

Abstract
Alpha toxin (AT) is the major virulence factor of Clostridium septicum that is a proteolytically activated pore-forming toxin belonging to the aerolysin-like family of toxins. AT is predicted to be a three-domain molecule based on functional and sequence similarity with aerolysin, for which the crystal structure has been solved. In the present study we have substituted the entire primary structure of AT with alanine or cysteine in order to identify those amino acids that comprise functional domains involved in receptor binding, oligomerization and pore formation. These studies revealed that receptor binding is restricted to domain 1 of the AT structure, whereas domains 1 and 3 are involved in oligomerization. These studies also revealed the presence of a putative functional region of AT proximal to the receptor-binding domain, but distal from the pore-forming domain that is proposed to regulate the insertion of the transmembrane β-hairpin of the prepore oligomer.
Clostridium septicum is a major cause of non-traumatic gas gangrene in humans, a fulminant form of myonecrosis that can progress to a fatal infection in less than 24 hours with reported mortality rates ranging from 67–100% (1–4). Of its many virulence factors, alpha toxin (AT) is the only lethal factor secreted by this organism (5) and it has recently been shown to be absolutely required for virulence of C. septicum (6). AT is classified as a pore-forming toxin and is a member of the aerolysin-like family of pore-forming toxins. AT and aerolysin share a great deal of structural and sequence similarity (72%) (5), which has allowed for the development of a molecular model of AT using the previously solved crystal structure of aerolysin (Fig. 1) (7, 8). Aerolysin is a two lobed protein in which the small lobe is comprised of domain 1 (D1) and the large lobe is comprised of domains 2–4 (D2–D4, Fig. 1). The primary structure of AT exhibits similarity with the large lobe of aerolysin but it lacks the small lobe structure of about 83 amino acids. Both toxins appear to follow a similar ordered path to form pores in cell membranes (5). Following secretion, they bind to receptors on the cell membrane where they are cleaved into their active form by cell surface proteases, usually furin. Once activated, the toxins oligomerize on the cell surface into a prepore complex followed by insertion of a transmembrane β-barrel into the membrane (8).
Figure 1.
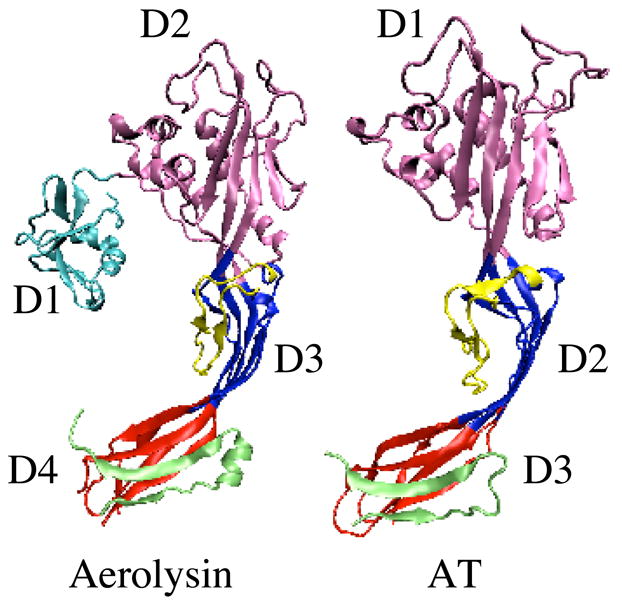
Crystal structure of aerolysin and molecular model of AT. Depicted are the α-carbon backbone structures for the aerolysin crystal structure (7) and the aerolysin-based molecular model of C. septicum AT (8). D1-D4, domains 1–4. The structures were prepared using MOLMOL (30).
AT and aerolysin bind to glycophosphatidylinositol (GPI)-anchored cell surface receptors and numerous receptors for both toxins have been identified. Aerolysin receptors include Thy-1 (CD90), contactin, the 47-kDa erythrocyte aerolysin receptor, and the variant surface glycoprotein (VSG) of Trypanosoma brucei (9, 10). AT receptors include the human folate receptor (hFR), the neuronal surface molecule contactin and Thy-1(9), as well as members of the SAG family on the surface of Toxoplamsa gondii (11). While aerolysin also binds contactin and Thy-1, it does not bind to hFR (9). In a similar manner, AT cannot bind to some identified aerolysin receptors. Thus, it appears that the two toxins bind to different sets of GPI-anchored proteins. Interestingly, the protein components of the AT- and aerolysin-binding GPI-anchored receptors lack sequence homology, however, all of the receptors identified for each toxin share a common property in their linkage to the membrane through a GPI anchor (12).
While the specific regions and residues of AT that mediate binding remain elusive, previous work suggests that residues in domains 1 and 2 of aerolysin appear to be involved in receptor binding (10, 13–15). Rossjohn et al. have shown that D1 of aerolysin contains a motif similar to the carbohydrate-binding domain of pertussis toxin, now named the aerolysin pertussis toxin domain (15). Fusion of this domain to the amino terminus of AT converts it to an aerolysin-like toxin such that it can bind to receptors previously only bound by aerolysin. Additional studies have found that the mutation of specific residues in D2 of aerolysin (D2 of aerolysin is analogous to D1 of AT, Fig. 1) affect receptor binding (10). These studies suggest that AT binding is mediated only via its D1 (D2 of aerolysin).
