U S A. 1996;93:3984–3989. doi: 10.1073/pnas.93.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pertwee RG. Cannabinoid receptors and pain. Prog Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- 78.Carrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yang W, Nithipatikom K, et al. Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol Pharmacol. 2004;65:999–1007. doi: 10.1124/mol.65.4.999. [DOI] [PubMed] [Google Scholar]
- 79.Cabral GA, Marciano-Cabral F. Cannabinoid receptors in microglia of the central nervous system: immune functional relevance. J Leukoc Biol. 2005;78:1192–1197. doi: 10.1189/jlb.0405216. [DOI] [PubMed] [Google Scholar]
- 80.Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- 81.Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, et al. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071:10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 82.Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, et al. The endocannabinoid anandamide protects neurons during CNS in ammation by induction of MKP-1 in microglial cells. Neuron. 2006;49:67–79. doi: 10.1016/j.neuron.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 83.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 84.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 85.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 86.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 87.Bisogno T, Melck D, Bobrov MYu, Gretskaya NM, Bezuglov VV, De Petrocellis L, et al. N-acyl-dopamines: novel synthetic CB(1) cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem J. 2000;351(Pt 3):817–824. [PMC free article] [PubMed] [Google Scholar]
- 88.Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Porter AC, Sauer JM, Knierman MD, Becker GW, Berna MJ, Bao J, et al. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther. 2002;301:1020–1024. doi: 10.1124/jpet.301.3.1020. [DOI] [PubMed] [Google Scholar]
- 90.Bisogno T, Ligresti A, Di Marzo V. The endocannabinoid signalling system: biochemical aspects. Pharmacol Biochem Behav. 2005;81:224–238. doi: 10.1016/j.pbb.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 91.Howlett AC, Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Porrino LJ. Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology. 2004;47(Suppl 1):345–358. doi: 10.1016/j.neuropharm.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 92.Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3:771–784. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- 93.Walter L, Stella N. Cannabinoids and neuroin ammation. Br J Pharmacol. 2004;141:775–785. doi: 10.1038/sj.bjp.0705667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alger BE. Endocannabinoid identification in the brain: studies of breakdown lead to breakthrough, and there may be NO hope. Sci STKE. 2005;309:pe51. doi: 10.1126/stke.3092005pe51. [DOI] [PubMed] [Google Scholar]
- 95.Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- 96.Stella N. Cannabinoid signaling in glial cells. Glia. 2004;48:267–277. doi: 10.1002/glia.20084. [DOI] [PubMed] [Google Scholar]
- 97.Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- 98.Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. 2003;140(5):790–801. doi: 10.1038/sj.bjp.0705467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Van der Stelt M, Di Marzo V. Cannabinoid receptors and their role in neuroprotection. Neuromol Med. 2005;7:37–50. doi: 10.1385/NMM:7:1-2:037. [DOI] [PubMed] [Google Scholar]
- 100.De Petrocellis L, Di Marzo V. Lipids as regulators of the activity of transient receptor potential type V1 (TRPV1) channels. Life Sci. 2005;77:1651–1666. doi: 10.1016/j.lfs.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 101.Croxford JL, Yamamura T. Cannabinoids and immune system: Potential for the treatment of inflammatory diseases? J Neuroimmunol. 2005;166:3–18. doi: 10.1016/j.jneuroim.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 102.Yu M, Ives D, Ramesha CS. Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J Biol Chem. 1997;272:21181–21186. doi: 10.1074/jbc.272.34.21181. [DOI] [PubMed] [Google Scholar]
- 103.Kozak KR, Rowlinson SW, Marnett LJ. Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glycerol prostaglandins by COX-2. J Biol Chem. 2000;27:33744–33749. doi: 10.1074/jbc.M007088200. [DOI] [PubMed] [Google Scholar]
- 104.Kozak KR, Crews BC, Morrow JD, Wang LH, Ma YH, Weinander R, et al. Metabolism of the endocannabinoid, 2-arachidonylglycerol and anandamide, into prostaglandin, thromboxane, and prostacyclin glycerol esters and ethanolamides. J Biol Chem. 2002;277:44877–44885. doi: 10.1074/jbc.M206788200. [DOI] [PubMed] [Google Scholar]
- 105.Kozak KR, Prusakiewicz JL, Marnett LJ. Oxidative metabolism of endocannabinoids by COX-2. Curr Pharmaceut Design. 2004;10:659–667. doi: 10.2174/1381612043453081. [DOI] [PubMed] [Google Scholar]
- 106.Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J Pharmacol Exp Ther. 1995;273:734–743. [PubMed] [Google Scholar]
- 107.Guzmán M, Sánchez C, Galve-Roperh I. Cannabinoids and cell fate. Pharmacol Ther. 2002;95:175–184. doi: 10.1016/s0163-7258(02)00256-5. [DOI] [PubMed] [Google Scholar]
- 108.Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 109.Isokawa M, Alger BE. Ryanodine receptor regulates endogenous cannabinoid mobilization in the hippocampus. J Neurophysiol. 2006;95:3001–3011. doi: 10.1152/jn.00975.2005. [DOI] [PubMed] [Google Scholar]
