

Abstract
Th cells can be subdivided into IFNγ-secreting Th1, IL-4/IL-5 secreting Th2, and IL-17 secreting Th17 cells. We have evaluated the capacity of fully differentiated Th1, Th2, and Th17 cells derived from a mouse bearing a transgenic TCR specific for the gastric parietal cell antigen, H/K ATPase, to induce autoimmune gastritis after transfer to immunodeficient recipients. We have also determined the susceptibility of the disease induced by each of the effector T cell types to suppression by polyclonal regulatory T cells (Treg) in vivo. Each type of effector cell induced autoimmune gastritis with distinct histological patterns. Th17 cells induced the most destructive disease with cellular infiltrates composed primarily of eosinophils accompanied by high levels of serum IgE. Polyclonal Treg could suppress the capacity of Th1 cells, moderately suppress Th2 cells, but could only suppress Th17 induced disease at early time points. The major effect of the Treg was to inhibit the expansion of the effector T cells. However, effector cells isolated from protected animals were not anergic and were fully competent to proliferate and produce effector cytokines ex vivo. The strong inhibitory effect of polyclonal Treg on the capacity of some types of differentiated effector cells to induce disease provides an experimental basis for the clinical use of polyclonal Treg in the treatment of autoimmune disease in man.
Keywords: Rodent, Th1/Th2 cells, autoimmunity, tolerance/suppression/anergy
Th cells are considered major players in the response against infectious organisms as well as in promoting systemic and organ-specific autoimmune diseases. In order to convey their full function, Th cells secrete a variety of cytokines and can be subdivided into three different types based on their cytokine signature: IFNγ-secreting Th1, IL-4- and IL-5-secreting Th2 (1) and IL-17- producing Th17 cells (2, 3). Over the past two decades, the Th1/Th2 paradigm prevailed and these two cell types were implicated in the pathogenesis of different autoimmune diseases. For instance, Th1 cells were thought to promote the pathology in Crohn’s disease (4), whereas Th2 mediated asthma (5). However, recent evidence suggests that a better understanding of the pathogenesis of autoimmune diseases requires that the dichotomous view of Th differentiation needs to be broadened (2). IL-17 producing Th17 cells appear to play a critical pathogenic role in autoimmunity, promoting organ damage in experimental autoimmune encephalitis (6), which has long been regarded as a typical Th1-mediated disease (3). In addition, Th17 cells appear to be key effector T cells in a variety of human autoimmune diseases including multiple sclerosis, rheumatoid arthritis, respiratory disease, systemic lupus erythematosus, psoriasis, systemic sclerosis and chronic inflammatory bowel disease (7). Nevertheless, the individual contributions of each of the effector T cell types to autoimmune disease remain difficult to dissect.
Autoimmune gastritis (AIG) is one of the few spontaneous animal models of organ-specific autoimmune disease in which the target Ag, the proton pump of the gastric parietal cell, the H/K-ATPase, has been identified (8, 9). In addition, murine AIG represents an animal model for pernicious anemia in man in which T and B cell responses also target the H/K-ATPase (10-12). We have generated a TCR transgenic (Tg) mouse (TxA23) whose T cells express a TCR from a clone derived from a 3 day thymectomized mouse that developed AIG. TxA23 T cells recognize a peptide from the H/K-ATPase α-chain and spontaneously develop severe AIG. Naive T cells from TxA23 mice transfer AIG into immunodeficient recipients, and the development of AIG can be prevented by co-transfer of polyclonal naturally occurring CD4+CD25+FoxP3+ regulatory T cells (Treg) (13, 14). Suppression correlated with a failure of the transferred autoreactive T cells to produce IFNγ and to differentiate into Th1 effector T cells (14).
The first aim of the present studies was to assess the capacity of Th1, Th2, and Th17 effector cells derived from naïve TxA23 T cells to induce AIG following transfer to immunodeficient recipients. As our previous studies focused on the capacity of polyclonal Treg to suppress the induction of AIG by naïve TxA23 cells, the second aim of this study was to determine the susceptibility of each of the three fully differentiated effector T cell types to suppression by polyclonal Treg in vivo. Although Treg have been shown to suppress cytokine production in vitro and IFNγ production in vivo (14), we also examined whether Treg could modulate cytokine production by fully differentiated effector cells in vivo. We demonstrate that each effector cell population induces AIG with a distinctive pathology. Polyclonal Treg readily suppressed the induction of disease mediated by Th1 effectors, while Th2-mediated disease was more resistant to suppression. In contrast, disease induced by Th17 cells was the most aggressive and was susceptible to suppression only at early time points after disease induction.
Materials and Methods
Mice and reagents
Female BALB/c and BALB/c nu/nu (4-8 wk old) were purchased from the National Cancer Institute animal facility and housed under specific pathogen-free conditions. TxA23 TCR-Tg mice have been described previously (15). All mice were maintained in our animal facility and cared for in accordance with institutional guidelines. H+/K+-ATPase α-chain peptides, PITAKAIAASVG, aa 630-641, was synthesized by the National Institute of Allergy and Infectious Diseases Peptide Synthesis Unit.
T cell purification and differentiation
Naive TxA23 T cells were isolated from the thymus of TxA23 mice expressing the surface marker Thy-1.1. As an enrichment step, thymocytes were depleted of CD8+ cells after incubation for 10 min with anti-CD8 beads MACS microbeads (Miltenyi Biotec) using the autoMACS deplete-sensitive program. Finally, cells were sorted for CD4+CD25- cells in our cell sorting facility. Sorting resulted in 99% to 100% purity of CD4+CD25-CD8- naive T cells. In some experiments, thymocytes from TxA23 Thy-1.1/Thy-1.2 heterozygous mice were used. Purified TxA23 CD4+CD25- thymocytes were stimulated with plate-bound anti-CD3 and anti-CD28 (2μg each/well) in on 24 well plates (0.25×106 cells/well) in complete RPMI medium (cRPMI) consisting of RPMI 1640 supplemented with 10% heat-inactivated FCS (Atlanta Biologicals), penicillin/streptomycin, 2 mM L-glutamine, 10 mM HEPES, 0.1 mM nonessential amino acids, and 1 mM sodium pyruvate (all from BioSource International). The culture medium was supplemented with specific recombinant cytokines and/or anti-cytokine-mAbs as follows: For Th1 differentiation, rhIL-12 (10ng/ml) and anti-IL-4 (10μg/ml, clone 11B11); for Th2 differentiation, rIL-4 (1000 U/ml), anti-IL-12 (10μg/ml; clone C17.8) and anti-IFNγ (10 μg/ml; clone XMG1.2) (16); for Th17 differentiation, TGFβ (2.5μg/ml), rIL-6 (10μl/ml), anti-IL-4, anti-IFNγ, anti-IL-12, and anti-IL-2 (clone S4B6, 10μg/ml). All cell cultures were split 1:2 on day 4 after activation and were further supplemented with rIL-2 (100 U/ml) or with medium alone in case of Th17 cultures (17). Optimal differentiation of Th17 cells was achieved after 1 week of differentiation in vitro. In order to optimize Th1 and Th2 differentiation, cultures were re-stimulated on day 8 with H/K-ATPase-αchain630-641 (10 μg/ml) in combination with APC (1×106, T cell-depleted BALB/c splenocytes that had been irradiated with 3000 rad) and cultured for an additional week under the same conditions as described above.