Prior studies using AT have identified the residues comprising two of its functional domains. The transmembrane domain (8) of AT is comprised of an amphipathic β-hairpin in D2 of the AT molecular model. Each monomer of the membrane oligomer contributes a single amphipathic β-hairpin to the formation of a transmembrane β-barrel. It has also been shown that the D3 propeptide, a short carboxy terminal peptide in D3 cleaved at the known AT activation site (R398), functions as an intramolecular chaperone that prevents premature oligomerization of AT (Fig. 1) (16). The focus of the present study was to identify the residues that comprise the other functional domains of AT. A combination of alanine and cysteine scanning of the primary structure of AT revealed residues that contribute to the formation of the receptor-binding site and that are involved in facilitating monomer-monomer interfaces. In addition, a functional region was identified in AT that we hypothesize is involved in conversion of the prepore oligomer to the inserted pore complex.
MATERIALS AND METHODS
Bacterial strains, plasmids, cell lines, and chemicals
The gene for AT was cloned into the pET-22(b)+ expression vector (Novagen, Madison, WI) (designated pBRS10) and placed into Escherichia coli BLR-DE3 cells for high-level expression as previously described (17). The SupT1 cell line was a generous gift of Dr. William Hildebrand (University of Oklahoma Health Sciences Center, Oklahoma City, OK). All chemicals were obtained from Sigma Chemical Company (St. Louis, MO) and all enzymes from Gibco BRL (Rockville, MD) unless otherwise specified.
Generation of alanine and cysteine point mutations in AT
All alanine and cysteine mutations were generated from either native AT (alanine mutants) or an active, cysteine-less derivative of AT, termed ATC86A (cysteine mutants) (8), using QuikChange site-directed mutagenesis (Stratagene). Procedures for the large-scale cysteine mutagenesis of the AT gene were carried out as previously described by us for the anthrax protective antigen (18).
Expression and purification of AT and derivatives
The growth and harvesting of E. coli BLR-DE3 expressing polyhistidine-tagged native AT and the various AT derivatives was performed according to Sellman et al. (17), with a few modifications. Lysis of the resuspended cell pellets was carried out in an EmulsiFlex-C5 high pressure homogenizer (Avestin, Ottawa, Ontario) at 15,000 p.s.i. The AT-containing fractions eluted from the cation-exchange column were pooled and placed in a Micro-ProDiCon System (Spectrum, Gardena, CA) equipped with a 10,000 MWCO membrane for simultaneous concentration and dialysis. For cysteine-substituted proteins, 1 mM dithiothreitol was included in the dialysis buffer. 10% glycerol was added to the concentrated toxin before storage at −80°C. Protein concentration was determined by absorbance at 280 nm using a molar extinction coefficient of 63,000 M−1 cm−1 (Tweten, unpublished data).
Receptor Blot Analysis
Purified hFR (64 ng) (a generous gift from Dr. Patrick Elwood, National Institutes of Health, Bethesda, MD) was separated on a 10% SDS-PAGE and blotted onto nitrocellulose in a Genie Electrophoretic blotter (Idea Scientific, Minneapolis, MN) according to manufacturer’s instructions. Following blocking overnight in PBS-Tween 20 (0.05%) containing 10% fetal calf serum, the blots were probed with 20 nM of either wild type AT or mutant toxin for 2 hours, followed by affinity-purified anti-AT antibody and a secondary antibody conjugated to horseradish peroxidase for colorimetric development using the solution 4-chloro-1-naphthol according to manufacturer’s instructions (Bio-Rad, Hercules, CA).
Cell Viability Assay
Activity of the various AT mutants on SupT1 cells was carried out as previously described (8). Samples were read at A450nm and the concentration of toxin that resulted in 50% cell death (tissue culture lethal dose 50% (TCLD50)) determined. Results from cells treated with mutant toxins were expressed as a percentage of the value obtained from cells treated with wild type AT.
Modification of AT with sulfhydryl-specific probes
ATC86A-T224C was modified with the sulfhydryl-specific derivative of the fluorescent dye 5-iodoacetamidofluoroscein (IAF) (Molecular Probes, Eugene, OR) as previously described (8). The extent of labeling was determined spectroscopically using an A492nm of 75,000−1 M−1 cm−1 for IAF (19). Protein concentration was determined by absorbance at 280 nm using a molar extinction coefficient of 63,000 M−1 cm−1.
For modification of cysteine sulfhydryls with N-ethylmaleimide (NEM) (Molecular Probes, Eugene, OR) a 20 M excess of NEM was added to 2 μg AT and brought to a total volume of 50 μl with double distilled water. The mixture was incubated at 37°C for 2 hours after which a 10 molar excess of dithiothreitol was added to inactivate any remaining NEM.
Competitive Binding Assay and Ki Determination
The ability of wild type toxin or point mutants to compete for binding with AT T224C-IAF to SupT1 cells was determined using a liquid phase binding assay as previously described by us (8), with each ligand concentration analyzed in triplicate. To determine the inhibition constants (Ki) for the competing toxin, the geometric mean fluorescence of the cells was plotted versus the concentration of competitor toxin. Competition curves were fitted from triplicate data points with GraphPad Prism software, nonlinear regression, one-site competition. Ki values were calculated using the Cheng and Prusoff equation with 95% confidence interval as described (20) with a Kd for ATT224C-IAF set at 92 nM (8).
SupT1 Membrane Preparation
Membranes from SupT1 cells were prepared as described (8). The membranes were resuspended in buffer C (50 mM HEPES, 0.5 M NaCl, pH 8.0) to an A600 of 10 and used immediately.
Activation and Oligomerization of AT on SupT1 Membranes
Activation and oligomerization of toxin in the presence of membranes was carried out as described (8). Colorimetric development of the bands was accomplished by developing the blot with the color development solution 4-chloro-1-naphthol according to manufacturer’s instructions (Bio-Rad, Hercules, CA).
