

Abstract
A number of biochemical processes rely on isoprenoids, including the post-translational modification of signaling proteins and the biosynthesis of a wide array of compounds. Photoactivatable analogues have been developed to study isoprenoid utilizing enzymes such as the isoprenoid synthases and prenyltransferases. While these initial analogues proved to be excellent structural analogues with good cross linking capability, they lack the stability needed when the goals include isolation of cross-linked species, tryptic digestion, and subsequent peptide sequencing. Here, the synthesis of a benzophenone-based farnesyl diphosphate analogue containing a stable phosphonophosphate group is described. Inhibition kinetics, photolabeling experiments, as well as x-ray crystallographic analysis with a protein prenyltransferase are described, verifying this compound as a good isoprenoid mimetic. In addition, the utility of this new analogue was explored by using it to photoaffinity label crude protein extracts obtained from Hevea brasiliensis latex. Those experiments suggest that a small protein, Rubber Elongation Factor, interacts directly with farnesyl diphosphate during rubber biosynthesis. These results indicate that this benzophenone-based isoprenoid analogue will be useful for identifying enzymes that utilize farnesyl diphosphate as a substrate.
Introduction
Farnesyl diphosphate (FPP) is an isoprenoid building block utilized in a number of cellular processes including the synthesis of cholesterol1, carotenoids2, and terpenes3, protein prenylation4, and rubber biosynthesis5. Protein prenylation is a post-translational modification targeted by anti-cancer drugs6–9 given that the farnesylation of mutant Ras proteins is required for transformation.10 Substrate analogues that covalently modify their cognate targets are useful for both identifying previously uncharacterized enzymes as well as for mapping the active sites of known proteins. Photoaffinity labeling is one technique that has proven useful in this regard. For that reason, we and others have developed a number of analogues of farnesyl- and geranylgeranyl diphosphates that can be used for this purpose.11–14
Initial photoactive isoprenoid analogues incorporated a diazotrifluoropropionate (DATFP) photophore into the isoprenoid chain.12,14 Despite being accurate mimetics of FPP, they suffer from low cross-linking efficiency and require prolonged short wavelength UV irradiation for photoactivation. Molecules that utilize benzophenone moieties proved to be an attractive alternative for a number of reasons. Besides being more chemically stable than diazo esters, they can be easily manipulated in ambient light and require longer UV wavelengths for activation, avoiding protein degradation.15 Given the significant overlap in the benzophenone and isoprenoid structures, analogues incorporating the benzophenone moiety as part of the isoprenoid chain were developed.11,13,16–19 X-ray crystallographic analysis of these molecules revealed that they are nearly superimposable with FPP in the active site of rat protein farnesyltransferase, confirming they are structurally analogous to FPP.13,20
In addition to providing information on a specific, known ligand or substrate, photoactivatable analogues of ligands can be used to isolate and identify unknown binding partners. By creating a photoaffinity labeling reagent structurally based on suberoylanilide hydroxamic acid, Webb and coworkers were able to identify a potential target for hybrid polar cytodifferentiation agents through isolation of labeled proteins and mass spectral analysis.21 Zhang and coworkers identified proteins that interact with platinum modified DNA using a photoreactive analogue of cisplatin.22 The critical component in studies of this type is the ability to detect cross-linked proteins in order to isolate and identify them. Photoaffinity analogues of isoprenoid pyrophosphates have often employed a 32P radiolabel for detection given the ease of synthesis of these compounds.16,18,20 However, because of the facile loss of the radiolabel due to the acid labile nature of the allylic diphosphate linkage, the use of such compounds has been impeded. The development of more stable photoreactive analogues is needed to successfully accomplish such studies.
In this paper we describe the synthesis of benzophenone-based farnesyl diphosphate analogue, 3b, containing a stable phosphonophosphate group that replaces the labile allylic diphosphate. A number of polyprenyl phosphonophosphates have been reported as isoprenoid diphosphate mimics in the design of inhibitors23,24, immunoregulators24,25, as well as affinity purification reagents.26,27 The stability of 3b in aqueous acid was investigated. Inhibition kinetics and photoaffinity labeling were studied using protein prenyltransferases as model systems to validate the utility of the new analogue. A co-crystal structure of 3b with protein farnesyltransferase (PFTase) illustrates the close complimentarity between the new probe and FPP as well as a previously synthesized photoactive FPP analog. Finally, 3b was used in photolabeling experiments with protein mixtures isolated from the latex of Hevea brasiliensis, the Brazilian rubber tree. One of the cross linked proteins previously designated as rubber elongation factor (REF) was identified by mass spectral analysis. These experiments suggest the REF interacts directly with isoprenoid diphosphates and clearly demonstrates the efficacy of this analogue for photoaffinity labeling experiments involving the isolation and identification of enzymes that employ isoprenoids as their substrates.
Results and Discussion
Synthesis and Stability of the Photoactive Analogue
In our initial design of a more stable photoactive analogue, we envisioned compound 3a, based on 2a, that would incorporate a C-P bond at C-1 in lieu of the C-O bond present in 2a and FPP (1, Figure 1).
Figure 1.
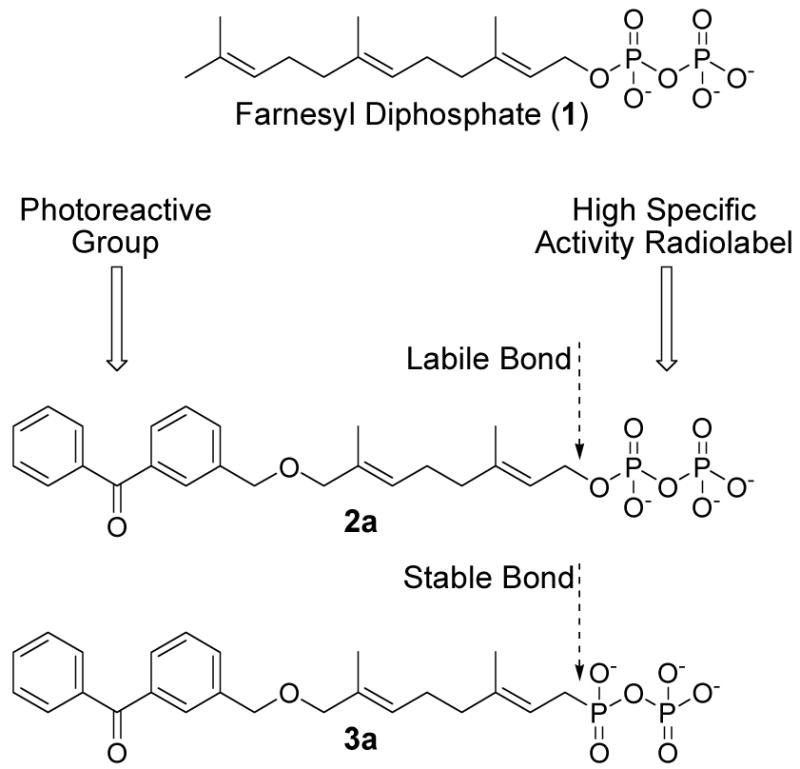
Benzophenone-containing analogues of farnesyl diphosphate
It was considered desirable to retain the ether linkage between the benzophenone moiety and the isoprenoid since that structural feature is a minimal perturbation relative to the natural molecule FPP; in earlier work, it had been shown that ether-containing FPP analogues bound to protein farnesyltransferase (PFTase) with higher affinity when compared to similar compounds incorporating ester or amide linkages13. Compound 4, an intermediate from an earlier synthesis, was reacted under Arbuzov conditions to yield the dimethylphosphonate (5) in excellent yield (Scheme 1) in order to prepare compound 3a.