- 110.Alger BE, Pitler TA, Wagner JJ, Martin LA, Morishita W, Kirov SA, et al. Retrograde signalling in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. J Physiol. 1996;496:197–209. doi: 10.1113/jphysiol.1996.sp021677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pitler TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron. 1994;13:1447–1455. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- 112.Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- 113.Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- 114.Alger BE. Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol. 2002;68:247–286. doi: 10.1016/s0301-0082(02)00080-1. [DOI] [PubMed] [Google Scholar]
- 115.Diana MA, Marty A. Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE) Br J Pharmacol. 2004;142:9–19. doi: 10.1038/sj.bjp.0705726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 117.Wilson RI, Nicoll RA. Endocannabinoid signaling in the brain. Science. 2002;296:678–682. doi: 10.1126/science.1063545. [DOI] [PubMed] [Google Scholar]
- 118.Kim J, Alger BE. Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci. 2004;7:697–698. doi: 10.1038/nn1262. [DOI] [PubMed] [Google Scholar]
- 119.Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Makara JK, Mor M, Fegley D, Szabó SI, Kathuria S, Astarita G, et al. Selective inhibition of 2-AG hydrolysis enhances endocannabinoid signaling in hippocampus. Nat Neurosci. 2005;8:1139–41. doi: 10.1038/nn1521. [DOI] [PubMed] [Google Scholar]
- 121.Safo PK, Regehr WG. Endocannabinoids control the induction of cerebellar LTD. Neuron. 2005;48:647–659. doi: 10.1016/j.neuron.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 122.Straiker A, Mackie K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J Physiol. 2005;569:501–517. doi: 10.1113/jphysiol.2005.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Szabo B, Schlicker E. Effects of cannabinoids on neurotransmission. Handb Exp Pharmacol. 2005;168:327–365. doi: 10.1007/3-540-26573-2_11. [DOI] [PubMed] [Google Scholar]
- 124.Katona I, Urbán GM, Wallace M, Ledent C, Jung KM, Piomelli D, et al. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M, et al. Localization of diacylglycerol lipase-alpha around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J Neurosci. 2006;26:4740–4751. doi: 10.1523/JNEUROSCI.0054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gopez JJ, Yue H, Vasudevan R, Malik AS, Fogelsanger LN, Lewis S, et al. Cyclooxygenase-2-specific inhibitor improves functional outcomes, provides neuroprotection, and reduces inflammation in a rat model of traumatic brain injury. Neurosurgery. 2005;56:590–604. doi: 10.1227/01.NEU.0000154060.14900.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Slanina KA, Schweitzer P. Inhibition of cyclooxygenase-2 elicits a CB1-mediated decrease of excitatory transmission in rat CA1 hippocampus. Neuropharmacol. 2005;49:653–659. doi: 10.1016/j.neuropharm.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 128.Nirodi CS, Crew BC, Kozak KR, Rorrow JD, Marnett LJ. The glyceryl ester of prostaglandin E2 mobilize calcium and activates signal transduction in RAW 264.7 cells. Proc Natl Acad Sci U S A. 2004;101:1840–1845. doi: 10.1073/pnas.0303950101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sang N, Zhang J, Chen C. PGE2-G, a COX-2 oxidative metabolite of 2-arachidonylglycerol, modulates inhibitory synaptic transmission in mouse hippocampal neurons. J Physiol (Lond) 2006;572:735–745. doi: 10.1113/jphysiol.2006.105569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sang N, Zhang J, Chen C. COX-2 oxidative metabolite of endocannabinoid 2-AG enhances excitatory glutamatergic synaptic transmission and induces neurotoxicity. J Neurochem. 2007;102:1966–1977. doi: 10.1111/j.1471-4159.2007.04668.x. [DOI] [PubMed] [Google Scholar]
- 131.Iunone T, Esposito G, Esposito R, Santamaria R, Di Rosa M, Izzo A. Neuroprotective effect of cannabidiol, a non-psychoactvie component from cannabis sativa, on b-amyloid-induced toxicity in PC12 cells. J Neurochem. 2004;89:134–141. doi: 10.1111/j.1471-4159.2003.02327.x. [DOI] [PubMed] [Google Scholar]
- 132.Khaspekov LG, Brenz Verca MS, Frumkina LE, Hermann H, Marsicano G. Lutz BInvolvement of brain-derived neurotrophic factor in cannabinoid receptor-dependent protection against excitotoxicity. Eur J Neurosci. 2004;19:1691–1698. doi: 10.1111/j.1460-9568.2004.03285.x. [DOI] [PubMed] [Google Scholar]
- 133.Marsicano G, Moosmann B, Hermann H, Lutz B, Behl C. Neuroprotective properties of cannabinoids against oxidative stress: role of the cannabinoid receptor CB1. J Neurochem. 2002;80:448–456. doi: 10.1046/j.0022-3042.2001.00716.x. [DOI] [PubMed] [Google Scholar]
- 134.Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- 135.Milton NG. Anandamide and noladin ether prevent neurotoxicity of the human amyloid-b peptide. Neurosci Lett. 2002;332:127–130. doi: 10.1016/s0304-3940(02)00936-9. [DOI] [PubMed] [Google Scholar]
- 136.Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, et al. Endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- 137.Panikashvili D, Mechoulam R, Beni SM, Alexandrovich A, Shohami E. CB1 cannabinoid receptors are involved in neuroprotection via NF-kappa B inhibition. J Cereb Blood Flow Metab. 2005;25:477–484. doi: 10.1038/sj.jcbfm.9600047. [DOI] [PubMed] [Google Scholar]
- 138.Puffenbarger RA, Boothe AC, Cabral GA. Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia. 2000;29:58–69. [PubMed] [Google Scholar]
- 139.Sarne Y, Mechoulam R. Cannabinoids: between neuroprotection and neurotoxicity Curr Drug Target-CNS. Neurol Dis. 2005;4:677–684. doi: 10.2174/156800705774933005. [DOI] [PubMed] [Google Scholar]
- 140.Sheng WS, Hu S, Cabral GA, Lokensgard JR, Peterson PK. Synthetic cannabinoid Win55,212–2 inhibits generation of in ammatory mediators by IL-1beta-stimulated human astrocytes. Glia. 2005;49:211–219. doi: 10.1002/glia.20108. [DOI] [PubMed] [Google Scholar]