RESULTS
Mutagenesis Strategy
The goal of the saturation mutagenesis of the AT molecule was to identify all functional domains of the toxin. For one of these domains, the receptor-binding domain, previous work carried out on aerolysin (10, 13–15) as well as experiments conducted in this study provided evidence to its probable location in D1. Based on this knowledge, scanning was initiated in this region and alanine was chosen for substitution due to its minimal side chain. Following the alanine mutagenesis within D1, the remainder of the molecule was substituted with cysteine. Substitutions were changed to cysteine since it could be used to substitute for alanine as well as all other amino acids in the cysteineless derivative of AT. It also allows positioning of a sulfhydryl-specific probe, if desired, at most locations within the protein structure. The previously characterized transmembrane domain was not subjected to mutagenesis in this study (8).
Mutation of Cysteine 86 in D1 Blocks Receptor-Binding Activity
Preliminary studies in our lab showed that mutation of the sole native cysteine of AT (C86) to leucine, or chemical modification of the cysteine sulfhydryl with N-ethylmaleimide (NEM), led to a complete loss of cytolytic activity (data not shown). To determine if the loss of activity by NEM modified AT (ATC86NEM) was due to a loss in receptor binding, ATC86NEM was tested for binding to purified hFR, a known receptor for AT (9). When immobilized purified hFR is probed with wild type AT (lane 1) it recognizes the 38 kDa hFR (Fig. 2A). In contrast, ATC86NEM did not recognize hFR (Fig. 2A) suggesting that the modified toxin is deficient in its receptor-binding capacity.
Figure 2.
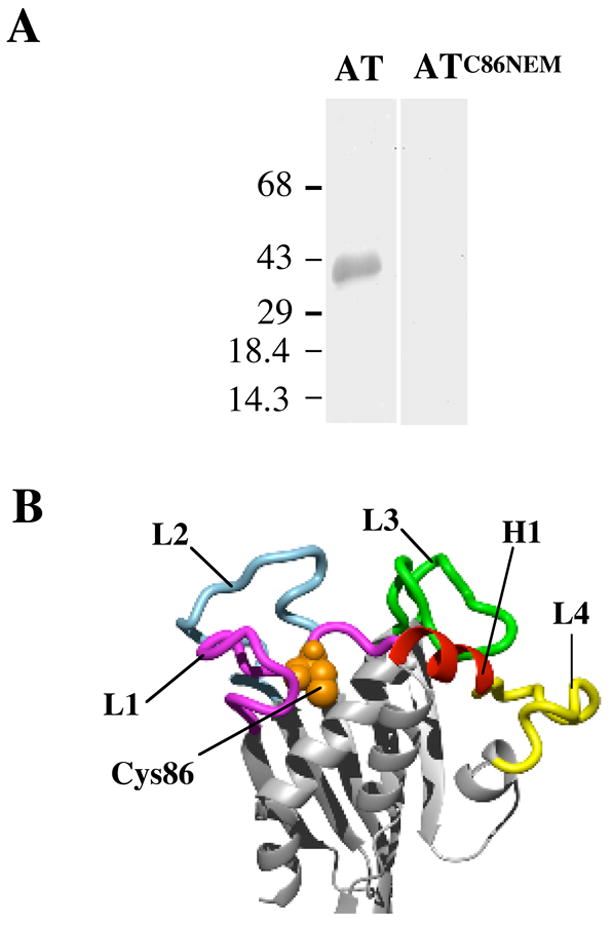
The involvement of D1 in receptor binding. (A) Modification of Cys86 with NEM abolishes binding to hFR. Purified hFR was separated on a 10% SDS-PAGE gel and blotted onto nitrocellulose. Following blocking, the blots were incubated with a solution of 20 nM wild type AT or AT that had been labeled with NEM (ATC86NEM). The blot was then incubated successively with anti-AT antibody and secondary antibody conjugated to alkaline phosphatase. The bands were visualized by the addition of a colorimetric substrate. The molecular mass of the protein markers is in kDa. (B) Molecular model of AT highlighting regions in D1 subjected to alanine scanning. The regions of D1 mutated are color-coded. L1-L4, loops 1–4. H1, helix 1. The position of Cys86 is shown in spacefill.
Identification of D1 residues that Contribute to Receptor Binding
The location of C86 in D1 of the molecular model of AT suggests that D1 contains the receptor-binding domain (RBD) of AT. The predicted secondary structure of D1 consists of five anti-parallel β-strands, seven α-helices and six loops (Fig. 2B). C86 resides on loop 1 in D1 of AT (L1, Fig. 2B). We substituted alanine for 10 residues on L1 with surface-exposed side chains or that were in the proximity to C86, 13 residues on L2, all residues of AT comprising L3 (16 residues), 4 residues on L4 and one residue on helix 6 (H6) (Fig. 2B). The effect of each mutation on lethal activity, receptor binding, proteolytic activation and oligomerization of the toxin were examined. Also, 3 of 5 tyrosine residues were mutated to threonine rather than alanine due to the instability of the alanine-substituted AT derivatives. All mutants and their location in D1 are listed numerically in Table 1.
Table 1. Relative lethal activity of alanine-substituted mutants in D1 of AT on SupT1 cells.