Scheme 1.
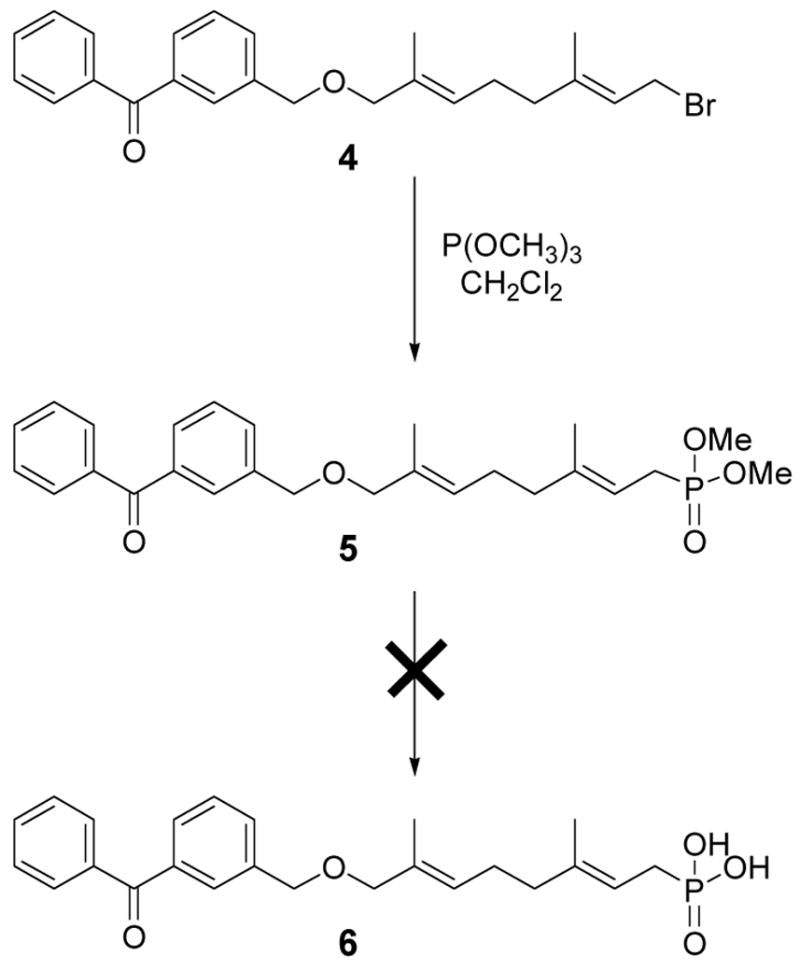
Original strategy for the synthesis of an analogue containing a stable phosphonate linkage.
Unfortunately, efforts to convert the phosphonate diester to the corresponding phosphonic acid (6) were not successful. Treatment of 5 with Me3SiCl or Me3SiBr resulted primarily in cleavage of the benzyl ether linkage with only small amounts of methyl ester cleavage occurring. Treatment of 5 under basic conditions (LiOH) resulted in incomplete saponification to the phosphonate monoester 6. Exposure of 5 to acidic conditions produced decomposition.
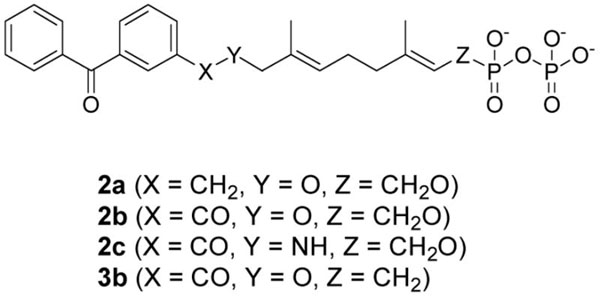
Based on the above observations, it was decided to redesign the target molecule to include an ester-linked benzophenone (Compound 3b, Scheme 2) in place of the original ether linkage. There was good evidence to believe that the ester-linked analogue would effectively bind since a related analogue, 2b (see Table 1), inhibited yeast PFTase with a KI of 380 nM.16 Compound 3b was prepared in six steps using the route shown in Scheme 2.
Scheme 2.

Synthesis of an analogue containing a stable phosphonate linkage.
Table 1.
IC50 Values for Inhibition of yPFTase by Benzophenone-Based Isoprenoid Diphosphate Photoaffinity Labeling Analogues.
Geraniol (7) was first converted to the corresponding bromide (8) using conditions described by Coates et al28, followed by Arbuzov reaction with P(OCH3)3 which proceeded in good yield to produce 9.23 The dimethyl phosphonate was then subjected to oxidation with SeO2 according to the procedure described by Umbreit and Sharpless29 to produce the allylic alcohol 10 in 78% yield. Apparently, the phosphonate moiety in 9 is sufficiently deactivating such that oxidation by SeO2 occurs predominantly at the alkene at the C-6 position and not at C-2. Acylation of 10 to form the desired ester-linked benzophenone-containing intermediate 11 proceeded in good yield. In contrast to our results obtained with the deprotection of compound 5, where cleavage of the ether linkage was observed, treatment of 11 with trimethylsilyl bromide and allyltrimethylsilane in CH2Cl2 resulted in clean deprotection of the phosphonate esters to produce 12 in excellent yield (92%) without any observable loss of the benzophenone moiety30. Finally, phosphonate 12 was converted to the desired phosphonophosphate 3b by activation to the corresponding imidazolide with carbonyl diimidazole followed by reaction with anhydrous phosphoric acid.31 Purification of 3b was performed by chromatographic separation using octyl-functionalized silica gel. For photoaffinity labeling experiments [32P]3b was prepared from phosphonate 12 using a similar procedure employing [32P]-H3PO4 followed by purification via a small reversed-phase column.
In earlier work, it was noted that the ester linkage between the benzophenone and isoprenoid moieties present in compounds similar to 3b are stable to the acidic conditions typically used in reversed-phase chromatography to separate and identify cross-linked peptides obtained from photoaffinity labeling experiments with proteins. Unfortunately, this is not the case for the C-O bond at C-1 between the isoprenoid and phosphate moieties in allylic diphosphates such as 2a and 2b. In such molecules, while the phosphate anhydride linkage between the α and β phosphates is relatively stable to acid, the bond between C-1 and O-1 is labile. As illustrated in Figure 2, treatment of geranyl diphosphate (GPP), which contains a C-O bond at C-1 like 2a, with a mixture of H2O and CH3CN containing 0.2% TFA (common conditions for peptide analysis) results in hydrolysis of the allylic diphosphate with a half life of approximately 2 hours.