Purification of Treg
A single cell suspension of spleen cells from BALB/c mice was stained with PE-anti-CD25 (PC61) for 15 min, washed, incubated for 15 min with anti-PE microbeads and then isolated using the autoMACS deplete-sensitive program. The positive fraction, which contained the CD25+ T cells, was further enriched for CD25+ T cells by using MACS LS columns. Purified CD4+CD25+ T cells were 91.4±2.8% FoxP3+ as measured by flow cytometry.
Cell transfer experiments
All cells were washed twice with PBS, and the cultured T cells were diluted into PBS such that an i.p. injection of 0.5 ml/mouse resulted in the transfer of 50,000 TxA23Thy-1.1+ T cells.
Flow cytometry
Cell surface stainings were performed according to standard procedures using antibodies against CD8, CD19, CD25, CD62L, Thy-1.1 and Thy-1.2 directly conjugated to fluorescein isothiocyanate (FITC) or phycoerythrin (PE) purchased from BD Pharmingen. Anti-CD4-Tri-Color was purchased from Caltag Laboratories. mAbs for intracellular stainings (anti-IL-2, anti-IL-4, anti-IL-5, anti-IL-10, anti-IL-17, anti-IFNγ, anti-TNFα) conjugated to PE or allophycocyanin (APC) were purchased from BD Pharmingen. PE- or FITC-labelled mAbs to FoxP3 were from eBioscience. All flow cytometry was performed on a FACScan or FACSCalibur and analyzed using CellQuest software (all from BD Biosciences) and FLOWJo (TreeStar).
Intracellular cytokine staining was performed with PE- or APC- labelled mAbs as described (17). In brief, cells were stimulated for 4h with phorbol myristate acetate (PMA) and ionomycin with Golgistop added after 2h. Cell stimulation was terminated by fixing in 4% formyl saline. Fixed cells were stained in 0.1% saponin permeablization buffer for 1h and finally analyzed on a FACS Calibur flow cytometer (Becton Dickinson).
In vitro T cell proliferation assay
Single-cell suspensions were prepared from gastric LN (gLN) or from cell cultures, washed in PBS containing 5% FCS and penicillin/streptomycin (100 U/ml and 10 μg/ml, respectively). The number of TxA23Thy-1.1+ cells was determined by cell counts and FACS analysis. TxA23 T cells (5×103) were added to a 96-well, U-bottom tissue culture plate with T-depleted irradiated splenocytes (1×105, 3000 rad) with or without H/K-ATPase-α-chain630-641 peptide (50 μg/ml) in cRPMI. After 72h, cultures were pulsed with 3H-TdR for 8h.
Gastric pathology
For each group, at least five stomachs were ligated and inflated by injection of fixative (4% paraformaldehyde). The stomachs were opened by cutting along the greater curvature, flattened with filter paper and embedded in paraffin. Sections were stained with either hematoxylin and eosin, or with the LUNA stain. CD3+ lymphocytes in gastric sections were stained with a rabbit anti-CD3 (Dako, Carpinteria, CA, Catalog #A-0452) at 1:300, using an antigen retrieval solution in a food steamer (Diva Solution-Catalog #DV2004G1, Biocare, Concord, CA). The extent of gastritis was graded on a scale of 1-6, depending on the extent of mononuclear cell infiltration and parietal and chief cell destruction (14). General descriptions for scores are as follows: 1.0, scattered lymphocytes throughout submucosa and muscularis; 1.5, one or two small dense blankets of lymphocytes; 2.0, two to four small dense clusters of lymphocytes in the submucosa/mucosa; 3.0, two or three areas with intermediate infiltration spanning one-third of mucosa; 4.0, large nodules of lymphocytic accumulation spanning half to all of mucosa; 4.5, large nodules of lymphocytic accumulation spanning half to all of mucosa beginning to show evidence of parietal and chief cell destruction (<25%); 5.0, heavy lymphocytic infiltration throughout mucosa, parietal and chief cell loss (25-75%), and replacement by foamy cells; and 6.0, total parietal and chief cell loss, no mucosal architecture, and many foamy cells. Thus, grade 1-4 indicate - potentially reversible - inflammation without damage of gastric parietal cells, whereas grade 4.5-6 indicate destructive AIG with loss of parietal cells and, finally, total destruction of the gastric mucosa. Stomach sections were scored blindly by two investigators. Anti-PCAb were detected by immunofluorescence on cryostat sections of normal BALB/c stomach as previously described (18). The method for isolating cells from stomach tissue has been previously described (19).
ELISAs
Ig ELISAs were performed as described (20). Briefly, plates were coated over night with 100 μl of anti-mouse IgE, anti-mouse IgG1 and anti-mouse IgG2a (all from Pharmingen). The next day, samples [1/50 or 1/500 dilutions of mouse serum or mouse Ig standards (Pharmingen)] were added and stained with biotin-anti mouse IgE/IgG1/IgG2a (all from Pharmingen) and, in a second step, with streptavidin-HRP (Pharmingen) (1/4,000 dilution in diluents/blocking buffer). Thereafter, TMB solution (Sigma, St. Louis, MO) and, finally, Stop Solution (2N H2SO4) was added. Plates were read at 450 nm, and the optical densities of the samples were then plotted on the linear part of the standard curve and multiplied by the dilution factor to calculate Ig concentration in serum samples. Cytokines were measured in supernatants of stimulated cells using cytometric bead assay (Pierce Biotechnology, Woburn, MA).
Statistical analysis
All group results are expressed as mean ± SD, if not stated otherwise. Student’s t-test, or Fisher’s exact test (2-tailed) were used for the comparison of group values and discriminatory parameters, where appropriate. For comparing group values that did not follow Gaussian distribution, Mann-Whitney’s test (2-tailed) was used. Welch’s correction was applied if variances were significantly different. Pearson and Spearman correlation coefficients were calculated for variables following, or not-following Gaussian distributions, respectively. P values less than 0.05 were considered significant.
Results
Generation of Th1, Th2 and Th17 effector populations
Naive CD4+Thy1.1+ T cells were isolated from thymi of TxA23 mice and cultured under specific priming conditions as described. Before injection, the in vitro differentiated cells were analyzed by intracellular cytokine staining. Th1 cultures were highly enriched for IFNγ producing cells (67.2±26.1%, mean±SD), while only a very small percentage produced IL-4 (1.9±2.0%), IL-5 (1.0±1.0%), or IL-17 (1.0±1.3%). Cells cultured under Th2 conditions produced IL-4 (41±13%) and IL-5 (33.4±23.2%), but almost no IFNγ (1.6±1.5%) or IL-17 (1.0±0.5%). Cells cultured under Th17 conditions were highly enriched for IL-17 producers (62.5±25.9%), and contained only low percentages of IFNγ (1.1±1.5%), IL-4 (1.7±0.7%) or IL-5 (0.9±0.9%) producing lymphocytes (Fig. 1A).