The tissue culture dose of each toxin that killed 50% of the cells (TCLD50) was determined for each mutant and compared to wild type AT.
| AT Mutant | Relative lethal activity (% of wild type) | AT Mutant | Relative lethal activity (% of wild type) |
|---|---|---|---|
| Loop 1 (L1) | Loop 3 (L3) | ||
|
| |||
| W74A | 0 | G289A | 4 |
| H79A | 2 | F290A | 0 |
| G82A | 0 | L291A | 0 |
| W85A | 100 | R292A | 1 |
| C86L | 0 | Y293Y | 2 |
| G87A | 17 | T294A | 100 |
| G88A | 0 | N296A | 0 |
| V94A | 3 | R298A | 3 |
| G95A | 6 | E299A | 15 |
| G97A | 2 | D300A | 2 |
| F98A | 0 | H301A | 0 |
|
|
|||
| Loop 2 (L2) | T302A | 27 | |
|
|
|||
| R117A | 13 | E303A | 39 |
| Y118T | 0 | D304A | 100 |
| N119A | 13 | R305A | 0 |
| N121A | 58 | P306A | 0 |
| D122A | 0 | ||
|
|
|||
| Y124T | 0 | Loop 4 (L4) | |
|
|
|||
| S126A | 100 | H329A | 3 |
| Y128T | 3 | Y335A | 5 |
| R129A | 2 | W338A | 1 |
| K131A | 78 | W340A | 0 |
| D132A | 3 | ||
|
|
|||
| R133A | 0 | Helix 6 (H6) | |
|
|
|||
| L134A | 3 | W342A | 0 |
Each point mutant in D1 was purified and evaluated for the ability to kill SupT1 cells, a human T lymphoblast cell line. We chose to use these cells as T cells have been shown to have a receptor for AT (9) and AT is cytolytically active on these cells. As shown in Table 1, thirty-three residues in D1 lost greater than 90% of their lethal activity on the SupT1 cells when mutated, with sixteen exhibiting no detectable lethal activity. Of those residues that lost at least 90% of the wild type lethal activity, nine are present in L1, eight in L2, eleven in L3, four in L4 and one in H6. Thus, mutants important for toxin activity are spread across the top of D1 of AT.
These mutants were initially examined for loss of receptor binding by a competition assay using flow cytometry. Changes in the relative Ki of each AT mutant were determined by adding increasing amounts of an unlabeled mutant toxin and a set amount of a fluorescently-tagged fully active form of AT to SupT1 cells. The amount of fluorescently-tagged toxin that remained bound to the SupT1 cells was assayed using flow cytometry (Figure 3). Results from these experiments were placed into PRISM software for non-linear regression, curve-fitting analysis. The graph obtained was used for Ki determination as described (20). Using this approach we were able to ascertain changes in the Ki for mutant toxins without having to introduce a probe into each mutant protein. The Ki was determined for each mutant and compared to that obtained for wild type toxin (Table 2).
Figure 3.

Competitive binding analysis of AT mutants using SupT1 cells. AT and ATH301A were added in increasing amounts (20 nM – 4000 nM) along with a constant amount of ATT224C-IAF (208 nM) to 106 SupT1 cells. Following incubation of the cells plus toxin, the cells were washed to remove non-specific binding before analysis on a FACS Calibur. Black – cells alone, Cyan – 20 nM; Orange – 100 nM; Blue – 200 nM; Green – 1000 nM; Red – 4000 nM.
Table 2. Ki values and percent binding of inactive alanine-substituted mutants in D1 of AT on SupT1 cells.
Ki values were determined from the mean fluorescence values obtained from the competitive binding experiments as shown in Figure 3. Binding of each mutant is expressed as the fold change as compared to the Ki (202.7 nM) of wild type AT (8). Ki (mu), Ki mutant AT; Ki (wt), Ki wild type AT.
| AT Mutant | Ki (nM) | Ki(mu)/Ki(wt) (fold change) | AT Mutant | Ki (nM) | Ki(mu)/Ki(wt) (fold change) |
|---|---|---|---|---|---|
| Loop 1 (L1) | Loop 3 (L3) | ||||
|
| |||||
| W74A | 5.8×104 | 2.9×102 | G289A | 1.6×103 | 8 |
| H79A | 9.8×102 | 5 | F290A | 1.3×109 | 6 ×106 |
| G82A | 2.2×105 | 1.1×103 | L291A | 1.4×104 | 6.9×101 |
| C86L | 1.6×109 | 7×106 | R292A | 1.2×103 | 6 |
| G88A | 9.4×102 | 5 | Y293T | 8.1×104 | 4.0×102 |
| V94A | 8.5×103 | 4.2×101 | N296A | 9.8×104 | 4.8×102 |
| G95A | 3.9×102 | 2 | R298A | 6.5×104 | 3.2×102 |
| G97A | 8.4×102 | 4 | D300A | 3.6×102 | 2 |
| F98A | 2.1×105 | 1×103 | H301A | 6.3×104 | 3.1×102 |
|
|
|||||
| Loop 2 (L2) | R305A | 1.3×109 | 6.4×106 | ||
|
|
|||||
| Y118T | 1.9×102 | 1 | P306A | 4.1×103 | 2×101 |
|
|
|||||
| D122A | 2.0×102 | 1 | Loop 4 (L4) | ||
|
|
|||||
| Y124T | 4.5×102 | 2 | H329A | 1.5×102 | 7×10−1 |
| Y128T | 5.8×102 | 3 | Y335A | 3.2×102 | 2 |
| R129A | 3.3×102 | 2 | W338A | 8.7×104 | 4.3×102 |
| D132A | 6.5×102 | 3 | W340A | 9.6×104 | 4.7×102 |
|
|
|||||
| R133A | 1.7×103 | 8 | Helix 1 (H1) | ||
|
|
|||||
| L134A | 2.9×102 | 1 | W342A | 1.6×109 | 8×106 |
Thirteen mutants display a 102 to 106 increase in their Ki compared to wild type AT (W74A, G82A, C86L, F98A, F290A, Y293T, N296A, R298A, H301A, R305A, W338, W340A and W342A) and three exhibit a 40–70 fold increase (V94A, L291A and Y335A). The majority of these residues are found in loops L1 and L3. An additional six mutants exhibited a 4–20 fold increase in their Ki, these are present throughout D1 and include H79A, G88A, G97A, R133A, G289A, and R292A. While L2 and L4 contained residues important for lethal activity on SupT1 cells, from the data in Table 2, the loss of activity induced by mutation of many of these residues does not appear to result from a significant change in the affinity for receptor.
