

Abstract
Signal transducers and activators of transcription (STATs) were originally discovered as components of signal transduction pathways. Persistent aberrant activation of STAT3 is a feature of many malignancies including prostate cancer and pancreatic cancer. One consequence of persistently-activated STAT3 in malignant cells is that they depend upon it for survival, thus STAT3 is an excellent molecular target for therapy. Previously we reported that single-stranded oligonucleotides containing consensus STAT3 binding sequences (13410 and 13411) were more effective for inducing apoptosis in prostate cancer cells than antisense STAT3 oligonucleotides. Control oligonucleotides (scrambled sequences) had no effect. Here we report that authentic STAT3 binding sequences, identified from published literature, were more effective for inducing apoptosis in prostate cancer cells and pancreatic cancer cells than was oligonucleotide 13410. Moreover, the authentic STAT3 binding sequences showed differing efficacies in the malignant cell lines depending upon whether the canonical STAT3 binding sequence was truncated at the 5′ or the 3′ end. Finally, expression of one STAT3-regulated gene was decreased following treatment, suggesting that STAT3 may regulate the same set of genes in the two types of cancer. We conclude that truncating the 5′ end left intact enough of the canonical STAT3 binding site for effective hybridization to the genome, whereas truncation of the 3′ end, which is outside the canonical binding site, may have affected binding of required cofactors essential for STAT3 activity, thereby reducing the capacity of this modified oligonucleotide to induce apoptosis. Additional experiments to answer this hypothesis are underway.
Keywords: STAT3, prostate cancer, pancreatic cancer
Introduction
Prostate cancer is the most diagnosed adenocarcinoma in American men, with over 186,000 cases estimated for 2008 (1). Due at least in part to its intrinsic chemoresistant nature, prostate cancer is the second leading cause of cancer death for American men, accounting for over 27,000 deaths last year and predicted to cause at least 28,000 in 2008 (1). Recently physicians documented that neither radical prostatectomy nor androgen-ablation therapy contributes to overall increased long-term survival (2, 3). The current selection of treatment options for prostate cancer patients is clearly not adequate: new therapeutic agents specifically designed to target prostate cancer are needed.
Pancreatic cancer is a deadly carcinoma, the incidence of which increases with age, and correlates with gender and race. Most cases of pancreatic cancer are ductal adenocarcinoma. Pancreatic cancer is the most lethal adenocarcinoma in America, with about 36,000 cases estimated for 2008 that will ultimately lead to about 34,000 deaths (1). Due at least in part to its intrinsic chemoresistant nature and inherent difficulties in early prognosis, pancreatic cancer is at present the fourth leading cause of cancer death for Americans (4). Conventional therapy (chemotherapy, radiation surgery, or combinations of these modalities) have little impact on the course of pancreatic cancer (4). The current selection of treatment options for pancreatic cancer patients is clearly not adequate: new therapeutic agents for pancreatic cancer are urgently needed.
Transcription factors are latent proteins that bind to the genome upon activation, either inducing or repressing gene expression. After activation, transcription factors bind to specific enhancer sequences on the genome upstream or near the promoter region of the gene regulated by the transcription factor. Signal transducers and activators of transcription (STAT) are part of the signal transduction pathway of many growth factors and cytokines and are activated by phosphorylation of tyrosine and serine residues by upstream kinases (5). For example, signaling by IL-6 generally induces phosphorylation of STAT3 (5). In benign cells, the signaling by STAT3 is under tight regulation, so that the signal is transient. However, aberrant signaling by STAT3 is found in many types of malignancies: multiple myeloma, head and neck cancer, breast cancer, prostate cancer, etc. (6–12). Malignant cells expressing persistently activated STAT3 become dependent on it for survival; disruption of activation or expression of STAT3 resulted in apoptosis (12). STAT3 binds to two known sequences, HSIE and GAS (13–15), through which its antiapoptotic and oncogenic effects are directed (16, 17). These sites contain the canonical STAT3 binding motifs TTC(N)2-4GAA or TT(N)4-6AA (16, 17).
Previously we showed that prostate cancer lines were sensitive to treatment with antisense STAT3 oligonucleotides; 48 hours after treatment, most of the cells were apoptotic, but few cells treated with sense oligonucleotide were affected (12). However, antisense as a therapeutic category has shown nonspecific activation of the innate immune system, primarily through unmethylated CpG motifs (18). Therefore, we created a new strategy for inhibiting STAT3 activity. Because STAT3 binds to discrete, known sequences on the genome, we synthesized two novel anticancer oligonucleotides based on the STAT3 consensus binding sequence, called 13410 and 13411 (19). We reported that both complementary strands of the binding sequence of STAT3 induced apoptosis in prostate cancer cells in vitro and furthermore reduced mean tumor volumes in vivo (19). In continuing this line of work, we compared the efficacy of oligonucleotide 13410 to that of several authentic STAT3 binding sequences we obtained from the published literature. We found that two published human STAT3 binding sequences induced significant apoptosis in prostate cancer and pancreatic cancer cell lines. Furthermore, the two STAT3 binding sequences induced apoptosis differentially in both types of cancer, depending upon whether we truncated the sequences at the 3′ end or the 5′ end. We think our data reveal that genomic sequences past the 3′ end of the canonical STAT3 binding sequence may contain co-factor binding sites necessary for STAT3 activity. Furthermore our data show that the 5′ end of the STAT3 canonical binding site may not be necessary for complete STAT3 activity.