FIGURE 1.

Characteristics of Th1, Th2 and Th17 cell lines. A. Cytokine production by the Th1, Th2 and Th17 cell lines upon re-stimulation with PMA and Ionomycin. B. In vitro T cell proliferation of the cells line stimulated with APC and peptide. C. Expression of Vα2, CD25, CD62L on the cell lines prior to transfer. D. Selective homing of CD4+Thy1.1+ T effectors to the gLN two weeks after transfer. E. Accumulation of T effectors in the gastric mucosa.
Differentiated Th1, Th2 and Th17 T effectors preferentially accumulate in the gastric LN and can also be isolated from gastric mucosa
After in vitro differentiation, all T effector population responded to their cognate antigen as measured by proliferation in vitro (Fig. 1B). All groups of T effectors also highly expressed the H/K-ATPase-specific Vα2 receptor, high levels of CD25, but low levels of CD62L (Fig. 1C). Following i.p. injection T effectors preferentially accumulated in the gastric lymph node (gLN) (Fig. 1D). As expected, CD4+Thy1.1+ cells could also be isolated from the inflamed gastric mucosa and represented a major fraction of all recovered cells (14). A typical example is given for a stomach flush at 6 weeks after injection (Fig. 1E).
In vitro differentiated Th1, Th2 and Th17 effectors induce AIG
All three Th cell types transferred AIG into nu/nu recipients. Two weeks after injection, only mild inflammation was seen in all groups and no parietal cell destruction was observed. After 4 weeks, gastritis with destruction of ≥25% of parietal cells was observed in all groups, but the incidence of destructive gastritis was highest in recipients of Th17 cells (80%), intermediate in mice that received Th2 cells (60%), and significantly less in recipients of Th1 cells (20%). However, after 6 weeks, 100% of mice in the Th17 group, 93% in the Th2 group, and 77% in the Th1 group showed high-grade AIG as defined by severe or complete parietal cell loss (score 5-6), epithelial hyperplasia, and massive inflammatory infiltrates (Table I, Fig. 2A-D). In addition, more than 50% of the mice in each group were positive for serum antibodies directed against parietal cells (anti-PC-Abs), a feature typical of murine and human AIG (21).
Table I.
Pathology of the stomach and spleen, 6 weeks after injection of Th1, Th2 and Th17 effector T cells
| T effector injected | parietal cell destruction (≥25-100%, score 5-6) | stomach infiltrates | epithelial hyperplasia | submucosal infiltrates | fore-stomach lesions | spleen |
|---|---|---|---|---|---|---|
| Th1 | 77% | Ly ++ Eos + GC - |
++ | + | + no Eos |
GCH +/++ PC +/++ |
| Th2 | 93% | Ly ++ Eos ++ GC + |
+++ | ++ | + no Eos |
GCH +/++ PC +/++ |
| Th17 | 100% | Ly + Eos +++ GC - |
+++ | +++ | +/++ Eos+ squamous cell hyperplasia, |
GCH -/+ PC +/++ |
Legend:parietal cell destruction (PCD) was scored as described above, with grade 5 indicating 25-50% PCD, grade 6 indicating 50-100% PCD. Cells within the stomach infiltrates, as well as fore-stomach and splenic lesions were rated either zero (-) or + to +++ for mild/moderate/high presence.
Abreviations: Eos=eosinophils, GC=macrophage giant cells, GCH=germinal center hyperplasia, Ly=lymphocytes, PC=plasmocytosis
FIGURE 2. Th1, Th2 and Th17 T effector cells induce AIG with distinct histological features.

A. Normal glandular stomach from a BALB/C nu/nu mouse. 6 weeks after transfer, Th1 (B), Th2 (C) and Th17 (D) effectors all induce highly destructive AIG (grade 5-6) characterized by epithelial hyperplasia, parietal cell destruction, and inflammatory cell infiltration. Eosinophils were rare and scattered in Th1 infiltrates (E), were present at higher numbers and forming clusters in Th2 infiltrates (E), but were most prominent in Th17-mediated AIG (F). Values for magnifications are approximate due to editing limitations.
The composition of inflammatory infiltrates differs in recipients of Th1, Th2, and Th17 cells
Inflammatory infiltrates were not only found in the gastric mucosa, but also submucosa, ranging from scattered infiltration to nodular foci of predominately CD3+ lymphocytes. Heavy submucosal infiltration was rarely seen in Th1-mediated disease, often observed in the Th2 group, and the rule in the Th17 group. Interestingly, the cellular composition of the inflammatory infiltrates in the glandular stomach, the site of parietal cells, consisted of mainly polymorphonuclear granulocytes with many eosinophils and some CD3+ lymphocytes in Th17 recipients. In Th2 recipients, the infiltrates were composed of lymphocytes, small clusters of eosinophils, and multinucleated macrophage giant cells, while in recipients of Th1 cells, the infiltrates mainly consisted of CD3+ lymphocytes, a few scattered eosinophils, but no giant cells (Table I, Fig. 2E, F). Most forestomachs in all groups showed mild to moderate signs of inflammation, squamous cell hyperplasia in all groups, and only in the Th17 group, eosinophilic infiltration. Interestingly, the spleens of all groups showed signs of increased lymphoid hyperplasia as indicated by germinal center hyperplasia and plasmacytosis (Table I).
Over time, the total number of cells in the gLN increased in all T effector groups, but with varying kinetics. The total number of cells per gLN rapidly increased in the Th17-injected group leading to a peak at 4 weeks (11.5±4.5×106 cells/gLN) that was 10-fold higher than in recipients of Th1 (1.4±5.4×106 cells/gLN) or Th2 (1.5±0.7×106 cells/gLN cells). At 6 weeks after injection, when almost all mice in all three Th groups had destructive AIG, there were still marked differences in gLN cell numbers with the highest numbers in Th17-injected mice (7.3±1.9×106), followed by the Th2 (2.7±1.2 ×106), and the Th1 recipients (1.4±0.4×106) (Fig. 3A). The increase in total cell number was paralleled by an increase in CD4+Thy1.1+ TxA23 cells. Recipients of Th17 cells had the highest numbers (Fig. 3B). However, the major cell type in the gLN in all groups consisted of CD19+ B cells, which accounted for 68-75% of cells in all groups. No major differences were observed in the frequency of CD11b+, CD11c+, or Gr-1+ cells (data not shown).
FIGURE 3.