Perturbations in the structure that affect folding could also be responsible for the observed loss of binding activity. Therefore, each of the mutant toxins that exhibited a change in receptor binding was examined for conformational integrity by cleavage with trypsin. Trypsin digestion is a sensitive means of detecting conformational changes in proteins (21). When digested with trypsin, all mutant toxins produced the peptide patterns similar to native AT (data not shown), suggesting that the decrease in receptor binding by these mutants is not due to folding-related defects, but rather to changes in the side chain structure.
Identification of Monomer-Monomer Contact Sites
Oligomerization of the AT monomers into pore complexes requires noncovalent interactions between specific residues of the complementary interfaces. Residues G95A, Y118T, D122A, Y124T, Y128T, R129A, D132A, L134A, D300A, H329A and Y335A exhibited a minimal change in binding to SupT1 cells, but lost ≥96% of their lethal activity, suggesting that these mutations primarily affected a step in the AT mechanism that follows receptor binding. Of these D1 residues, three appeared to be affected in oligomerization. Two mutants, H329A and Y335A, can be activated with trypsin similar to wild type toxin, but are greatly reduced in formation of SDS-resistant oligomers. Both residues are located on L4, one of the two regions of D1 that did not harbor mutations that significantly affected receptor binding. D300A does not produce membrane-bound oligomer, however, it appears to be misfolded since trypsin digestion results in a product that migrates faster than activated wild type AT (Fig. 4). Therefore, D1 contains regions that contribute to receptor binding and oligomerization of AT.
Figure 4.

Activation and oligomerization of various D1 mutants on SupT1 membranes. Purified AT or the various mutants were activated with trypsin, added to SupT1 cells and incubated at 37°C. The membranes were isolated from the toxin treated cells and the membrane proteins separated by SDS-PAGE and transferred to nitrocellulose. Monomeric and oligomeric AT were identified using anti-AT antibody.
In addition, three mutations in D3 (S178C, Y250C, and V254C) and one in D2 (G369C) were identified that retained less than 1% native lethal activity on SupT1 cells (Table 3) and did not significantly affect receptor binding (Table 3). Similar to residues H329A and Y335A in D1, the D3 cysteine mutants (S178C, Y250C, and V254C) were completely deficient in oligomerization (data not shown). These mutant proteins exhibited the same tryptic peptide pattern as for wild type toxin (data not shown), suggesting that the proteins were folded properly. The loss of the capacity to oligomerize by these mutants appears to be due to the perturbation of crucial monomer-monomer contact sites in D3 that are required for oligomerization.
Table 3. Relative lethal activity, Ki values, and percent binding of cysteine-substituted residues in D2 and D3 of AT on SupT1 cells.
Relative lethal activity was determined as outlined in Table 1. Ki values were calculated as described in Table 2.
| AT Mutant | Relative lethal activity (% of wild type) | Ki (nM) | Ki(mu)/Ki(wt) (fold change) |
|---|---|---|---|
|
Domain 2
| |||
| G369C | 0 | 414 | 2 |
|
| |||
|
Domain 3
| |||
| S178C | 0 | 167 | 0.8 |
| Y250C | 1 | 81 | 0.4 |
| V254C | 0 | 72 | 0.4 |
The G369C mutation in D2 does not significantly affect binding or oligomerization of AT (Table 3 and data not shown). This mutant also appears to be folded properly as its trypsin cleavage pattern was similar to wild type AT (data not shown). G396 is, however, located near the predicted hinge region of the AT transmembrane β-hairpin (8), suggesting that the placement of the cysteine side chain at this location may have constrained a conformational change that is necessary for its proper orientation and insertion into the membrane.
A Putative Molecular Switch that Controls the Conversion of the Prepore to Pore Complex
Mutants G95A, Y118T, D122A, Y124T, Y128T, R129A, D132A, and L134A are striking in that all of them exhibit less then 3% of the lethal activity of native AT (with most not exhibiting any detectable activity), but were mostly unaffected in receptor binding activity and were proteolytically activated and converted to the SDS-resistant membrane-bound oligomer (Fig. 4). Except for G95, all of these residues are located within the L2 loop in D1, which is juxtaposed to the regions of AT shown to comprise the receptor-binding site. These mutants can form the prepore oligomer, but apparently cannot convert the prepore to the pore complex. These observations strongly suggest that residues within the L2 loop of D1 are important in regulating the insertion of the transmembrane β-hairpin located within D2 (8).
DISCUSSION
Each residue of the primary structure of C. septicum AT, except for the previously identified transmembrane β-hairpin structure (8), was scanned using a combination of alanine and cysteine substitution in order to map out the functional domains of AT. These results show that receptor-binding function is restricted to residues within D1. This was not true of residues whose substitution affected oligomerization: mutations in both domains 1 and 3 were found to affect oligomer formation, showing that multiple contact regions exist between the monomers of AT in the pore oligomer. Interestingly, mutations in loop L2 of D1 were found to prevent the pore formation, but not the formation of the prepore oligomer (8). Hence, it appears plausible that loop L2 regulates prepore to pore conversion. The locations of these functional domains are depicted on the molecular model (Fig. 6) and primary structure (Fig. 6) of AT.