Figure 2.
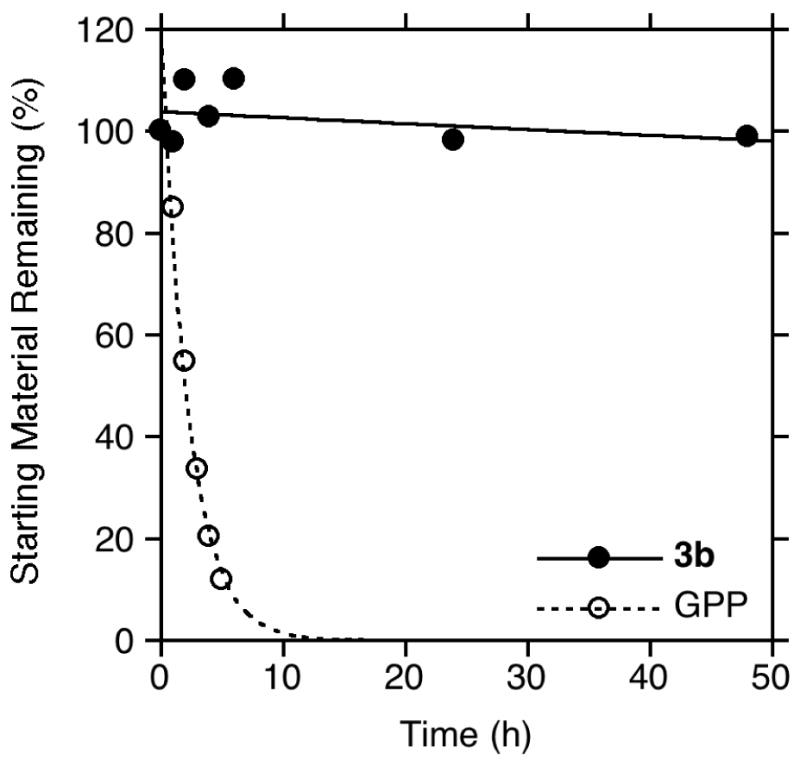
Stability of phosphonophosphate 3b compared to geranyl diphosphate (GPP).
This significant rate of degradation via phosphate loss makes it impractical to use 32P-labeled allylic diphosphates for photoaffinity labeling applications when tryptic digestion and subsequent peptide sequencing are desired. In contrast, treatment of phosphonophosphate 3b under the same conditions results in less than 5% hydrolysis even after 50 hours of reaction. This enhanced stability significantly increases the utility of this benzophenone-containing analogue.
Inhibition Kinetics
Given the inherent stability of the C-P bond at position C-1, phosphonophosphate 3b was tested as a competitive inhibitor of yPFTase. The rate of yPFTase-catalyzed farnesylation of N-Dansyl-GCVIA was measured in the presence of a fixed concentration of FPP and varying concentrations of 3b. An IC50 value of 960 nM was calculated for 3b, which is comparable to previously synthesized benzophenone-based FPP analogues (see Table 1). The similarity in values between the ether-linked analogue 2a and 3b was unexpected, given the previously observed reduction in binding when a carbonyl group is involved in linkage between the benzophenone moiety and the isoprenoid.
X-ray Crystallographic Studies
In contrast to our earlier work that always employed analogues containing an allylic diphosphate, compound 3b possesses a phosphonate linkage that is one carbon out of register relative to FPP. Crystals of 3b bound to rPFTase were obtained and the structure of the resulting complex was determined via x-ray diffraction experiments to investigate the effect of this modification. The structure of 3b bound to rPFTase is shown in Figure 3; for comparison the structure of FPP bound to rPFTase is shown in a superposition (panel A). Figure 4 highlights the key amino acid residues involved in the binding.
Figure 3.
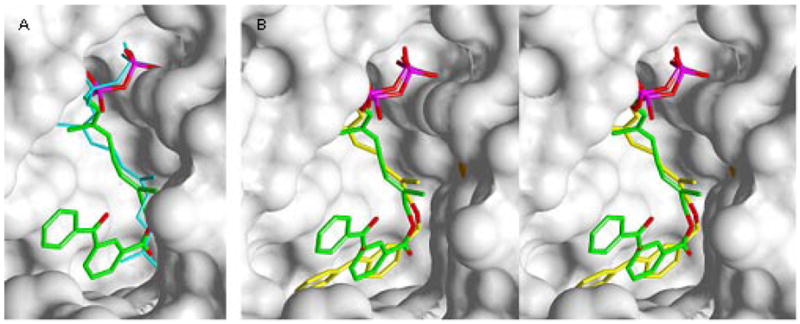
Crystallographic Analysis of Analogue 3b bound to rPFTase. A (left image): Superposition of 3b and FPP bound to PFTase. B (center and right images): Stereoview of a superposition of 3b and a related diphosphate analogue 2a. Analogue 3b is shown in green (carbon), red (oxygen) and purple (phosphorous). FPP is shown in blue. Analogue 2a is shown in yellow (carbon), red (oxygen) and purple (phosphorous). The solvent accessible protein surface is white with the Zn atom shown in red.
Figure 4.

Stereoview of 3b bound to rPFTase. Residues highlighted in red are from the α subunit; black are from the β subunit. The catalytic zinc is shown as a red sphere.
Electron density maps clearly show that 3b is bound in the rPFTase active site cavity. The presence of the higher molecular weight phosphate atoms in the molecule provides helpful information in defining the binding mode. Comparison of the protein in the structure of the rPFTase:3b complex and the rPFTase:FPP:SCH66336 ternary complex shows a 0.2 Å r.m.s. deviation for all protein atoms, indicating that no significant movement of the protein side-chains has occurred.32,33 Interestingly, the two phosphate P atoms from FPP and those from 3b are essentially superimposable suggesting that the phosphonophosphate substitution present in the analogue is not deleterious. Due to the absence of the intervening oxygen atom in 3b (the oxygen attached to C-1 in FPP) and the presence of the bulky benzophenone group, there small are perturbations in the structure of the isoprenoidal portion of the analogue; the average r.m.s. deviation for all atoms in the diphosphate unit and the first two isoprene units is 0.8 Å. However, these changes are in general small, indicating close complementarity between the natural substrate, FPP, and 3b. A superposition of 3b and 2a, a related diphosphate-based analogue with an ether-linked benzophenone, is shown in Figure 3, panel B. As was observed above with FPP, there is substantial overlap between these two structures particularly in the diphosphate unit and the first two isoprene units. However, there is a significant difference in the position of the benzophenone group in 3b relative to 2a. Clearly, the presence of the more conformationally restricted ester linkage in 3b alters the orientation of the benzophenone compared to that of the more flexible ether-linked analogue 2a. This structural difference may account for the greater inhibitory potency observed with 2a compared with 3b. Nevertheless, it can be concluded that beyond the greater size of the benzophenone groups that causes them to protrude beyond the FPP binding site, analogues such as 3b are good mimics of FPP and that the use of a phosphonophosphate group does not cause any significant changes in the mode of binding to PFTase.