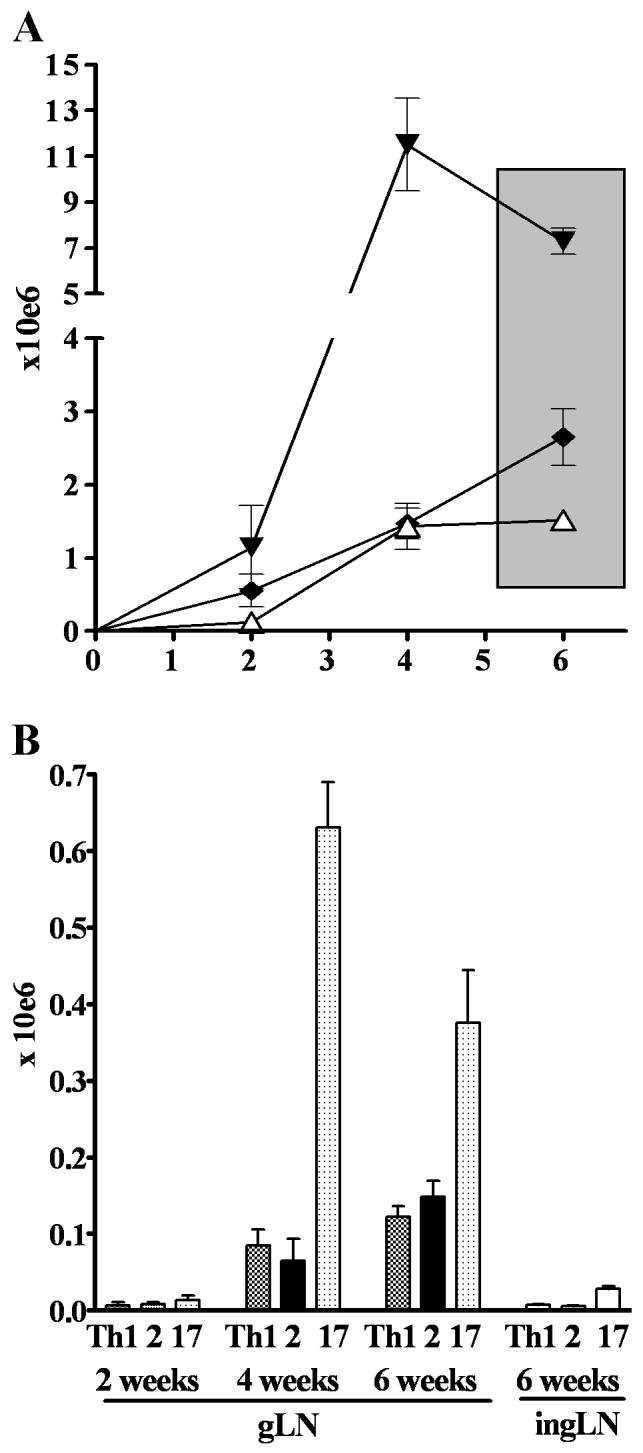
Kinetics of increase in total cell numbers (A) and CD4+Thy-1.1+ effector cells (B) in the gLN following transfer of Th1 (white triangle), Th2 (black diamond), and Th17 (black triangle) effectors. Very few effector cells could be detected in the inguinal LN.
Serum Ig levels
Total serum IgE, IgG1 and IgG2a were analyzed in several groups of recipient mice (n=20 for each group). As expected, serum IgE levels were higher in Th2-injected mice (7.6±9.6 mg/ml) than in Th1-injected mice (2.6±1.8 mg/ml), but were, surprisingly, highest in Th17-mediated disease (12.0±2.0 mg/ml). IgG2a and IgG1 levels varied widely between all experimental groups and no consistent differences were seen between groups (data not shown).
Suppression of AIG by Tregs
Significant signs of AIG were first seen four weeks after transfer in recipients of all three effector cell types. The least destructive disease was seen in Th1 recipients, while the most aggressive disease was seen in Th17 recipients. Co-transfer of polyclonal Tregs reduced the incidence of high-grade destructive gastritis and disease severity (as indicated by a reduction of the gastritis score) in each group (Fig. 4A). After 6 weeks, when most mice had developed severe AIG, Tregs prevented destructive AIG in 84% of Th1-injected and 70% of Th2-injected mice, but were not effective in suppressing disease in Th17-mediated AIG (only 9% with non-destructive disease). In addition, Treg significantly reduced disease severity in Th1- and Th2-mediated AIG, but not in Th17-injected mice (Fig. 4B). The effects of Tregs on anti-PC-Abs were also analyzed at 6 weeks after injection. Tregs reduced the percentage of anti-PC-Abs positive mice in Th1- and Th2-injected mice by more than a half, but only had minimal effects on anti-PC-Ab in Th17 recipients (data not shown).
FIGURE 4.
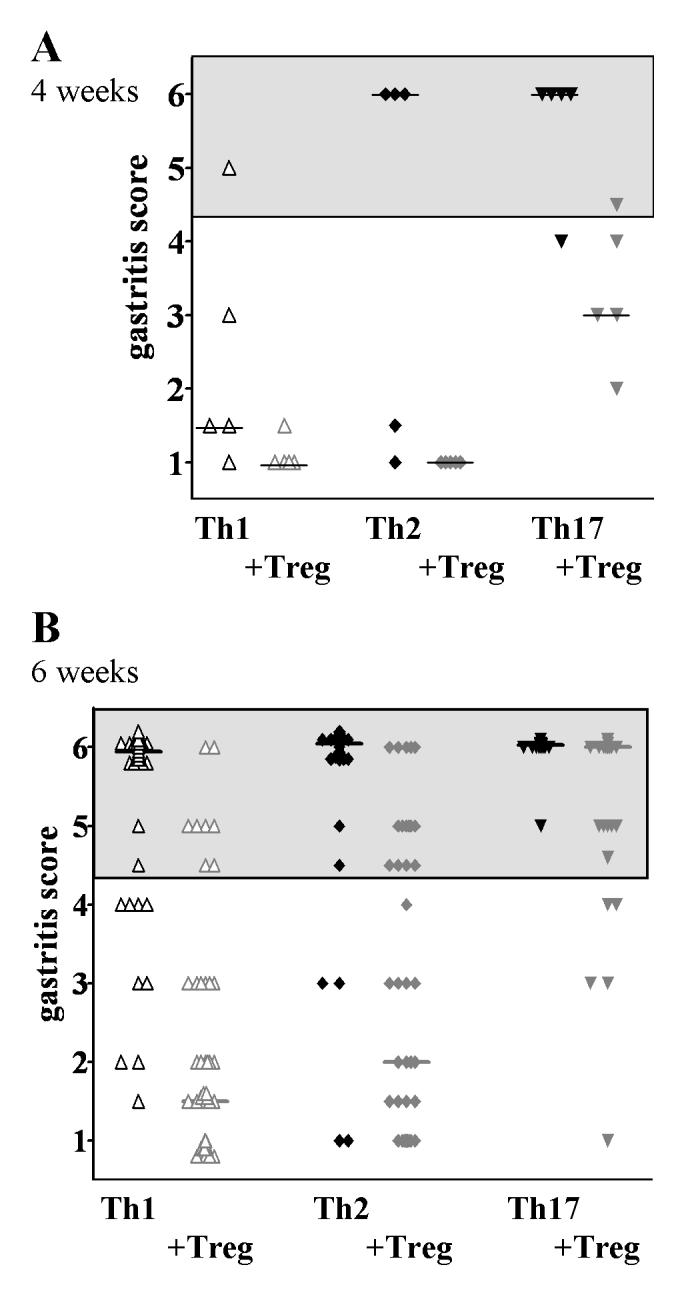
Inhibition of destructive gastritis by co-transfer of polyclonal Treg 4 weeks (A) and 6 weeks (B) after transfer. 6 weeks after transfer, Treg reduced the number of irreversibly diseased mice and significantly decreased the mean gastritis score in Th1-AIG (from 5.5±1.1 to 2.0±1.6, p<0.0001) and in Th2-AIG (from 5.7±1.1 to 2.7±1.9, p<0.0001), but had no effect on Th17-mediated disease (from 5.9±0.2 to 5.1±1.3, p=n.s.). Grey areas indicate significant destructive gastritis. Bars indicate the median gastritis score.