Materials and Methods
Oligonucleotides
Oligonucleotides were synthesized using phosphorothiorate chemistry by the Molecular Resources Facility at University of Medicine and Dentistry-New Jersey Medical School (Newark, NJ). The ribose moieties for 5 bp at both 5′ and 3′ ends were modified with 2′-O-methoxyl groups to increase stability of the oligonucleotides and to provide higher hybridization affinity (20, 21). For determinations of transfection efficiencies, a fluorescent oligonucleotide (13778a; Table 1) labeled with FITC was synthesized and included in every transfection experiment. Sequences of oligonucleotides used are given in Table 1. Two of the oligonucleotides, B and D, which spanned the 3′ end of the STAT3 consensus binding sequence, were modified to span the 5′ end of the consensus binding sequence (Sequences BB and DD).
Table 1.
Putative STAT3 Inhibiting Oligonucleotide Sequences
| Name | Sequence | Reference |
|---|---|---|
| 13410a | 5′-TCCCGTAAATCCCTA-3′ | (1); truncated 13410 |
| Aftable | 5′-TCCCCGTAAATCCCT-3′ | (2) |
| B | 5′-TCCGAGAAATCCCTA-3′ | (3) |
| BB | 5′-TTCCGAGAAATCCCT-3′ | (3) |
| C | 5′-TCTGGGAATTCCTAG-3′ | (5) |
| D | 5′-TCTGGGAAATCCCTA-3′ | (6) |
| DD | 5′-TTCTGGGAAATCCCT-3′ | (6) |
Legend: The sources and sequences for the oligonucleotides used in the experiments are listed.
Cells
DU145 cells were the gift of Dr. James Turkson (University of South Florida, Tampa, FL). They were grown in either DMEM/Ham’s F12 (Invitrogen, Carlsbad, CA) plus 10% newborn bovine serum (Hyclone, Logan, UT). LNCaP cells were obtained from the ATCC (Manassas, VA); they were maintained in RPMI 1640 (Invitrogen) plus fetal bovine serum (Hyclone). PANC-1 cells were the gift of Dr. James Freeman, University of Texas Health Sciences Center, San Antonio (22, 23) and were grown in the same medium as DU-145 cells. Primary human dermal fibroblasts were obtained from LifeLine Cell Technology (Walkersville MD) and grown in their proprietary medium. Cell viabilities were determined using fluorescein diacetate (Sigma Chemical Co., St. Louis, MO) and a Universal RIII fluorescence microscope (Zeiss, Jena, Germany).
Transfection of Oligonucleotides into Cells
LipofectAMINE 2000 transfection reagent (Invitrogen) was used to transfect oligonucleotides into the prostate cancer lines as described previously (19). Briefly, cells plated in six-well plates were grown to 50% confluence. Oligonucleotides were diluted in Opti-MEM I (Invitrogen) appropriately. Next, LipofectAMINE 2000 was diluted in Opti-MEM I (2 μL LipofectAMINE 2000 to 250 μL Opti-MEM I per well). The diluted LipofectAMINE 2000 was allowed to incubate for 5 minutes at room temperature; next, 250 μL of it per well were mixed with 250 μL of diluted oligonucleotide. The liposome-oligonucleotide mixture remained at room temperature for 20 minutes before the 500 μL of liposome-oligonucleotide mixture were added to each well of cells. Cells were incubated with the mixture for 6 hrs at 37°C; then 1.5 mL of medium per well containing 30% serum was added to each well. Additional medium containing 10% serum was added each of the following days until the experiment was ended.
Apoptosis Determinations
FITC-Annexin V and propidium iodide staining (Abcam, Malvern PA; Sigma, St. Louis MO) were used to measure the induction of apoptosis by oligonucleotides after incubation for 24. Harvested cells were washed twice in buffer; then, 5 × 10 5 cells in 1 mL of buffer, containing at least 40 mmol/L Ca 2+, were put into each tube (Falcon Plastics, Franklin Lakes, NJ). Five μL of FITC-Annexin V and either propidium iodide or 7-amino-actinomycin D (7-AAD; Sigma Chemicals, St. Louis, MO) were put into each tube as well. Fluorescence was quantified using a FACScan flow cytometer (Becton Dickinson, San Jose, CA) on at least 10,000 events using CellQuest Pro software (Becton Dickinson) and an Apple Macintosh G4 dual coprocessor computer running OS X 10.3.9 (Apple, Inc., Cupertino CA). The JC-1 dye assay was used for measuring the reduction in mitochondrial transmembrane potential during apoptosis (24, 25). Briefly, 10 6 cells in 1 mL were stained with 1 μL of JC-1 (Molecular Probes, Eugene, OR) at 1 mg/mL in DMSO, according to manufacturer’s instructions at various times following treatment with active oligonucleotides or control oligonucleotides. After incubating for 15 min at 37°C, cells were analyzed for decreased orange fluorescence by flow cytometry.
CD46 Expression Assay
DU-145 or PANC-1 cells were transfected with oligonucleotides as described above. 24 hr later, cells were harvested using trypsin, washed, then stained with FITC-anti-human CD46 or matching isotype control (Pharmingen) for 1 hr using 1 μL of Ab per 106 cells. After thoroughly washing the cells, fluorescence was measured using a FACScan flow cytometer.