Figure 6.

Alignment between AT and aerolysin. Residues are color-coded based on functional mutation as follows: red – 0% binding; blue – 1–30% binding; dark green – oligomerization deficiency. Light green residues are those in the transmembrane domain and cyan is the site of activation. Boxes indicate conserved residues.
The GPI anchor structure, which is rich in carbohydrates, appears to be a major determinant of alpha toxin and aerolysin binding (12). Although each toxin can recognize and bind to some common GPI-anchored proteins, it appears that each can also recognize a different subset of GPI-anchored receptors (12). GPI structure can vary in eukaryotic cells (22) and it appears that differences in the carbohydrate structure of the GPI anchor plays a role in the specificity of aerolysin and AT (23). The receptor-binding domain of AT is particularly rich in aromatic residues. Of the 16 residues that were mapped to the receptor-binding site, 8 are aromatic residues (including a histidine). The presence of the aromatic residues in the binding site of AT is consistent with the observation that aromatic residues are frequently found to participate in carbohydrate binding sites of proteins, and their orientation and position can alter the carbohydrate specificity (24). Of note, 4 of the 8 aromatic residues are tryptophans, which have been shown to form stacking interactions with sugar rings in the carbohydrate binding site of various proteins (25–27). Of the four tryptophans in AT, three are conserved in aerolysin and are closely clustered within a region of D1 of AT and the analogous D2 of aerolysin. It is reasonable that variation in the orientation of these residues and the overall structure of D1 may contribute to the differing specificities of AT and aerolysin for GPI-anchored proteins that serve as receptors for each toxin (9, 28). This scenario is also consistent with the observation that differences in the carbohydrate structure of the GPI anchor are recognized by each toxin (23). In addition, there are 3 basic residues in the binding site that could form ionic interactions with the negatively charged phosphates of the GPI anchor.
Mutation of residues G95, Y118, Y124, Y128, R129, D132 and L143, located in loop L2 of D1 (Fig. 2B), abolished >96% of the lethal activity of AT (Table 1). The L2 loop is juxtaposed to those regions of AT that form the receptor-binding domain. One mutant, Y118T, is particularly interesting because it exhibits a slightly higher affinity than native AT for the receptor, remains unaffected in activation and oligomerization, but lacks lethal activity. Therefore we hypothesize that this mutant and the others in L2 affect the insertion of the distal transmembrane β-hairpin in D2. We propose that L2 interacts with the membrane surface and/or with the receptor in such a way as to trigger one or more conformational changes in the structure of AT that lead to the insertion of the transmembrane β-hairpin in D2 of AT. This scenario is similar to that observed for the unrelated pore-forming toxin intermedilysin (ILY) from the family of cholesterol-dependent cytolysins. Residues in domain 4 of ILY, which contains the receptor binding domain of ILY, also appear to control the insertion of the transmembrane hairpins (TMHs) in the distal domain 3 and lock ILY into the prepore oligomer (29). The L2 loop of AT may play a similar role in its prepore to pore transition and could represent a common mechanism by which β-barrel pore-forming toxins control the conversion of the prepore oligomer to the pore complex.
Cysteine scanning of the AT structure also identified a residue in D2 important for pore formation by AT that is not located within the membrane spanning β-hairpin of AT (8). Mutation of G369 resulted in the complete loss in lethal activity on SupT1 cells, without affecting receptor binding or oligomerization to any significant effect. Again, this residue appears to affect the insertion of the transmembrane β-hairpin and formation of the β-barrel pore. Based on the molecular model, G369 is present on a β-strand near the transmembrane domain (Fig. 5). We have previously shown that the transmembrane β-hairpin must rotate away from the D2 β-sheet in order to insert into the membrane (8). The location of G369 in D2 near the predicted hinge region for the transmembrane β-hairpin suggests that the substitution of the native glycine with a side chain-containing amino acid may interfere with its proper movement, thereby preventing the formation of a functional β-barrel.
Figure 5.
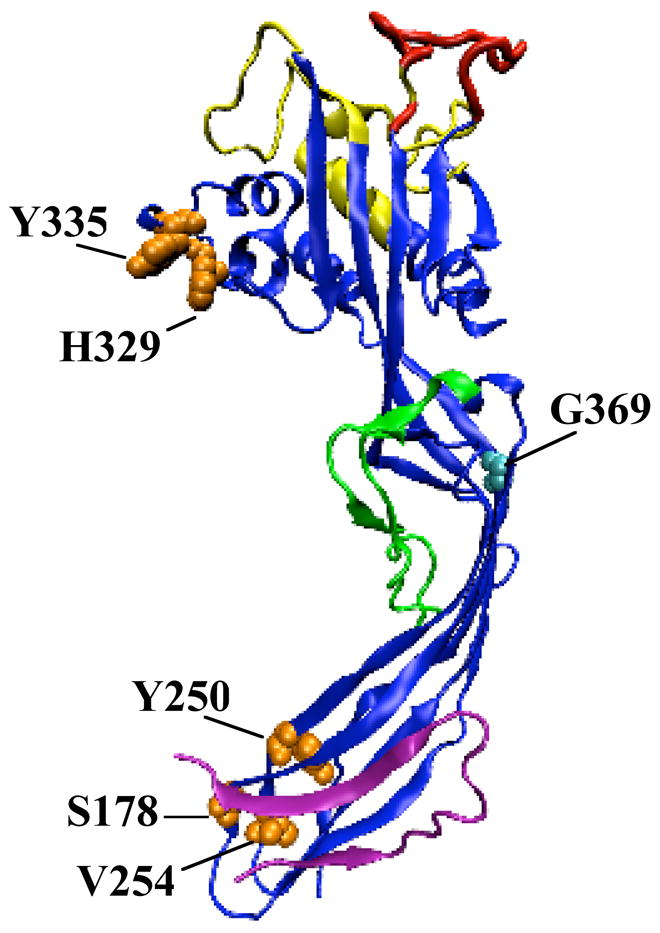
Molecular model of AT highlighting the functional domains. Color coding is as follows: receptor-binding domain - yellow; putative prepore to pore regulatory loop - red; monomer-monomer contact residues - orange (space-filled), transmembrane β-hairpin – green (8); propeptide – mauve; G369 – cyan (space-filled).