In order to examine whether the suppression of disease persisted over the long term, we also examined mice at 20 weeks after injection: All mice that had received effectors alone had destructive gastritis (grade 5-6). All mice in both the Th2 Treg treated and the Th17 Treg treated groups also developed high-grade destructive gastritis, while 60% of the mice in the Th1 Treg treated group were still protected from parietal cell loss (not shown).
Treg decrease total cell numbers and numbers of T effectors in the gLN
Co-transfer of Treg significantly reduced the total number of cells in the gLN and the numbers of the injected CD4+Thy1.1+ effectors both at four weeks and six weeks after transfer in recipients of Th1 and Th2 effectors (Fig. 5A, B). While the total cell number and number of CD4+Thy1.1+ effectors were also reduced in Th17-injected mice at four weeks, only a minimal, non-significant reduction was observed after six weeks (Fig. 5A, B, Table II). No effect of Treg on cell numbers could be observed in the non-draining inguinal LN (not shown).
FIGURE 5.

Co-transfer of Treg (gray bars) decreases the total cell number (A) and the numbers of CD4+Thy1.1+ T effectors (B) in the gLN. Please note the different scales for Th1, Th2, and Th17 graphs in this figure. * denotes significant differences (p<0.05 for 5A and p<0.01 for 5B, respectively) .
Table II.
Succeptibility of Teffector cells to Treg mediated suppression in vivo at 2, 4 and 6 weeks after injection
| weeks | Teffector | +Tregs | +Tregs | ||||
|---|---|---|---|---|---|---|---|
| total cells/gLN | total cells/gLN | Δ | Thy1.1+ cells/gLN | Thy1.1+ cells/gLN | Δ | ||
| 2 | Th1 | 120 | 101 | -16% | 6.35 | 0.42 | -93% |
| 2 | Th2 | 552 | 510 | -8% | 7.88 | 0.63 | -92% |
| 2 | Th17 | 1,143 | 837 | -27% | 13.33 | 5.70 | -57% |
| 4 | Th1 | 1,430 | 427 | -70% | 84.90 | 9.21 | -89% |
| 4 | Th2 | 1,540 | 564 | -63% | 64.37 | 23.52 | -63% |
| 4 | Th17 | 11,510 | 3,745 | -67% | 632.00 | 130.90 | -77% |
| 6 | Th1 | 1,450 | 736 | -49% | 122.20 | 16.55 | -86% |
| 6 | Th2 | 2,650 | 1,230 | -54% | 147.90 | 42.61 | -71% |
| 6 | Th17 | 7,290 | 5,690 | -22% | 375.80 | 246.10 | -35% |
Legend: Δ= delta (Treg-mediated reduction of cell numbers per gLN expressed in %). All other values are given as cells per gLN × 1000.
Suppression by Tregs depends on the T effector:Treg ratio
In the previous experiments, T effector (5×104) and Tregs (1×106) were injected at a ratio 1:20. To determine if a change in the T effector:Treg ratio would result in more effective prevention of AIG, we co-transferred 15,000 or 5,000 of the more resistant to Treg mediated suppression Th2 and Th17 effectors with 1×106 Tregs resulting in Teff/Treg ratios of 1:67 and 1:200, respectively. Destructive gastritis was seen in 70% of the mice following transfer of 15,000 Th2 effectors, but transfer of 5,000 Th2 effectors induced destructive AIG in only a small percentage of mice (30%). Co-transfer of Treg prevented destructive gastritis in all recipients at six weeks (Fig. 6A). Transfer of either 15,000 or 5,000 Th17 induced highly destructive gastritis in all mice. However, co-transfer of 1×106 Treg was somewhat effective in preventing AIG as 40% of the mice injected with 15,000 Th17 cells and 60% of the mice injected with 5,000 Th17 cells were protected from destructive AIG (Fig. 6B).
FIGURE 6.
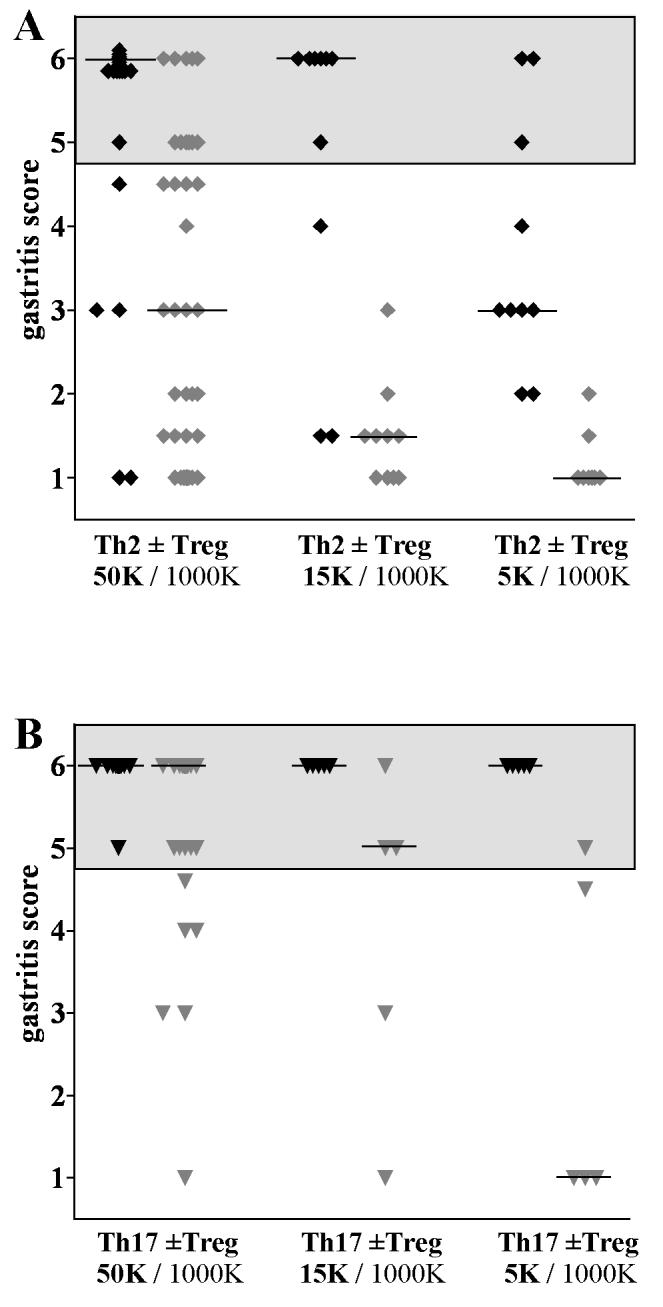
Suppression by Tregs depends on the T effector:Treg ratio. Disease scores in mice that received different numbers of Th2 (A) and Th17 (B) effectors with or without a constant number (1×106) Treg. Black symbols are mice injected with T effectors only; gray sumbols are mice co-injected with Treg. Grey areas indicate significant destructive gastritis. Bars indicate the median gastritis score.