Statistical Analysis
The graphing program Kaleidagraph 4.1 (Synergy Software, Reading, PA) and the statistical program InStat3 (GraphPad Software, San Diego, CA) were used for data analyses.
Results
Two Anti-STAT3 Sequences Induced Apoptosis in Pancreatic and Prostate Cancer Cell Lines
Our original STAT3-inhibiting oligonucleotide, 13410, was a phosphorothiorated 24-mer that was 2′-O-methoxylated at the 5 bases at each end (19). Because we are actively exploring the use of peptide nucleic acids (PNAs) having STAT3 binding sequences as potential anti-cancer therapy, we truncated the sequence of 13410 to a 15-mer called 13410a. The reason it is necessary to truncate oligonucleotide sequences in this way is that the synthesis of PNAs becomes very inefficient when the sequence is longer than 20 bases. Therefore, one of the original goals of these studies was to ascertain which sequence or sequences in oligonucleotide form (oligonucleotides being much easier to synthesize than PNAs) might be superior to 13410a for inducing apoptosis in cancer cell lines. Therefore, all oligonucleotides used in these studies were 15-mers.
Figure 1 shows the comparison of the concentration response curves for the 15-mer oligonucleotides in DU-145 cells; 13778a induced very little apoptosis at 1000 nM, whereas 13410a and Sequence D had IC50’s of approximately 40 and 580 nM, respectively. Extending these studies to PANC-1 cells, we found that 500 nM of 13410a induced apoptosis in approximately 43% of cells; however the same concentration of Sequence B induced more than 60% apoptosis (Figure 2). We observed that 13778a induced only 18% apoptosis at 1000 nM, whereas 13410a and Sequence B induced 40% and 65%, respectively (Figure 2A). The IC50 for Sequence B on PANC-1 cells was calculated to be 427 nM, whereas the IC50 of Sequence D was calculated to be 57 nM (Figure 2B). Sequence B, like Sequence D, has the same sequence at the 3′ end as 13410a, but differs from both Sequence D and 13410a at the 5′ end. To examine the importance of the 5′ end of the STAT3 binding site, we synthesized oligonucleotides BB and DD, which retained the consensus STAT3 binding sequence on the 5′ ends, but were truncated at the 3′ ends. We found that 500 nM of either Sequence BB or Sequence DD induced much less apoptosis than the corresponding oligonucleotides B and D. Sequence BB induced only 17% apoptosis and Sequence DD induced only 18.5% apoptosis, compared to 51% for Sequence B and 73% for Sequence D at 500 nM (Figure 2). We conclude from these data that integrity of the oligonucleotide sequence extending beyond the 3′ end of the canonical STAT3 binding sequence must be very important, if not required, for STAT3 activity.
Figure 1.
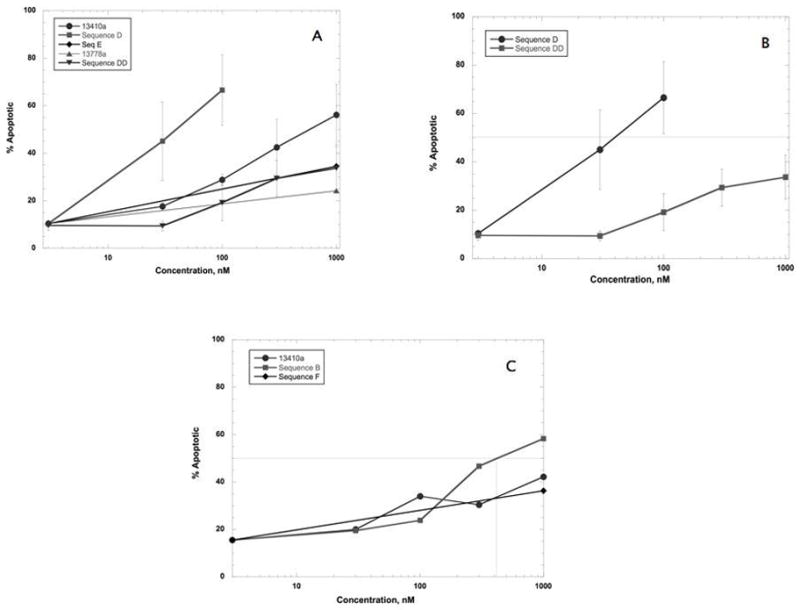
Effect of STAT3-inhibiting oligonucleotides on DU-145 cells. DU145 cells were transfected with oligonucleotides at the concentrations indicated. 48 hrs later, cells were harvested, washed, and stained with FITC-annexin V and PI. Percent apoptosis was quantified by subtracting the % viable population from the total. The concentration of oligonucleotide that induced 50% apoptosis (IC50,) is noted where appropriate. IC50 values were obtained using the Identify tool in the Kaleidagraph software package. The average data from 3 experiments ± SD are shown. A: Comparison of active sequences 13410a and Seq D to control sequences E and 13778a. IC50 values were calculated using the Identify tool from the Kaleidagraph software package. B: Comparison of Sequences D to DD, illustrating the importance of the purines at the 3′ end on STAT3-inhibiting efficacy.
Figure 2.