By the complete saturation mutagenesis of the primary structure of C. septicum AT we have identified the domains and specific residues that are necessary for receptor-binding and oligomerization. We have further identified residues that we hypothesize to control the prepore to pore conversion and may represent a common mechanism used by pore-forming toxins for regulating this step in their cytolytic mechanism. Taken together, these studies show that D1 is a complex, multifunctional domain that initiates binding to receptor, contains part of the monomer-monomer interface required for membrane oligomer formation, and may control prepore to pore conversion. Studies herein also demonstrated that D3 contributes residues to the monomer-monomer interface, in addition to containing the known activation site and regulating oligomerization via the propeptide. Overall, these studies provide a deeper understanding of the functional domains of AT and the aerolysin-like family of toxins and how these domains contribute to the formation of the pore.
Footnotes
This work was supported in part by National Institutes of Health Grant AI37657 (to R.K.T.).
Abbreviations are as follows: AT, alpha toxin; TMH, transmembrane β-hairpin; GPI, glycophosphatidylinositol; RBD, receptor-binding domain; D1–D4, domains 1–4; hFR, human folate receptor; NEM, n-ethylmaleimide; IAF, 5-iodoacetamidofluoroscein; ILY, intermedilysin.
References
- 1.Arenas RB, Seaman DS, McLaughlin CM, Sweeney T, Ciardiello K. Clostridium septicum myonecrosis in association with colonic malignancy [Review] Connecticut Medicine. 1988;52:709–10. [PubMed] [Google Scholar]
- 2.Cheng YT, Huang CT, Leu HS, Chen JS, Kiu MC. Central nervous system infection due to Clostridium septicum: A case report and review of the literature. Infection. 1997;25:171–174. doi: 10.1007/BF02113607. [DOI] [PubMed] [Google Scholar]
- 3.Corey EC. Nontraumatic gas gangrene: case report and review of emergency therapeutics. J Emerg Med. 1991;9:431–436. doi: 10.1016/0736-4679(91)90214-z. [DOI] [PubMed] [Google Scholar]
- 4.Stevens DL, Musher DM, Watson DA, Eddy H, Hamill RJ, Gyorkey F, Rosen H, Mader J. Spontaneous, nontraumatic gangrene due to Clostridium septicum. Rev Infect Dis. 1990;12:286–296. doi: 10.1093/clinids/12.2.286. [DOI] [PubMed] [Google Scholar]
- 5.Tweten RK, Sellman BR. Clostridium septicum Pore-Forming and Lethal Toxin. In: Alouf J, Freer J, editors. Bacterial Toxins: A Comprehensive Sourcebook. Academic Press; London: 1999. pp. 345–352. [Google Scholar]
- 6.Kennedy CL, Krejany EO, Young LF, O’Connor JR, Awad MM, Boyd RL, Emmins JJ, Lyras D, Rood JI. The alpha-toxin of Clostridium septicum is essential for virulence. Mol Microbiol. 2005;57:1357–66. doi: 10.1111/j.1365-2958.2005.04774.x. [DOI] [PubMed] [Google Scholar]
- 7.Parker MW, Buckley JT, Postma JPM, Tucker AD, Leonard K, Pattus F, Tsernoglou D. Structure of the Aeromonas toxin proaerolysin in its water-soluble and membrane-channel states. Nature. 1994;367:292–295. doi: 10.1038/367292a0. [DOI] [PubMed] [Google Scholar]
- 8.Melton JA, Parker MW, Rossjohn J, Buckley JT, Tweten RK. The identification and structure of the membrane-spanning domain of the Clostridium septicum alpha toxin. J Biol Chem. 2004;279:14315–14322. doi: 10.1074/jbc.M313758200. [DOI] [PubMed] [Google Scholar]
- 9.Gordon VM, Nelson KL, Buckley JT, Stevens VL, Tweten RK, Elwood PC, Leppla SH. Clostridium septicum alpha toxin uses glycosylphosphatidylinositol-anchored protein receptors. J Biol Chem. 1999;274:27274–27280. doi: 10.1074/jbc.274.38.27274. [DOI] [PubMed] [Google Scholar]
- 10.MacKenzie CR, Hirama T, Buckley JT. Analysis of receptor binding by the channel-forming toxin aerolysin using surface plasmon resonance. J Biol Chem. 1999;274:22604–9. doi: 10.1074/jbc.274.32.22604. [DOI] [PubMed] [Google Scholar]
- 11.Wichroski MJ, Melton JA, Donahue CG, Tweten RK, Ward GE. Clostridium septicum alpha-toxin is active against the parasitic protozoan Toxoplasma gondii and targets members of the sag family of glycosylphosphatidylinositol-anchored surface proteins. Infect Immun. 2002;70:4353–61. doi: 10.1128/IAI.70.8.4353-4361.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diep DB, Nelson KL, Raja SM, Pleshak EN, Buckley JT. Glycosylphosphatidylinositol anchors of membrane glycoproteins are binding determinants for the channel-forming toxin aerolysin. J Biol Chem. 1998;273:2355–60. doi: 10.1074/jbc.273.4.2355. [DOI] [PubMed] [Google Scholar]