Effector T cells recovered from animals protected by Treg remain functional in vitro
Although Treg induced significant suppression of effector cell expansion and destructive AIG following co-transfer of Th1 or Th2 cells, significant numbers of effector T cells could still be detected in protected mice. In order to determine if exposure to Treg in vivo modified effector T cell function, cells were recovered from the gLN 6 weeks after transfer and 5,000 CD4+Thy1.1+ T effectors were re-challenged with their cognate peptide presented by irradiated APCs. Th1, Th2, and Th17 effectors proliferated after re-stimulation with no differences between cells from animals that received effectors alone or effectors and Treg (Fig. 7A). Co-transfer of Treg also had no effect on the capacity of the effector cells to produce cytokines after stimulation with PMA and Ionomycin, but changes in the pattern of cytokine secretion were observed. A high percentage (53%) of effectors from the Th1 recipients remained IFNγ-producers, but a similar percentage (56%) of IFNγ-producers was seen in Th2 recipients accompanied by a reduction in IL-4 producers (8%) compared to 41% in the starting population. In the Th17 groups, 18% of the effector T cells were IL-17 producers after 6 weeks, but 22% also produced IFNγ (Fig. 7B), in contrast to very low percentages of IFNγ producers in the starting population. Cells derived from both recipients of Th2 and Th17 cells produced low, but similar, levels of IL-5 indicating that the enhanced eosinophilic infiltrates in the Th17 recipients were not secondary to higher levels of IL-5 production by Th17 cells (Fig. 7C).
FIGURE 7.

Proliferative responses to peptide and APC (A) and cytokine production by T effectors following PMA/ionomycin restimulation (B) or by cytometric bead assay of supernatants following APC and peptide restimulation (C) of recovered from the gLN 6 weeks after transfer without (filled bars) or with Treg (open bars).
Discussion
CD4+Foxp3+ Tregs play critical roles in the prevention of organ-specific autoimmune diseases. We have previously demonstrated that co-transfer of polyclonal Treg can prevent AIG induced by transfer of naive TCR Tg T cells from the TxA23 mouse strain whose TCR is specific for a peptide derived from the α-chain of the H/K ATPase (14). In subsequent studies, we have also demonstrated that stimulation of naïve TxA23 cells in the presence of TGFβ results in the conversion of these potential effector T cells to antigen-specific Foxp3+ Treg that are potent inhibitors of AIG induced by transferred naïve TxA23 cells (22). Most studies of Treg function in models of organ-specific autoimmune diseases have focused on prevention of disease induced by naïve T cells rather than on treatment of ongoing disease or prevention of disease induced by fully differentiated effector T cells. The goals of the present studies were to compare the capacity of fully differentiated Th1, Th2, Th17 cells derived from the A23 TCR Tg mouse to induce AIG upon transfer to nu/nu recipients and to determine the susceptibility of these fully differentiated effector T cell populations to control by co-transfer of polyclonal Treg derived from normal BALB/c mice.
Although TxA23 mice spontaneously develop AIG characterized by a predominant Th1 pattern of cytokine secretion, we were readily able to induce differentiation of thymocytes from the TxA23 mouse to Th1, Th2 and Th17 cells lines. After transfer, all 3 effector T cells populations preferentially accumulated in the gLN and the gastric mucosa. Several studies have shown that in vitro differentiated Th1 and Th2 effector cells are capable of promoting organ-specific autoimmune disease (23). Th1-mediated inflammatory infiltrates consisted mainly of CD3+ T lymphocytes, whereas Th2-mediated infiltrates also contained a large number of granulocytes, primarily eosinophils, and were associated with elevated serum-IgE levels. We now show that all three types of T effector cells induced destructive AIG in the vast majority of recipients, but with different histological patterns independent of disease severity. As expected, Th1-mediated infiltrates mainly consisted of CD3+ lymphocytes and Th2-mediated infiltrates had a higher number of granulocytes and eosinophils. Surprisingly, the highest number of infiltrating polymorphonuclear leukocytes including a very high percentage of eosinophils was observed in the Th17-injected mice. Disease induced by Th17 cells seemed to be the most aggressive type (24), since destructive AIG was seen in all animals after 4 weeks and only 5,000 Th17 effectors could induce destructive AIG in 100% of recipients. This enhanced aggressiveness of the disease induced by Th17 cells may be secondary to their increased proliferative responses in vivo or perhaps to enhanced homing to the target organ. It also remains possible that the expansion of the Th17 cells in vivo and their resistance to suppression by Treg are in part mediated by environmental factors in the gastric mucosa or in the gastric lymph node such as high levels of IL-23 production by the gastric mucosa.
Previous studies of the composition of cellular infiltrates associated with Th17 cells have demonstrated that they were composed of granulocytes, predominantly neutrophils (25, 26). It is unclear if the predominantly eosinophilic infiltration observed in our experiments is a unique characteristic Th17-induced AIG or if studies of other Th17-associated diseases overlooked the presence of eosinophils, as they are hard to distinguish from neutrophils without special staining. It is also possible that the protocol we used to generate Th17 cell lines, which included the use of anti-IL-2, generated the production of other members of the IL-17 family in addition to IL-17A (27), such as IL-17E that stimulates eotaxin production and thus eosinophilic infiltration. Alternatively, IL-17A itself is able to trigger eotaxin release by other cells in some circumstances, as shown for smooth airway muscle cells (28).
It is also of interest that the cell lines used to induce AIG exhibited variable patterns of stability after transfer and exposure to autoantigen in vivo. All of the lines exhibited the appropriate restricted pattern of cytokine expression when tested prior to transfer. Th1 cells for the most part maintained their phenotype when isolated from animals 6 weeks after transfer, while cells isolated from Th2 recipients contained a significant number of IFNγ producing cells after 6 weeks. It is possible that the IFNγ producing cells were derived from non-committed precursors in the starting population that responded to environmental signals in this AIG model that seems to favor Th1 differentiation (14). Alternatively, it remains possible that some plasticity exists in the committed Th2 population that facilitates conversion to Th1 cells upon receipt of environmental cues as has been shown in certain infectious disease models (29). A significant number of the recovered cells from the Th17 injected mice continued to produce IL-17, but also a similar percentage of IFNγ producing cells was recovered; interestingly, as examined in a limited study, many of these cells were IL-17/IFNγ double producers (data not shown). Since a significant number of IFNγ producing cells are seen in almost all purported Th17-mediated diseases (30), it remains possible that the IFNγ producing cells in the Th17 recipients represent Th17 cells that have further differentiated to produce both IL-17 and IFNγ. The lack of stability in cytokine profile over 6 weeks in vivo had little effect on the striking differences in the inflammatory infiltrates or on the susceptibility of the animals to suppression by Treg.