Photolabeling of Protein Prenyltransferases with [32P]3b
To verify the ability of phosphonophosphate 3b to act as a photoaffinity label of protein prenyltransferases, the radiolabeled analogue [32P]3b was synthesized using a method developed by Bartlett and co-workers34, adapted to incorporation of a radiolabel, and purified using a method previously described.35 Photolysis of the prenyltransferases with [32P]3b at a concentration 10-fold higher than the IC50 value determined for yPFTase was performed with 300–400 nm light15 for 2 hours. Preferential labeling of the β subunit was observed for both yeast protein farnesyltransferase (yPFTase) and human protein geranylgeranyltransferase type I (hPGGTAse-I) (Figure 5: yPFTase and [32P]3b and Figure 6: hPGGTase-I and [32P]3b; lane 2′ in each). Upon addition of the natural substrate (FPP for yPFTase, GGPP for hPGGTase-I) in the photolysis reactions containing [32P]3, protection from labeling was observed (Figure 5: yPFTase and [32P]3b and Figure 6: hPGGTase-I and [32P]3b; lane 3′ in each). Phosphorimaging analysis was used to quantify the relative labeling efficiencies of each subunit for each enzyme. For yPFTase, the β subunit was labeled 4.8-fold over the α subunit. Inclusion of the natural substrate FPP, 1, in the photolysis reaction resulted in a substantial (2.3-fold) decrease in β subunit labeling. In the case of hPGGTase-I photolabeling by [32P]3b, the β subunit was labeled 24-fold over the α subunit, while inclusion of the physiologically relevant substrate GGPP resulted in a substantial decrease in β subunit labeling (8.7-fold). The observed labeling specificity is in agreement with results obtained from other photoactive analogues with yPFTase and hPGGTase-I, all implicating β subunit involvement in prenyl group recognition.13,16
Figure 5.
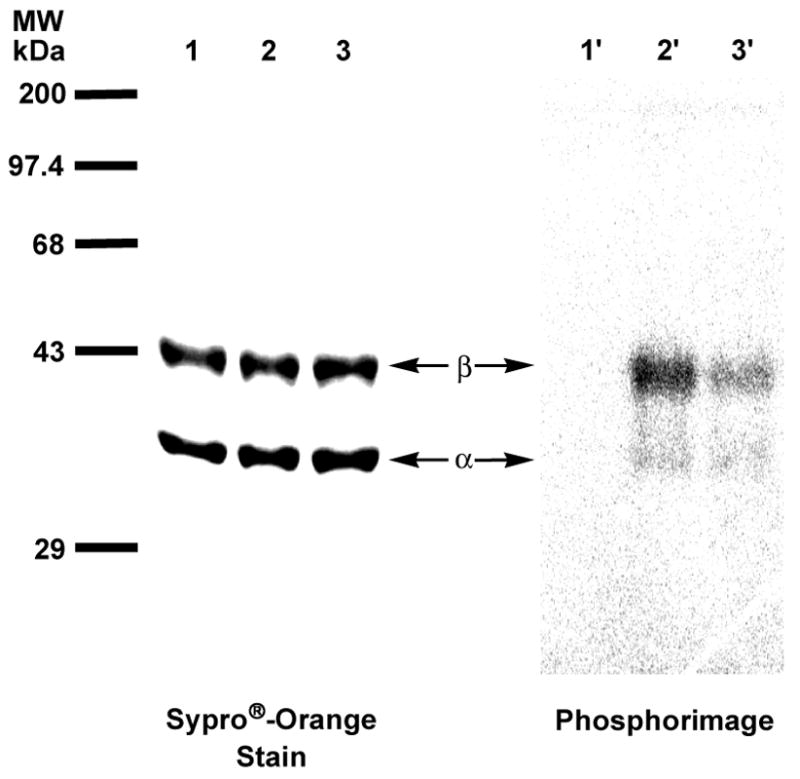
Analysis of photolabeling of pure yPFTase with [32P]3 by SDS-PAGE. Lanes 1 and 1′contain samples of yPFTase and [32P]3b that were not irradiated. Lanes 2 and 2′ contain samples of yPFTase and [32P]3 irradiated at 350 nm. Lanes 3 and 3′ contain samples of yPFTase and [32P]3b irradiated in the presence of FPP. Lanes 1, 2 and 3 show proteins identified with Sypro®-Orange staining. Lanes 1′, 2′ and 3′ show the radiolabeled proteins identified with phosphorimaging.
Figure 6.
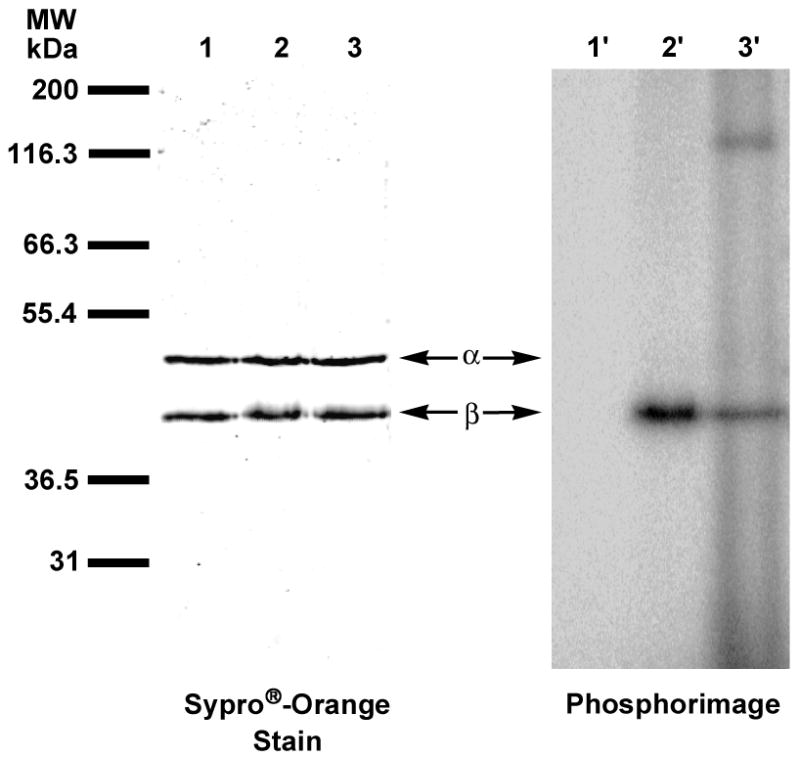
Analysis of photolabeling of pure hPGGTase-I with [32P]3b by SDS-PAGE. Lanes 1 and 1′contain samples of hPGGTase-I and [32P]3b that were not irradiated. Lanes 2 and 2′ contain samples of hPGGTase-I and [32P]3b irradiated at 350 nm. Lanes 3 and 3′ contain samples of hPGGTase-I and [32P]3 irradiated in the presence of GGPP. Lanes 1, 2 and 3 are showed proteins identified with Sypro®-Orange staining. Lanes 1′, 2′ and 3′ show the radiolabeled proteins identified with phosphorimaging.