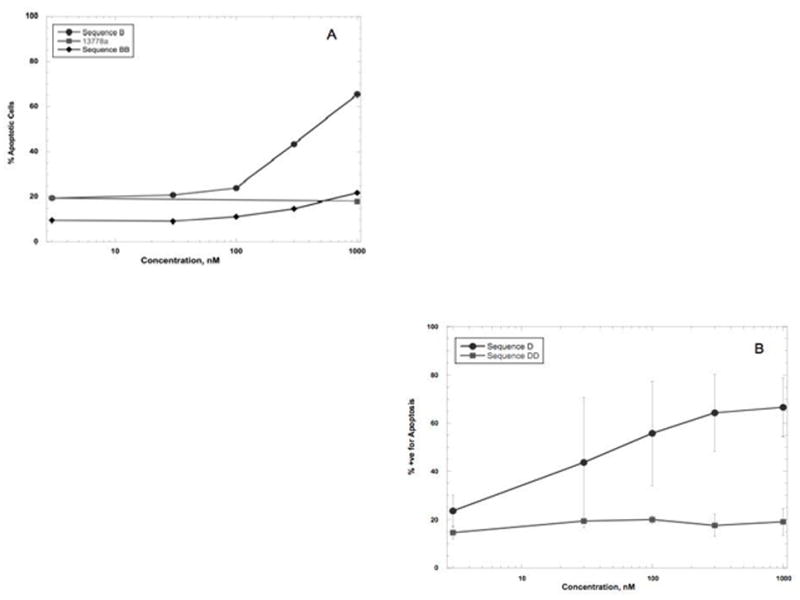
Effect of STAT3-inhibiting oligonucleotides on PANC-1 cells. PANC-1 cells were transfected with oligonucleotides at the concentrations indicated. 48 hrs later, cells were harvested, washed, and stained as in the legend to Figure 1. Percent apoptosis was quantified by subtracting the % viable population from the total. The concentration of oligonucleotide that induced 50% apoptosis (IC50) is noted where appropriate. The average data from 3 experiments ± SD are shown. A: Comparison of effect of Sequences B to BB on induction of apoptosis. Sequence B has an IC50 of aproximately 270 nM, while Sequence BB has an IC50 calculated to be greater than 4400 nM. C: Effect of Seq D on PANC-1 cells.
To confirm the results presented above, we used the JC-1 dye to measure the inhibition of mitochondrial transmembrane potential (12). As shown in Figure 3 Panels A and B, the orange fluorescence of aggregated (oxidized) JC-1 by DU-145 cells was significantly decreased by 80% in the presence of 500 nM Sequence D or by 30% in the presence of 500 nM of Sequence B. Figure 3 Panels C and D shows the results in PANC-1 cells. The orange fluorescence of aggregated JC-1 was significantly decreased by nearly 45% in the presence of 500 nM Sequence D or by 30% in the presence of 500 nM of Sequence B. Scrambled sequence oligonucleotide 13778a had no effect on JC-1 oxidation even at 1000 nM (Figure 3). Thus, by a measurement of the physiology of apoptosis, we noted that Sequence D was more potent than Sequence B in the 2 types of cancer cell lines used.
Figure 3.
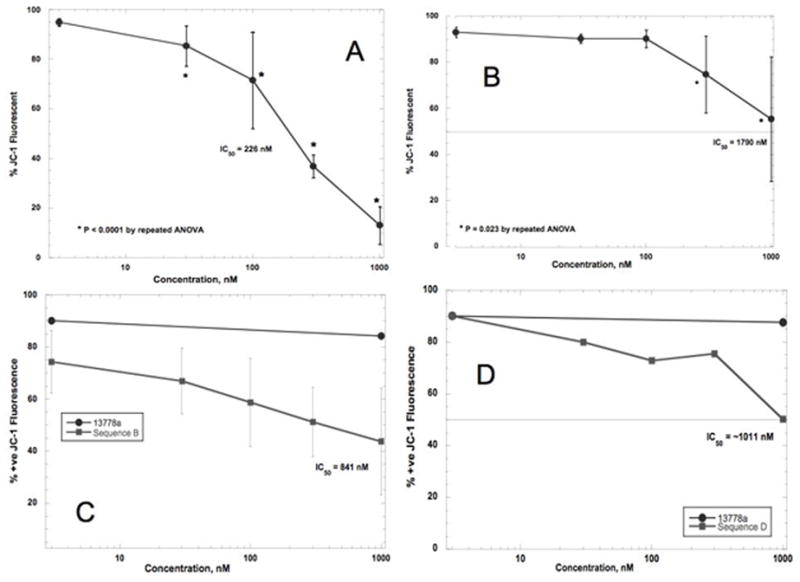
Effect of STAT3-inhibiting oligonucleotides on JC-1 oxidation. A: Effect of Seq D on DU-145 cells. Using JC-1, the IC50 was calculated to be about 226 nM. Differences among treated cells and untreated cells were highly significant (P = 0.0001 by repeated ANOVA). B: Effect of Seq B on DU145 cells. The IC50 was calculated to be about 1790 nM. Differences among treated cells and untreated cells were less significant than for Seq D but still significant (P = 0.023 by repeated ANOVA). C: Effect of Seq B on PANC-1 cells. The IC50 was calculated to be about 841 nM. Differences among values were very significant by repeated ANOVA (P = 0.0058). D: Effect of Seq D on PANC-1 cells. The IC50 was calculated to be about 1011 nM; the differences among values were not significant (P = 0.1225 by repeated ANOVA).