In our previous studies, we demonstrated that the induction of AIG by naïve TXA23 CD4+ thymocytes could be readily suppressed by polyclonal Treg from normal BALB/c mice (9). We used the same Treg:Teffector ratios in the present study and showed that polyclonal Treg could markedly suppress the capacity of Th1 cells, moderately suppress Th2 cells, but could only suppress Th17-induced AIG at early time points. We and others have also shown that antigen-specific Treg are much more potent inhibitors of the induction of organ-specific autoimmunity (22, 31) and have proposed that antigen-specific Treg prevent disease by inhibiting T cell activation and expansion by acting on autoantigen-presenting dendritic cell (DC) (22, 32-35). It remains unclear how polyclonal Treg exert their inhibitory effects. Our previous studies with naïve T cells demonstrated that they failed to inhibit the initial effector cell expansion in the draining LN, but did have effects on Th1 differentiation. In sharp contrast, in the present studies, significant inhibition of the effector cell expansion was seen when polyclonal Treg were co-transferred with Th1 and Th2 effectors and the inhibition of expansion was an excellent correlate of protection from destructive AIG. If one assumes that Treg mediated suppression is independent of the type of effector T cells present, it is possible that Th17 effectors, and to a lesser extent Th2 effectors, are able to overcome suppression by proliferating to a greater extent than Th1 effectors and potentially overcome suppression by outnumbering the Tregs for access to DC.
There are several important differences between our previous studies with naïve effectors and the differentiated effector cells used here. Naïve effectors could easily be detected in the gLN as early as 3 days after transfer and significant expansion could be demonstrated by day 5 (14). In contrast, the fully differentiated effector cells could hardly be detected in the gLN after one week (not shown). This result is not surprising as the effector populations expressed only low levels of CD62L that is required for entry into lymph nodes. It remains possible that the effector cells were first stimulated by their target antigen in the stomach and then acquired the ability of traffic to the gLN (36). The co-transferred Treg express high levels of CD62L and could easily be detected in all lymph nodes at early time points after transfer (14 and data not shown). It remains conceivable that a small percentage of the polyclonal Treg population expressing receptors for antigens derived from the target organ were retained in the gLN, could expand, and then be prepared to exert inhibitory effects on DC resulting in inhibition of the expansion of the susceptible effector populations upon their arrival in the gLN. A primary effect of Treg on T cell expansion is also consistent with our data, demonstrating that the effector cells that can be isolated from the gLN of protected animals are not anergic and are fully competent to proliferate and produce effector cytokines ex vivo. This result is also consistent with studies in the 3 day thymectomy model of oophoritis in which the continuous presence of Treg was required for protection from disease (37). Taken together, these findings are most consistent with a model where Tregs in vivo are not shutting off effector cytokine by T cells directly, but are regulating their expansion by acting on APC.
Cellular biotherapy with Treg is now being considered as a potential therapy for organ-specific autoimmunity. While most studies in experimental animal models suggest that antigen-specific Treg act in bystander fashion and are the most potent inhibitors of disease, it may be very difficult to isolate antigen-specific Foxp3+ T cells from patients. The present studies demonstrating a strong inhibitory effect of polyclonal Treg on the capacity of some types of differentiated effector cells to induce disease provide an experimental basis for the clinical use of polyclonal Treg in human disease. It remains to be determined if the mechanism of action of the polyclonal Treg is similar or different from those proposed for the antigen-specific Treg.
Acknowledgements
We thank Dr. J. Milner for advise and help with the ELISA, Sarah Tanksley for cell sorting, and the team of the animal facility for helping maintain our mouse colony.
Abbreviations used in this paper
- AIG
(autoimmune gastritis)
- DC
(dendritic cell)
- IgE
(immunoglobulion E)
- TCR
(T cell receptor)
- Th
(T helper lymphocytes)
- Treg
(CD4+CD25+FoxP3+ regulatory T cells)
Footnotes
This work was supported funds from the intramural program of the NIAID and by a Max Kade Foundation Postdoctoral Research Exchange Grant (to G.H.S).
Disclosures
The authors have no financial conflict of interest.
References
- 1.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu.Rev.Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 2.McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27:17–23. doi: 10.1016/j.it.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 3.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat.Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 4.Strober W, Ludviksson BR, Fuss IJ. The pathogenesis of mucosal inflammation in murine models of inflammatory bowel disease and Crohn disease. Ann.Intern.Med. 1998;128:848–856. doi: 10.7326/0003-4819-128-10-199805150-00009. [DOI] [PubMed] [Google Scholar]
- 5.Umetsu DT, McIntire JJ, Akbari O, Macaubas C, DeKruyff RH. Asthma: an epidemic of dysregulated immunity. Nat.Immunol. 2002;3:715–720. doi: 10.1038/ni0802-715. [DOI] [PubMed] [Google Scholar]
- 6.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J.Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 7.Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin.Exp.Immunol. 2007 doi: 10.1111/j.1365-2249.2007.03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suri-Payer E, Amar AZ, McHugh R, Natarajan K, Margulies DH, Shevach EM. Post-thymectomy autoimmune gastritis: fine specificity and pathogenicity of anti-H/K ATPase-reactive T cells. Eur.J.Immunol. 1999;29:669–677. doi: 10.1002/(SICI)1521-4141(199902)29:02<669::AID-IMMU669>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 9.Callaghan JM, Khan MA, Alderuccio F, van Driel IR, Gleeson PA, Toh BH. Alpha and beta subunits of the gastric H+/K(+)-ATPase are concordantly targeted by parietal cell autoantibodies associated with autoimmune gastritis. Autoimmunity. 1993;16:289–295. doi: 10.3109/08916939309014648. [DOI] [PubMed] [Google Scholar]