The applicability of this probe for labeling specific targets in a mixture of proteins was demonstrated by the photolysis of a partially purified sample of hPGGTase-I with [32P]3b. Of the crude mixture of proteins, the β subunit is the major labeled species, while no labeling of the α subunit is observed (Figure 7). In this crude mixture, a large number of proteins are clearly visible by Sypro®-orange staining. Interestingly, the phosphorimage (Fig. 7, lane 1′) reveals three major labeled species with the β subunit of hPGGTase-I as the most intensely labeled protein. Thus, although 3b is not absolutely specific for hPGGTase-I, it obviously manifests substantial selectivity in this complex mixture. Based on the promising results obtained with 3b labeling in crude extracts, we decided to explore the ability of this probe to identify an unknown target.
Figure 7.
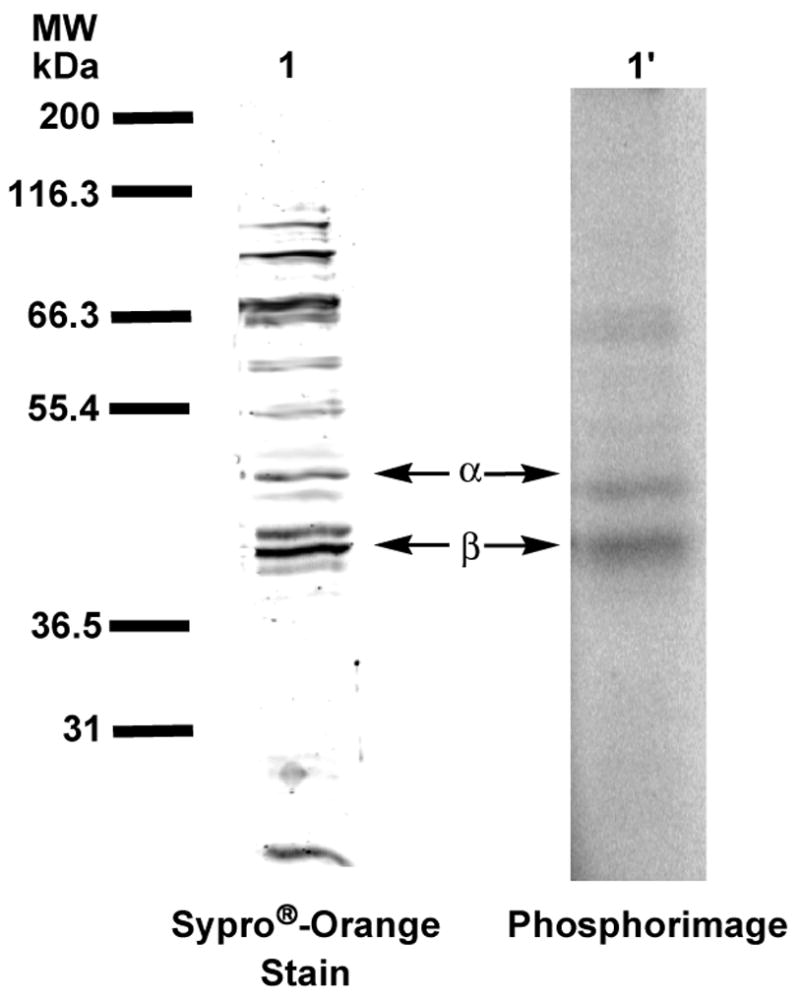
Analysis of crude hPGGTase-I with [32P]3b by SDS-PAGE. Lanes 1 and 1′ contain samples of crude cell lysate after ion-exchange chromatography, irradiated at 350 nm in the presence of [32P]6. Lane 1 shows protein identified with Sypro®-orange staining. Lane 1′shows radiolabeled protein identified with phosphorimaging.
Photolabeling of Hevea brasiliensis washed rubber particles with [32P]3b
H. brasiliensis, the Brazilian rubber tree, is among the many species of angiosperms known to produce rubber of high molecular weight and is the predominant source of commercially available natural rubber. The latex obtained from these trees has been shown to contain enzymatically-active rubber particles that, when purified and incubated with an allylic diphosphate initiator and isopentenyl diphosphate as the monomer, are able to carry out biosynthesis of cis-1,4-polyisoprene36. Despite intense research, the proteins involved in rubber biosynthesis have yet to be unequivocally identified and reported. Based on the observation that FPP can act as an initiator of rubber biosynthesis in washed rubber particles (WRPs), it was decided to utilize 3b to identify FPP binding components in the complex mixture of proteins present in H. brasiliensis WRPs. Photolysis of suspensions of WRPs containing 10 μM [32P]3b was performed for 6 hours. Electrophoretic separation of the reaction mixture and visualization of the proteins indicated the presence of a wide range of species. The WRPs from H. brasiliensis are clearly a complex mixture. Interestingly, phosphorimaging analysis of the photolysis mixture gave a very different picture. Two major labeled species were observed as well as several minor bands (Figure 8, Lane 1′). The presence of FPP during the labeling reaction reduced the amount of radiolabel throughout the gel (Figure 8, Lane 2′).
Figure 8.
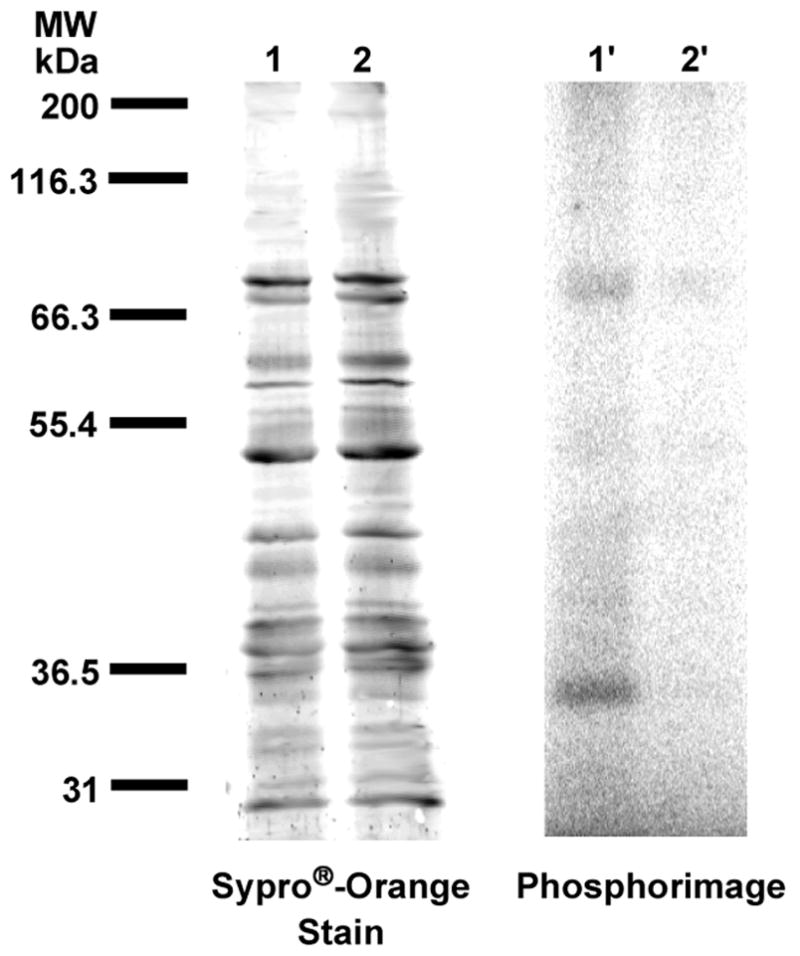
Analysis of photolabeling of H. brasiliensis washed rubber particles with [32P]3b by SDS-PAGE. Lanes 1 and 1′ contain samples of WRPs and [32P]3b irradiated at 350 nm. Lanes 2 and 2′ contain samples of WRPs and [32P]3b irradiated in the presence of FPP. Lanes 1 and 2 show proteins identified with Sypro®-orange staining. Lanes 1′and 2′ show radiolabeled proteins identified with phosphorimaging.