Anti-STAT3 Oligonucleotides Did Not Induce Apoptosis in Primary Cells
We postulated that targeting STAT3 for cancer treatment would spare benign cells the deleterious effects of most cancer treatments. To test this hypothesis, we previously published data indicating that benign immortalized prostate cell lines (BPH-1 and NRP-152) were not killed by oligonucleotide 13410 (12). But benign immortalized cells, because they contain viral sequences, are not the same as the normal cells of the body (24, 26–28). Therefore, to examine the potential effect of STAT3-inhibiting oligonucleotides on bystander cells in the body, we used primary human dermal fibroblasts from LifeLine Cell Technology for the following series of experiments. Fibroblasts were transfected with oligonucleotides; apoptosis was determined by annexin V/PI staining and flow cytometry 48 hr later. The results shown in Table 2 indicate that concentrations of 13410a or Sequence D that induced significant apoptosis in DU-145 or PANC-1 cells induced no apoptosis in human dermal fibroblasts. These data indicate that the oligonucleotides have specificity for the targeted binding sequence of STAT3, and do not kill bystander cells indiscriminiately.
Table 2.
Anti-STAT3 Oligonucleotides Did Not Induce Apoptosis in Normal Human Fibroblasts
| ON | % Viable | ON | % Viable |
|---|---|---|---|
| none | 92.9 ± 3.3 | Sequence B | 90.8 ± 3.5 |
| 13410a | 92 ± 3 | Sequence BB | 90.5 ± 2.1 |
| 13778a | 89.6 ± 2.1 | Sequence D | 89.8 ± 0.3 |
| Sequence DD | 91.8 ± 0.7 |
Legend: Oligonucleotides were made as described in Materials and Methods. Oligonucleotides were transfected at 500 nM into normal human dermal fibroblasts cells using Lipfectamine 2000; apoptosis was determined at 48 hr by flow cytometry, after staining with FITC-annexin V and PI. Data are given as averages ± SD.
Anti-STAT3 Oligonucleotides Inhibited Expression of a STAT3-Regulated Gene, CD46
STAT3 is a known regulator of several anti-apoptotic and cell cycling genes. Recently it was shown to regulate the expression of CD46, a cell surface protein expressed by many tumors that helps tumor cells escape complement-mediated cytolysis (25, 29). Therefore, we followed up on this observation to see if CD46 was inhibited by the STAT3 inhibitors when given to PANC-1 and DU-145 cells. We found that our anti-STAT3 oligonucleotides inhibited CD46 expression; the results are presented in Figure 4. Figures 4A and C shows that approximately 90% of untreated DU-145 cells expressed CD46. Treatment with 13410 significantly reduced expression by about 75% (Figure 4B) whereas treatment with 13778 had no effect on CD46 expression. Transfection with the truncated form of 13410, 13410a, decreased CD46 expression from 91% to 45%, while Sequence D inhibited CD46 expression on DU-145 cells by 50% (Figure 4D).
Figure 4.
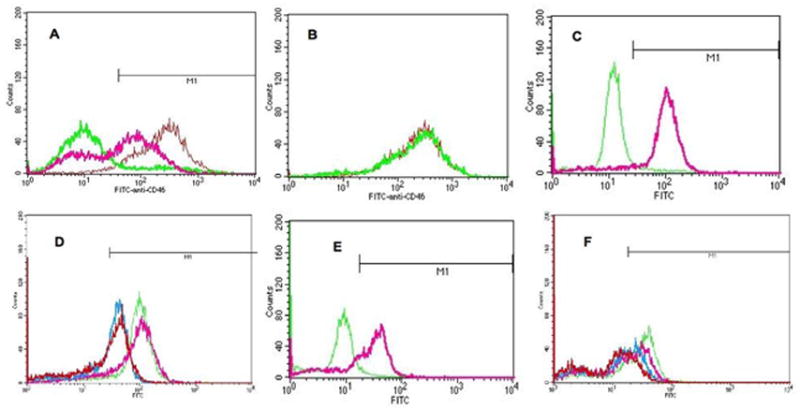
Effect of STAT3-inhibiting oligonucleotides on STAT3-regulated CD46 expression. Oliogonucleotides were transfected as described in Materials and Methods. 24 hr later, cells were harvested, then stained with FITC-anti-human CD46 or FITC-isotype control (both, Pharmingen). Fluorescence was quantified on a FACScan flow cytometer. A: 24 hr after transfection with 13410 (thick pink line), approximately 75% of the DU-145 cells stained less intensely with anti-CD 46, and 22% of cells did not stain at all with anti-CD46. Red line = DU-145 cells treated with 13778; green line = isotype control. Approximately 90% of DU-145 cells stained positive for CD46 in these experiments (region defined by M1). B: Untreated (Red line) and 13778-treated (green line) DU-145 cells stained with anti-CD46 Ab exhibited the same fluorescence intensity. C: green line, untreated DU-145 cells; pink line, 13778a; blue line, 13410a inhibited CD46 by approximately 45%; red line, Sequence D inhibited CD46 by approximately 50%. D: Effect of 13410a and Sequence D on CD46 expression in PANC-1 cells. Green line, untreated cells; pink, 13778a; blue, 13410a inhibited CD46 by approximately 40%; red, Sequence D inhibited CD46 expression by approximately 50%.