- 10.Karlsson FA, Burman P, Loof L, Mardh S. Major parietal cell antigen in autoimmune gastritis with pernicious anemia is the acid-producing H+,K+-adenosine triphosphatase of the stomach. J.Clin.Invest. 1988;81:475–479. doi: 10.1172/JCI113344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gleeson PA, Toh BH. Molecular targets in pernicious anaemia. Immunol.Today. 1991;12:233–238. doi: 10.1016/0167-5699(91)90036-S. [DOI] [PubMed] [Google Scholar]
- 12.D’Elios MM, Bergman MP, Azzurri A, Amedei A, Benagiano M, De Pont JJ, Cianchi F, Vandenbroucke-Grauls CM, Romagnani S, Appelmelk BJ, Del Prete G. H(+),K(+)-atpase (proton pump) is the target autoantigen of Th1-type cytotoxic T cells in autoimmune gastritis. Gastroenterology. 2001;120:377–386. doi: 10.1053/gast.2001.21187. [DOI] [PubMed] [Google Scholar]
- 13.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J.Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- 14.DiPaolo RJ, Glass DD, Bijwaard KE, Shevach EM. CD4+CD25+ T cells prevent the development of organ-specific autoimmune disease by inhibiting the differentiation of autoreactive effector T cells. J.Immunol. 2005;175:7135–7142. doi: 10.4049/jimmunol.175.11.7135. [DOI] [PubMed] [Google Scholar]
- 15.McHugh RS, Shevach EM, Margulies DH, Natarajan K. A T cell receptor transgenic model of severe, spontaneous organ-specific autoimmunity. Eur.J.Immunol. 2001;31:2094–2103. doi: 10.1002/1521-4141(200107)31:7<2094::aid-immu2094>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 16.Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, Killeen N, Urban JF, Jr., Guo L, Paul WE. Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat.Immunol. 2004;5:1157–1165. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 17.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’shea JJ. Interleukin-2 Signaling via STAT5 Constrains T Helper 17 Cell Generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Suri-Payer E, Cantor H. Differential cytokine requirements for regulation of autoimmune gastritis and colitis by CD4(+)CD25(+) T cells. J.Autoimmun. 2001;16:115–123. doi: 10.1006/jaut.2000.0473. [DOI] [PubMed] [Google Scholar]
- 19.Alderuccio F, Toh BH, Gleeson PA, van Driel IR. A novel method for isolating mononuclear cells from the stomachs of mice with experimental autoimmune gastritis. Autoimmunity. 1995;21:215–221. doi: 10.3109/08916939509008018. [DOI] [PubMed] [Google Scholar]
- 20.Milner JD, Ward JM, Keane-Myers A, Paul WE. Lymphopenic mice reconstituted with limited repertoire T cells develop severe, multiorgan, Th2-associated inflammatory disease. Proc.Natl.Acad.Sci.U.S.A. 2007;104:576–581. doi: 10.1073/pnas.0610289104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alderuccio F, Sentry JW, Marshall AC, Biondo M, Toh BH. Animal models of human disease: experimental autoimmune gastritis--a model for autoimmune gastritis and pernicious anemia. Clin.Immunol. 2002;102:48–58. doi: 10.1006/clim.2001.5134. [DOI] [PubMed] [Google Scholar]
- 22.DiPaolo RJ, Brinster C, Davidson TS, Andersson J, Glass D, Shevach EM. Autoantigen-specific TGFbeta-induced Foxp3+ regulatory T cells prevent autoimmunity by inhibiting dendritic cells from activating autoreactive T cells. J.Immunol. 2007;179:4685–4693. doi: 10.4049/jimmunol.179.7.4685. [DOI] [PubMed] [Google Scholar]
- 23.Lafaille JJ, Keere FV, Hsu AL, Baron JL, Haas W, Raine CS, Tonegawa S. Myelin basic protein-specific T helper 2 (Th2) cells cause experimental autoimmune encephalomyelitis in immunodeficient hosts rather than protect them from the disease. J.Exp.Med. 1997;186:307–312. doi: 10.1084/jem.186.2.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bettelli E, Korn T, Kuchroo VK. Th17: the third member of the effector T cell trilogy. Curr.Opin.Immunol. 2007 doi: 10.1016/j.coi.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiseter BS, Miller GT, Happ MP, Kasaian MT. Treatment of murine experimental autoimmune encephalomyelitis with a myelin basic protein peptide analog alters the cellular composition of leukocytes infiltrating the cerebrospinal fluid. J.Neuroimmunol. 1998;91:156–170. doi: 10.1016/s0165-5728(98)00171-4. [DOI] [PubMed] [Google Scholar]
- 26.Stamp LK, James MJ, Cleland LG. Interleukin-17: the missing link between T-cell accumulation and effector cell actions in rheumatoid arthritis? Immunol.Cell Biol. 2004;82:1–9. doi: 10.1111/j.1440-1711.2004.01212.x. [DOI] [PubMed] [Google Scholar]
- 27.Kawaguchi M, Adachi M, Oda N, Kokubu F, Huang SK. IL-17 cytokine family. J.Allergy Clin.Immunol. 2004;114:1265–1273. doi: 10.1016/j.jaci.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 28.Rahman MS, Yamasaki A, Yang J, Shan L, Halayko AJ, Gounni AS. IL-17A induces eotaxin-1/CC chemokine ligand 11 expression in human airway smooth muscle cells: role of MAPK (Erk1/2, JNK, and p38) pathways. J.Immunol. 2006;177:4064–4071. doi: 10.4049/jimmunol.177.6.4064. [DOI] [PubMed] [Google Scholar]
- 29.Krawczyk CM, Shen H, Pearce EJ. Functional plasticity in memory T helper cell responses. J.Immunol. 2007;178:4080–4088. doi: 10.4049/jimmunol.178.7.4080. [DOI] [PubMed] [Google Scholar]
- 30.Suryani S, Sutton I. An interferon-gamma-producing Th1 subset is the major source of IL-17 in experimental autoimmune encephalitis. J.Neuroimmunol. 2007;183:96–103. doi: 10.1016/j.jneuroim.2006.11.023. [DOI] [PubMed] [Google Scholar]
- 31.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J.Exp.Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang Q, Adams JY, Tooley AJ, Bi M, Fife BT, Serra P, Santamaria P, Locksley RM, Krummel MF, Bluestone JA. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat.Immunol. 2006;7:83–92. doi: 10.1038/ni1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur.J.Immunol. 2000;30:1538–1543. doi: 10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 34.Misra N, Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Cutting edge: human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J.Immunol. 2004;172:4676–4680. doi: 10.4049/jimmunol.172.8.4676. [DOI] [PubMed] [Google Scholar]
- 35.Veldhoen M, Moncrieffe H, Hocking RJ, Atkins CJ, Stockinger B. Modulation of dendritic cell function by naive and regulatory CD4+ T cells. J.Immunol. 2006;176:6202–6210. doi: 10.4049/jimmunol.176.10.6202. [DOI] [PubMed] [Google Scholar]
- 36.Guarda G, Hons M, Soriano SF, Huang AY, Polley R, Martin-Fontecha A, Stein JV, Germain RN, Lanzavecchia A, Sallusto F. L-selectin-negative CCR7-effector and memory CD8+ T cells enter reactive lymph nodes and kill dendritic cells. Nat.Immunol. 2007;8:743–752. doi: 10.1038/ni1469. [DOI] [PubMed] [Google Scholar]
- 37.Samy ET, Parker LA, Sharp CP, Tung KS. Continuous control of autoimmune disease by antigen-dependent polyclonal CD4+CD25+ regulatory T cells in the regional lymph node. J.Exp.Med. 2005;202:771–781. doi: 10.1084/jem.20041033. [DOI] [PMC free article] [PubMed] [Google Scholar]