Mass spectral analysis of photolabeled proteins from Hevea brasiliensis washed rubber particles
The identify the cross-linked proteins from the photolabeling of H. brasiliensis WRPs with [32P]3b was determined by performing a second set of photolysis reactions. Samples were fractionated by isoelectric focusing followed by SDS-PAGE. Surprisingly, the higher molecular weight proteins shown to be labeled previously were not observed. Instead, a series of proteins (10–20 kDa) appeared to be labeled (see Supporting Information). These labeled protein bands were extracted from the gel, subjected to tryptic digestion, and analyzed by MALDI-MS/MS. A peak list of the data was then generated and searched using the MASCOT database. A 14.6 kDa protein, Rubber Elongation Factor, was identified (Table 2) based on the masses of eight different peptides. Together these represent approximately 69% of the complete primary sequence. A number of as yet unidentified peptides were also observed at low abundance.
Table 2.
Tryptic peptides determined by MS/MS Analysis
| Amino Acid Residues | Calculated Mass | Observed Mass | Sequence | Ion Score | Confidence Interval % |
|---|---|---|---|---|---|
| 17–40 | 2689.3547 | 2689.3516 | YLGFVQDAATYAVTTFSNVYLFAK | 120 | 100.000 |
| 41–58 | 1849.0065 | 1849.0023 | DKSGPLQPGVDIIEGPVK | 135 | 100.000 |
| 43–58 | 1605.8846 | 1605.8828 | SGPLQPGVDIIEGPVK | 111 | 100.000 |
| 68–77 | 1109.599 | 1109.6146 | FSYIPNGALK | 70 | 99.999 |
| 78–92 | 1621.8795 | 1621.8759 | FVDSTVVASVTIIDR | 24 | 82.268 |
| 93–99 | 753.4869 | 753.4849 | SLPPIVK | 41 | 99.591 |
| 100–110 | 1158.6477 | 1158.6460 | DASIQVVSAIR | 82 | 100.000 |
| 118–128 | 1088.5946 | 1088.5912 | SLASSLPGQTK | 34 | 97.974 |
Given the apparent contradiction in the observed labeled proteins, the originally observed 35 and 70 kDa labeled proteins (as shown in Figure 7) were isolated, subjected to tryptic digestion, and analyzed by MALDI-MS/MS. Upon searching of the MASCOT database, it was found that both the 35 kDa and the 70 kDa proteins could be attributed to REF. The appearance of the 14.6 kDa REF protein at these higher molecular weights appears to be due to oligomerization. Czuppon et al. have previously found that REF exists as a homotetramer of 58 kDa when isolated from raw latex.37 A study done by Yeang and coworkers showed that another rubber particle protein, Hev b3, is prone to aggregation upon prolonged storage.38
Rubber Elongation Factor (REF), also known as the latex allergen Hev b138, is a known constituent in H. brasiliensis latex compromising 10–60% of the total protein.39 While it is possible that REF can exist as carrier or binding partner in rubber biosynthesis, it has been directly implicated in playing a larger role. A study done by Dennis and coworkers showed that treatments of WRPs to either hydrolyze or remove REF completely abolishes prenyltransferase activity. However, attempts to reconstitute rubber biosynthesis in REF depleted WRPs with solubilized REF were unsuccessful, making the exact role of REF in rubber biosynthesis uncertain.40 In our experiments, the cross-linking of REF in WRP suspensions with a photoactivatible analogue of FPP suggests that REF can interact with isoprenoid diphosphates. This interaction may be key for rubber biosynthesis. However, even if this is the case, it is unlikely that REF is the sole protein responsible for rubber synthesis. We are currently using compound 3b and related analogues to identify other components, including the unidentified peptides noted above, present in WRPs that contribute to the catalytic machinery of rubber biosynthesis.
Conclusion
Compound 3b is a photoactive phosphonophosphate-containing analogue of FPP with enhanced stability that can be efficiently prepared in six steps from geraniol. Kinetic and structural studies with 3b and PFTase demonstrate that this probe is a good mimic of isoprenoid diphosphates. The greater stability of 3b has allowed us to use it to label and identify a specific protein that may be involved in rubber biosynthesis. These findings suggest that 3b and analogues thereof will be useful tools for probing the structure and function of FPP utilizing enzymes.
Experimental Section
General Methods and Materials
See the Supporting Information.
Dimethyl Geranylphosphonate (9)
A mixture of 8 (35 g, 0.16 mol) and trimethyl phosphite (22 g, 0.18 mol) was heated to reflux for 4 h. After cooling to rt, the mixture was chromatographed on silica gel (hexanes/ethyl acetate, 5:1 to 1:2) to give the product as a colorless oil (36 g, 91%). 1H-NMR (CDCl3, 300 MHz): 5.15 (1H, m), 5.03 (1H, m), 3.72 (3H, s), 3.69 (3H, s), 2.55 (2H, dd, J = 7.8, 21.9 Hz), 2.03 (4H, m), 1.64 (3H, s), 1.63 (3H, d, J = 5.7 Hz) and 1.57 (3H, s). 13C-NMR (CDCl3, 75 MHz): 140.4 (d, J = 14.3 Hz), 131.6, 123.8, 112.0 (d, J = 11.2 Hz), 52.6, 52.5, 39.6 (d, J = 2.9 Hz), 25.6, 25.4 (d, J = 142.0 Hz), 17.6, 16.2 and 16.1. 31P-NMR (CDCl3, 121 MHz): 31.7. HRMS (ESI) calcd for C12H24O3P [M + H]+ 247.1463; found 247.1453.