- 13.Green MJ, Buckley JT. Site-directed mutagenesis of the hole-forming toxin aerolysin: studies on the roles of histidines in receptor binding and oligomerization of the monomer. Biochemistry. 1990;29:2177–80. doi: 10.1021/bi00460a031. [DOI] [PubMed] [Google Scholar]
- 14.Wilmsen HU, Buckley JT, Pattus F. Site-directed mutagenesis at histidines of aerolysin from Aeromonas hydrophila - a lipid planar bilayer study. Mol Microbiol. 1991;5:2745–2751. doi: 10.1111/j.1365-2958.1991.tb01983.x. [DOI] [PubMed] [Google Scholar]
- 15.Rossjohn J, Buckley JT, Hazes B, Murzin AG, Read RJ, Parker MW. Aerolysin and pertussis toxin share a common receptor-binding domain. EMBO J. 1997;16:3426–3434. doi: 10.1093/emboj/16.12.3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sellman BR, Tweten RK. The propeptide of Clostridium septicum alpha toxin functions as an intramolecular chaperone and is a potent inhibitor of alpha toxin-dependent cytolysis. Mol Microbiol. 1997;25:429–440. doi: 10.1046/j.1365-2958.1997.4541820.x. [DOI] [PubMed] [Google Scholar]
- 17.Sellman BR, Kagan BL, Tweten RK. Generation of a membrane-bound, oligomerized pre-pore complex is necessary for pore formation by Clostridium septicum alpha toxin. Mol Microbiol. 1997;23:551–558. doi: 10.1046/j.1365-2958.1997.d01-1876.x. [DOI] [PubMed] [Google Scholar]
- 18.Mourez M, Yan M, Lacy DB, Dillon L, Bentsen L, Marpoe A, Maurin C, Hotze E, Wiggelsworth Pimental R, Ballard JD, Collier RJ, Tweten RK. Mapping dominant-negative mutations of anthrax protective antigen by scanning mutagenesis. PNAS. 2003;100:13803–13808. doi: 10.1073/pnas.2436299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haugland RP. Handbook of fluorescent probes and research chemicals. Vol. 6. Molecular Probes, Inc; Eugene, OR: 1996. [Google Scholar]
- 20.Motulsky H. GraphPad PRISM 2.0 Users Guide. GraphPad Software, Inc.; San Diego, CA: 1994. [Google Scholar]
- 21.Tweten RK, Barbieri JT, Collier RJ. Diphtheria toxin: Effect of substituting aspartic acid for glutamic acid-148 on ADP-ribosyl transferase activity. J Biol Chem. 1985;260:10392–10394. [PubMed] [Google Scholar]
- 22.Hwa KY. Glycosyl phosphatidylinositol-linked glycoconjugates: structure, biosynthesis and function. Adv Exp Med Biol. 2001;491:207–14. doi: 10.1007/978-1-4615-1267-7_15. [DOI] [PubMed] [Google Scholar]
- 23.Abrami L, Velluz MC, Hong Y, Ohishi K, Mehlert A, Ferguson M, Kinoshita T, Gisou van der Goot F. The glycan core of GPI-anchored proteins modulates aerolysin binding but is not sufficient: the polypeptide moiety is required for the toxin-receptor interaction. FEBS Lett. 2002;512:249–54. doi: 10.1016/s0014-5793(02)02274-3. [DOI] [PubMed] [Google Scholar]
- 24.Muraki M. The importance of CH/pi interactions to the function of carbohydrate binding proteins. Protein Pept Lett. 2002;9:195–209. doi: 10.2174/0929866023408751. [DOI] [PubMed] [Google Scholar]
- 25.Vazquez-Ibar JL, Guan L, Svrakic M, Kaback HR. Exploiting luminescence spectroscopy to elucidate the interaction between sugar and a tryptophan residue in the lactose permease of Escherichia coli. PNAS. 2003;100:12706–11. doi: 10.1073/pnas.1835645100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muraki M, Morii H, Harata K. Chemically prepared hevein domains: effect of C-terminal truncation and the mutagenesis of aromatic residues on the affinity for chitin. Protein Eng. 2000;13:385–9. doi: 10.1093/protein/13.6.385. [DOI] [PubMed] [Google Scholar]
- 27.Ponyi T, Szabo L, Nagy T, Orosz L, Simpson PJ, Williamson MP, Gilbert HJ. Trp22, Trp24, and Tyr8 play a pivotal role in the binding of the family 10 cellulose-binding module from Pseudomonas xylanase A to insoluble ligands. Biochemistry. 2000;39:985–91. doi: 10.1021/bi9921642. [DOI] [PubMed] [Google Scholar]
- 28.Diep DB, Nelson KL, Lawrence TS, Sellman B, Tweten RK, Buckley JT. Expression and properties of an aerolysin-Clostridium septicum alpha toxin hybrid protein. Mol Microbiol. 1999;31:785–94. doi: 10.1046/j.1365-2958.1999.01217.x. [DOI] [PubMed] [Google Scholar]
- 29.Polekhina G, Giddings KS, Tweten RK, Parker MW. Insights into the action of the superfamily of cholesterol-dependent cytolysins from studies of intermedilysin. PNAS. 2005;102:600–605. doi: 10.1073/pnas.0403229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–5. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]