Similar results were found when PANC-1 cells were transfected with 13410a or Sequence D. PANC-1 cells expressed CD46 at somewhat lower levels than did DU-145 cells levels; approximately 67% of untreated PANC-1 cells expressed CD46 (Figure 4E). Treatment with either 13410a or Sequence D reduced expression of CD46 (Figure 4F) from 67% to 35% and 33%, respectively. Once again, 13778a had no effect on CD46 expression (Figure 4F). We concluded from these data that our STAT3-inhibiting oligonucleotides also inhibited STAT3-regulated genes other than those involved in apoptosis.
Discussion
STAT3 inhibitors merit extensive study in the context of cancer therapy because they have the potential to induce apoptosis selectively in cancer cells, sparing benign cells. Our previous data showed that a novel STAT3 inhibitor, 13410, spared benign immortalized prostate cells and hormone-sensitive prostate cancer cells while inducing apoptosis in hormone-refractory prostate cancer cells (19). Here we extended those studies to evaluate the effect of related novel STAT3 inhibitors on prostate cancer cells and on benign primary cells. We observed that our novel STAT3 inhibitors did not induce apotosis in in primary fibroblasts (Table 2). We expect based on these data that bystander cells would not be affected adversely by anti-STAT3 treatment, thus this treatment modality offers a tremendous advantage to the patient over conventional anti-cancer chemotherapy.
Furthermore, we found that altering the sequence of 13410 by truncating it to the 15-mer 13410a decreased its efficacy somewhat, but that using an inhibitor with the sequence of an authentic human STAT3 binding sequence, Sequence D, increased the efficacy (Figures 1 and 2). The IC50 of 13410a was 410 nM, but that of Sequence D was 40 nM, making Sequence D 10-fold more potent for induction of apoptosis in DU-145 cells. At this juncture, we have no explanation for why the IC50 of Sequence D measured by inhibition of JC-1 oxidation was observed to be 226 nM (Figure 3) other than to postulate that the 2 assays, annexin V binding/PI uptake and JC-1 oxidation, are fundamentally different enough that perhaps the only correlation one ought to expect is rank-order potency, which is what we observed.
Moreover we demonstrated that this set of novel STAT3 inhibitors decreased the expression of the STAT3-regulated gene product, CD46. CD46 is a cell-surface glycoprotein, one of those involved in protection of tumor cells against complement-mediated cytotoxicity. Recent work by Buettner and colleagues demonstrated that CD46 is regulated by STAT3 (29). We use CD46 as a STAT3 target gene because of the ease with which is is detected and quantified. We found that at least 90% of DU-145 cells expressed CD46, but that the proportion of cells expressing CD46 decreased significantly by about 50% or more when the cells were transfected with 13410, 13410a, or Sequence D (Figure 4A-D). Similar results were observed for PANC-1 cells, which normally express CD46 somewhat less than DU-145 cells (about 67% of PANC-1 cells expressed CD46; Figure 4E). When transfected with 13410a or Sequence D, the level of expression was decreased by about 50% (Figure 4F).
The most striking aspect of our data is the huge difference in efficacy observed when anti-STAT3 sequences were altered at the 3′ end. Apparently the 5′ T of the canonical STAT3 binding sequence is not as important for anti-STAT3 activity as those bases beyond the 3′ end of the canonical STAT3 binding sequence (Figure 1). In comparing Sequences D to DD and B to BB, we observed a more than 10-fold difference in IC50 between the pairs of oligonucleotides. We hypothesize that the sequences extending beyond the 3′ end of the STAT3 binding sites may be binding sites for cofactors required for STAT3 activity. Among the known STAT3 cofactors are CBP/p300 (30, 31), NcoA/SRC1a (32), MITF (33), cellular oncogenes such as c-jun and c-fos (34), and undoubtedly more besides the ones named. STAT1:STAT2 heterodimers are thought to recruit coactivators through divergent sequences outside of their canonical binding sequences (35); there is no a priori reason to suppose that STAT3 cofactor binding might similarly be affected by sequence changes fairly close to the STAT3 binding site in situ. Experiments addressing this hypothesis are planned for the future.
We are using oligonucleotides to test for optimum STAT3-inhibiting sequence, but in practice, we intend to develop a PNA STAT3-inhibitor. We believe PNAs are better because coupled to the correct cell-penetrating peptide, they offer more complete in vivo uptake than do oligonucleotides; they form stable triple helices, which are essential for inhibiting STAT3: genome binding; and their stability means little degradation (36–39). Thus we are creating a test PNA for proof-of-principle, to ascertain whether a PNA bearing Sequence D, the most potent sequence we’ve identified to date, could induce apoptosis in tumor cells. These data give us the rationale to continue to study STAT3 binding sequences as the basis for experimental inhibitors and to synthesize PNAs for testing in the future in clinical trials.
Acknowledgments
The authors acknowledge the efforts of the New Jersey Medical School Molecular Resources Facility, and in particular the help of Dr. Robert Donnelly.
This work was supported by NIH grant CA 121782 and a Research & Development Merit Award from the Department of Veterans Affairs (BEB), as well as NIH grant AI142529 (VP).