Dimethyl (8-Hydroxy-3,7-dimethylocta-2,6-dienyl)phosphonate (10)
To a suspension of SeO2 (31 mg, 0.27 mmol), salicylic acid (190 mg, 1.3 mmol) and 90% t-Bu-OOH (4.5 ml) in CH2Cl2 (20 ml) was added 9 (2.5 g, 10 mmol) in CH2Cl2 (5.0 mL) at rt. The mixture was diluted with Et2O after being stirred for 21 h, washed with dilute Na2SO3 solution and brine, then dried over Na2SO4. The solvents were evaporated and the residue was chromatographed on silica gel (hexanes/ethyl acetate, 1:1 to 0:1) to give the alcohol as a colorless oil (1.3 g, 78% yield based on the converted starting material), together with the recycled starting material (0.95 g). 1H-NMR (CDCl3, 300 MHz): 5.26 (1H, m), 5.08 (1H, m), 3.91 (2H, s), 3.71 (3H, s), 3.67 (3H, s), 2.98 (1H, br s), 2.51 (2H, dd, J = 8.1, 21.9 Hz), 2.11 (4H, m), 1.60 (3H, s), 1.59 (3H, m). 13C-NMR (CDCl3, 75 MHz): 139.9 (d, J = 14.6 Hz), 135.5, 124.6, 112.4 (d, J = 11.4 Hz), 68.6, 52.6, 52.5, 39.1 (d, J = 3.1 Hz), 25.2 (d, J = 3.9 Hz), 25.1 (d, J = 140.3 Hz), 15.9 (d, J = 2.9 Hz), 13.6. 31P-NMR (CDCl3, 121 MHz): 31.8. IR (NaCl, cm−1): 3400 (br), 2955 (m), 1719 (m), 1683 (m). HRMS (ESI) calcd for C12H24O4P [M + H]+ 263.1412; found 263.1419.
Dimethyl (E,E) ((8-(3-Benzoyl)benzoyloxy)-3,7-dimethylocta-2,6-dienyl)phosphonate (11)
To a solution of 10 (2.2 g, 8.3 mmol), 3-benzoyl-benzoic acid (2.3 g, 10 mmol) in anhydrous CH2Cl2 (100 mL) was added DCC (2.1 g, 10 mmol) and DMAP (50 mg, 0.40 mmol) at rt. The resulting suspension was stirred for 20 h at room temperature and then filtered through Celite. The filtrate was concentrated and chromatographed on silica gel (hexanes/ethyl acetate, 2:1 to 1:2) to give the ester as a viscous oil (3.3 g, 82%). 1H-NMR (CDCl3, 300 MHz): 8.43 (1H, m), 8.24 (1H, dt, J = 7.8, 1.8 Hz), 7.97 (1H, dt, J = 7.8, 1.5 Hz), 7.79 (1H, m), 7.77 (1H, m), 7.45–7.62 (4H, m), 5.50 (1H, m), 5.17 (1H, m), 4.70 (2H, s), 3.72 (3H, s), 3.69 (3H, s), 2.55 (2H, dd, J = 7.8, 21.9 Hz), 2.16 (2H, m), 2.09 (2H, m), 1.70 (3H, s), 1.65 (3H, d, J = 3.3 Hz). 13C-NMR (CDCl3, 75 MHz): 195.8, 165.6, 139.9 (d, J = 14.3 Hz), 137.9, 137.0, 134.0, 133.2, 132.8, 131.0, 130.6, 130.1, 130.0, 129.4, 128.5, 128.4, 112.5 (d, J = 11.1 Hz), 70.9, 52.6, 52.5, 38.9 (d, J = 2.6 Hz), 26.1 (d, J = 3.4 Hz), 25.4 (d, J = 140.0 Hz), 16.2 (d, J = 2.4 Hz), 14.0. 31P-NMR (CDCl3, 121 MHz): 31.6. IR (NaCl, cm−1): 3065 (w), 2955 (s), 2854 (m), 1731 (s), 16675 (s) 1602 (s), 1579 (m). HRMS (ESI) calcd for C26H32O6P [M + H]+ 471.1936; found 471.1945.
Ammonium (E,E) 8-(3-Benzoylbenzoyloxy)-3,7-dimethylocta-2,6-dienylphosphonate (12)
To a solution of 11 (1.0 g, 2.1 mmol) in anhydrous CH2Cl2 (30 ml) was added allyltrimethylsilane (0.50 ml, 3.1 mmol) and bromotrimethylsilane (0.90 ml, 6.8 mmol) at rt. The mixture was stirred for 38 h and concentrated in vacuo. The residue was dissolved in H2O (4.0 mL), neutralized with NH4HCO3, then lyophilized to afford the phosphonic acid in its ammonium form as a white powder (0.87 g, 92%). 1H-NMR (25 mM NaHCO3 in D2O, 500 MHz): 7.80 (1H, s), 7.60 (1H, s), 7.27 (1H, d, J = 6.0 Hz), 7.15 (2H, m), 7.00 (2H, m), 6.89 (2H, m), 5.06 (1H, s), 5.01 (1H, d, J = 5.5 Hz), 4.15 (2H, s), 2.10 (2H, d, J = 15.0 Hz), 1.67 (4H, m), 1.28 (3H, s), 1.17 (3H, s). 13C-NMR (25 mM NaHCO3 in D2O, 75 MHz): 195.1, 165.3, 136.8, 136.4 (d, J = 13.0 Hz), 136.0, 133.8, 132.7, 129.9, 129.8, 129.6, 129.2, 128.1, 117.1, 70.9, 38.6, 28.9 (d, J = 131.2 Hz), 25.9, 15.3, 13.2. 31P-NMR (25 mM NaHCO3 in D2O, 121 MHz): 22.8. IR (NaCl, cm−1): 2971 (w), 2922 (w), 1720 (s), 1655 (s), 1602 (w). HRMS (ESI) calcd for C24H26O6P [M − H]− 441.1473; found 441.1481. The ammonium counter-ions within this compound are not observed during mass spectral analysis.
Ammonium (E,E) P-8-(3-Benzoylbenzoyloxy)-3,7-dimethylocta-2,6-dienylphosphonophosphate (3b)
A mixture of the phosphonate 12 (100 mg, 230 μmol) and 1,1′-carbonyldiimidazole (60 mg, 370 μmol) in anhydrous DMF (4.0 mL) was stirred for 5 h at rt. To this clear solution was added directly 98% phosphoric acid solid (~ 50 mg, 510 μmol). The solvent was removed under high vacuum after being stirred for 27 h. The residue was dissolved in H2O (1.0 mL), and loaded on an octyl-functionalized silica gel column (18 cm × 1 cm). The column was washed with a step gradient of H2O and MeOH (10 mL per step, 10% MeOH per step) from 0% to 70% MeOH. Fractions were collected and those containing product as determined by TLC were pooled and lyophilized. The lyophilized powder was dissolved in H2O, passed through a short, strong acidic ion exchange column, then washed with H2O. The resulting solution was neutralized with NH4HCO3 and lyophilized to give the product in it’s ammonium form as a white powder (28 mg, 23%). 1H-NMR (D2O, 300 MHz): 7.77 (1H, s), 7.75 (1H, d, J = 7.5 Hz), 7.50 (1H, d, J = 7.5 Hz), 7.09–7.31 (6H, m), 5.16 (1H, m), 5.03 (1H, m), 4.26 (2H, s), 2.33 (2H, dd, J = 7.5, 21.5 Hz), 1.78 (4H, m), 1.39 (3H, d, J = 3.3 Hz) and 1.31 (3H, s). 13C-NMR (D2O, 75 MHz): 197.5, 166.4, 138.2 (d, J = 14.0 Hz), 136.6, 135.7, 134.3, 133.4, 130.6, 129.8, 129.5, 129.4, 128.7, 128.4, 115.5 (d, J = 9.7 Hz), 71.1, 38.5, 28.4 (d, J = 135.2 Hz), 25.7, 15.4, 13.1. 31P-NMR (D2O, 121 MHz, pD 7.0, NH4+): 16.4 (d, J = 26.4 Hz), −9.6 (d, J = 26.4 Hz). IR (NaCl, cm−1): 2925 (w), 1715 (m), 1655 (m), 1101 (m). (HRMS (ESI) calcd for C24H27O9P2 [M − H]− 521.1136; found 521.1174. The ammonium counter-ions within this compound are not observed during mass spectral analysis.