Abbreviations
- STAT
signal transducers and activators of transcription
- 7-AAD
7-amino actinomycin D
- Ab
antibody
- DMSO
dimethyl sulfoxide
- FITC
fluorescein isothiocyanate
- PNA
peptide nucleic acid
Footnotes
The authors declare no conflicts of interest.
References Cited
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57(1):43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Holmberg L, Bill-Axelson A, Helgesen F, et al. A randomized trial comparing radical prostatectomy with watchful waiting in early prostate cancer. N Engl J Med. 2002;347(11):781–9. doi: 10.1056/NEJMoa012794. [DOI] [PubMed] [Google Scholar]
- 3.Cooperberg MR, Grossfeld GD, Lubeck DP, Carroll PR. National practice patterns and time trends in androgen ablation for localized prostate cancer. J Natl Cancer Inst. 2003;95(13):981–9. doi: 10.1093/jnci/95.13.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie K, Wei D, Huang S. Transcriptional anti-angiogenesis therapy of human pancreatic cancer. Cytokine Growth Factor Rev. 2006 doi: 10.1016/j.cytogfr.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Ihle JN. STATs and MAPKs: obligate or opportunistic partners in signaling. BioEssays. 1996;18(2):95–8. doi: 10.1002/bies.950180204. [DOI] [PubMed] [Google Scholar]
- 6.Grandis JR, Drenning SD, Chakraborty A, et al. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J Clin Invest. 1998;102(7):1385–92. doi: 10.1172/JCI3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catlett-Falcone R, Landowski TH, Oshiro MM, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–15. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 8.Barton BE, Murphy TF, Adem P, Watson RA, Irwin RJ, Huang HS. IL-6 signaling by STAT3 participates in the change from hyperplasia to neoplasia in NRP-152 and NRP-154 rat prostatic epithelial cells. BMC Cancer. 2001;1:19. doi: 10.1186/1471-2407-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Epling-Burnette PK, Liu JH, Catlett-Falcone R, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest. 2001;107(3):351–62. doi: 10.1172/JCI9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Bokhoven A, Varella-Garcia M, Korch C, et al. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57(3):205–25. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 11.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–54. [PubMed] [Google Scholar]
- 12.Barton BE, Karras JG, Murphy TF, Barton AB, Huang HF. STAT3 Activation in Prostate Cancer: Direct STAT3 Inhibition Induces Apoptosis in Prostate Cancer Lines. Mol Cancer Ther. 2004;3(1):11–20. [PubMed] [Google Scholar]
- 13.Seidel H, Milocco L, Lamb P, Darnell JJ, Stein R, Rosen J. Spacing of palindromic half sites as a determinant of selective STAT (signal transducers and activators of transcription) DNA binding and transcriptional activity. Proc Natl Acad Sci USA. 1995;92(7):3041–5. doi: 10.1073/pnas.92.7.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong M, Henriksen MA, Takeuchi K, et al. Implications of an antiparallel dimeric structure of nonphosphorylated STAT1 for the activation-inactivation cycle. Proc Natl Acad Sci U S A. 2005 doi: 10.1073/pnas.0501063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong Z, Wen Z, Darnell JE., Jr Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264(5155):95–8. doi: 10.1126/science.8140422. [DOI] [PubMed] [Google Scholar]
- 16.Darnell JE., Jr Validating Stat3 in cancer therapy. Nat Med. 2005;11(6):595–6. doi: 10.1038/nm0605-595. [DOI] [PubMed] [Google Scholar]
- 17.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98 doi: 10.1016/s0092-8674(00)81959-5. (3) 295-3-3. [DOI] [PubMed] [Google Scholar]
- 18.Gursel I, Gursel M, Yamada H, Ishii KJ, Takeshita F, Klinman DM. Repetitive elements in mammalian telomeres suppress bacterial DNA-induced immune activation. J Immunol. 2003;171:1393–400. doi: 10.4049/jimmunol.171.3.1393. [DOI] [PubMed] [Google Scholar]
- 19.Barton BE, Murphy TF, Shu P, Huang HF, Meyenhofen M, Barton AB. Novel Single-Stranded Oligonucleotides that Inhibit STAT3 Induce Apoptosis In Vitro and In Vivo in Prostate Cancer Cell Lines. Mol Cancer Ther. 2004;3(10):1183–91. [PubMed] [Google Scholar]
- 20.Guvakova MA, Yakubov LA, Vlodavesky I, Tonkinson JL, Stein CA. Phosphothiorate oligodeoxynucleotides bind to basic fibroblast growth factor, inhibit its binding to cell surface receptors, and remove it from low affinity binding sites on extracellular matrix. J Biol Chem. 1995;270:2620–7. doi: 10.1074/jbc.270.6.2620. [DOI] [PubMed] [Google Scholar]
- 21.Goethe JW, Bittova M, Vogel JU, Kotchekov R, Doerr HW, Cinati J., Jr Antisense oligonucleotide ISIS 2922 targets IE-expression and prevents HCMV-IE-induced suppression of TSP-1 and TSP-2 expression Nucleosides Nucleotides. Nucleic Acids. 2001;20(4–7):1425–8. doi: 10.1081/NCN-100002569. [DOI] [PubMed] [Google Scholar]
- 22.Venkatasubbarao K, Choudary A, Freeman JW. Farnesyl transferase inhibitor (R115777)-induced inhibition of STAT3(Tyr705) phosphorylation in human pancreatic cancer cell lines require extracellular signal-regulated kinases. Cancer Res. 2005;65(7):2861–71. doi: 10.1158/0008-5472.CAN-04-2396. [DOI] [PubMed] [Google Scholar]
- 23.DeArmond D, Brattain MG, Jessup JM, et al. Autocrine-mediated ErbB-2 kinase activation of STAT3 is required for growth factor independence of pancreatic cancer cell lines. Oncogene. 2003;22(49):7781–95. doi: 10.1038/sj.onc.1206966. [DOI] [PubMed] [Google Scholar]
- 24.Hayward SW, Wang Y, Cao M, et al. Malignant transformation in a nontumorigenic prostatic epithelial cell line. Cancer Res. 2001;61:8135–42. [PubMed] [Google Scholar]
- 25.Rushmere NK, Knowlden JM, Gee JMW, et al. Analysis of the level of mRNA expression of the membrane regulators of complement, CD59, CD55, and CD46, in breast cancer. Int J Cancer. 2004;108:930–6. doi: 10.1002/ijc.11606. [DOI] [PubMed] [Google Scholar]
- 26.Danielpour D. Transdifferentiation of NRP-152 rat prostatic basal epithelial cells toward a luminal phenotype: regulation by glucocorticoid, insulin-like growth factor-I and transforming growth factor-beta. J Cell Science. 1998;112:169–79. doi: 10.1242/jcs.112.2.169. [DOI] [PubMed] [Google Scholar]
- 27.Lucia MS, Sporn MB, Roberts AB, Stewart LV, Danielpour D. The role of transforming growth factor-β1, -β2, -β3 in androgen-responsive growth of NRP-152 rat prostatic epithelial cells. J Cell Physiol. 1998;175:184–92. doi: 10.1002/(SICI)1097-4652(199805)175:2<184::AID-JCP8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Sudlivsky D, Zhang B, et al. A human prostatic epithelial model of hormonal carcinogenesis. Cancer Res. 2001;61:6064–71. [PubMed] [Google Scholar]
- 29.Buettner R, Huang M, Gritsko T, et al. Activated Signal Transducers and Activators of Transcription 3 Signaling Induces CD46 Expression and Protects Human Cancer Cells from Complement-Dependent Cytotoxicity. Mol Cancer Res %R 101158/1541-7786MCR-06-0352. 2007;5(8):823–32. doi: 10.1158/1541-7786.MCR-06-0352. [DOI] [PubMed] [Google Scholar]
- 30.Culig Z, Comuzzi B, Steiner H, Bartsch G, Hobisch A. Expression and function of androgen receptor coactivators in prostate cancer. J Steroid Biochem Mol Biol. 2004;92(4):265–71. doi: 10.1016/j.jsbmb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Wallner L, Dai J, Escara-Wilke J, et al. Inhibition of Interleukin-6 with CNTO328, an Anti-Interleukin-6 Monoclonal Antibody, Inhibits Conversion of Androgen-Dependent Prostate Cancer to an Androgen-Independent Phenotype in Orchiectomized Mice. Cancer Res. 2006;66(6):3087–95. doi: 10.1158/0008-5472.CAN-05-3447. [DOI] [PubMed] [Google Scholar]
- 32.Barre B, Avril S, Coqueret O. Opposite Regulation of Myc and p21waf1 Transcription by STAT3 Proteins. J Biol Chem %R 101074/jbcM210422200. 2003;278(5):2990–6. doi: 10.1074/jbc.M210422200. [DOI] [PubMed] [Google Scholar]
- 33.Joo A, Aburuatani H, Morii E, Iba H, Yoshimura A. STAT3 and MITF cooperatively induce cellular transformation through upregulation of c-fos expression. Oncogene. 2004;23:726–34. doi: 10.1038/sj.onc.1207174. [DOI] [PubMed] [Google Scholar]
- 34.Levy DE, Lee C-k. What does Stat3 do? J Clin Invest. 2002;109(9):1143–8. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulson M, Pisharody S, Pan L, Guadagno S, Mui AL, Levy DE. Stat protein transactivation domains recruit p300/CBP through widely divergent sequences. J Biol Chem. 1999;274(36):25343–9. doi: 10.1074/jbc.274.36.25343. [DOI] [PubMed] [Google Scholar]
- 36.Koppelhus U, Nielsen PE. Cellular delivery of peptide nucleic acid (PNA) Adv Drug Delivery Rev. 2003;55:267–80. doi: 10.1016/s0169-409x(02)00182-5. [DOI] [PubMed] [Google Scholar]
- 37.Nielsen PE. Addressing the challenges of cellular delivery and bioavailability of peptide nucleic acids (PNA) Quarterly Rev Biophys. 2006;38(4):345–50. doi: 10.1017/S0033583506004148. [DOI] [PubMed] [Google Scholar]
- 38.Mae M, Langel U. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr Opin Pharmacol. 2006;6(5):509–14. doi: 10.1016/j.coph.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Zhilina ZV, Ziemba AJ, Nielsen PE, Ebbinghaus SW. PNA-nitrogen mustard conjugates are effective suppressors of HER-2/neu and biological tools for recognition of PNA/DNA interactions. Bioconjug Chem. 2006;17(1):214–22. doi: 10.1021/bc0502964. [DOI] [PubMed] [Google Scholar]