(E,E) β-P-(3-Benzoylbenzoyloxy)-3,7-dimethylocta-2,6-dienylphosphonophosphate ([32P]3b)
A mixture of phosphonate 1 (4.0 mg, 8.0 μmol) and 1,1′-carbonyldiimidazole (4.0 mg, 24 μmol) in anhydrous DMF (250 μL) was stirred for 12 h at rt. 32P-labeled phosphoric acid was then prepared by lyophilizing 250 μL of 1% (v/v) phosphoric acid and 250 μL of 10 mCi/mL 32P-phosphoric acid over P2O5 overnight. Activated phosphonate in DMF was then added via syringe to the flask containing the dry 32P-labeled phosphoric acid (4.0 mg, 40 μmol) and left to stir for 24 h at rt, and monitored by TLC (6:3:1 2-propanol/NH4OH/H2O). Solvent was then removed under a stream of N2(g). The residue was dissolved in 25 mM NH4HCO3 (3 mL) and applied to a Sep-Pak® column equilibrated in the same solution. The column was washed with a step gradient of 25 mM NH4HCO3 and increasing CH3CN (10% increase per step, 2 mL per step). Fractions were collected and analyzed by TLC (6:3:1 2-propanol/NH4OH/H2O). Under these conditions, the desired product 2 eluted at 30–40% CH3CN/25 mM NH4HCO3. Product fractions were pooled, lyophilized, dissolved in 1.0 mL 25 mM NH4HCO3. UV analysis (ε258 24100 mol−1cm−1) and liquid scintillation counting were used to determine the solution concentration and specific activity (0.55 mM and 334 Ci/mol). Solution was used for protein cross-linking experiments without further purification.
Acid Stability Study of GPP
500 μL of 1.0 mM solution of GPP in 50:50 25 mM NH4HCO3/CH3CN containing 0.2 % TFA was allowed to sit at rt. 50 μL aliquots were removed at prescribed intervals (h) and analyzed by reverse-phase HPLC. A gradient of solvent A (25mM NH4HCO3) and solvent B (CH3CN) and detecting at 214 nm (flow rate, 0.7 mL/min) was employed. Elution was performed by a 40 min linear gradient of 20–80% B. Integration of the peak corresponding to GPP was performed using HPLC software. Hydrolysis half life was determined from a plot of remaining starting material (%) as a function of time.
Acid Stability Study of 3b
The same procedure as described for GPP was performed with a 0.30 mM solution of 3b in in 50:50 25 mM NH4HCO3/CH3CN containing 0.2 % TFA.
Purification of yPFTase and hPGGTase-I
yPFTase was purified as described by Mayer et al.41 and published in our earlier work.16 hPGGTase-I was purified using modifications of published procedures.12,42,43
Enzyme Assays
A continuous fluorescence assay was employed to monitor yPFTase activity as described by Gaon et al.16 modified from the original published procedures.44,45
Enzyme Inhibition Experiments
The concentration of 3b was varied (0, 0.4, 0.6, 1.5, 3, 4.5, 6 μM) while the natural susbtrate, 1, and yPFTase were maintained at a fixed concentrations (10 μM and 11 nM). Enzymatic rates were obtained from linear regression analysis of the time-dependent fluorescence emission data using the fluorimeter software and the IC50 was calculated from a plot of the enzymatic rate versus concentration of 3b.
X-Ray Crystallography Studies
Crystals of the rPFTase:3b complex was prepared by soaking 3b into preformed crystals using methods previously described.32 X-ray diffraction data for the PFTase:3b complex was collected on a Rigaku rotating anode generator equipped with osmic mirrors and a Raxis-IV image plate detector. With the detector set at 150 mm, data were collected in 323 contiguous 0.30° oscillation images each exposed for 8 minutes. The data extend to 2.2 Å resolution and have a Rmerge of 4.6% with a 5.8-fold multiplicity. The structure was refined using CNX2002 (Accelrys Inc.) to an Rfactor of 18.3% and an Rfree of 21.9%.
Photolysis Reaction of Protein Prenyltransferases with [32P]3b
All reactions (100 μL) were performed in silinized quartz test tubes (10 × 75 mm) and contained 52 mM Tris-HCl (pH 7.5), 5.8 mM DTT, 12 mM MgCl2, 12 μM ZnCl2, 25 mM NH4HCO3, 10 μM [32P]3b (~10 fold above IC50 value) and 400 nM pure enzyme (yPFTase, hPGGTase-I) or 21.2 μg of partially purified (after ion-exchange FPLC) protein sample containing hPGGTase-I. For substrate protection experiments, FPP or GGPP was added to a final concentration of 100 μM. Reactions were photolyzed for 2 h at 4°C using a Rayonet Mini-Reactor fitted with 6– 350 nm bulbs and a spinning platform. Samples were spun to ensure an even exposure to the light. Loading buffer (100 μL) was then added to each sample and samples were heated to 100°C for 3 min followed by analysis with 12% Tris-Glycine SDS-polyacrylamide gel electrophoresis. Gels were stained with Sypro®-orange and subjected to 32P-phosphorimaging at rt.
Preparation of Hevea Brasiliensis Washed Rubber Particles
H. brasiliensis WRPs used in this study were prepared as previously described by R. Krishnakumar et al.46 and Cornish et al.47
Photolysis Reaction of Hevea Brasiliensis Washed Rubber Particles with [32P]3b
All reactions (~100 μL) were performed in silinized quartz test tubes (10 × 75 mm) and contained 0.1 M Tris-HCl (pH 7.5), 1.25 mM MgSO4, 5 mM DTT, 10 μM [32P]3b and one H. brasiliensis WRP (35–40 μL/particle, 4.51 μg protein/μL). For substrate protection experiments, FPP was added to a final concentration of 100 μM. Reactions were photolyzed for 6 h using the apparatus described above. Loading buffer (50 μL) was then added to each sample and samples were heated to 100°C for 3 min followed by gel analysis as described above.
Tryptic Digest of Labeled Proteins from Photolysis Reaction of Hevea Brasiliensis Washed Rubber Particles with [32P]3b
Proteins from previously described photolysis reaction (no SDS-PAGE analysis) were extracted with 5 mL of sequential extraction reagent (5.0 M urea, 2.0 M thiourea, 2.0% CHAPS, 2% SB-3–10, 40 mM Tris, and 0.2% Bio-Lyte 3 to 10 ampholyte). Samples were incubated in this buffer for 1 h at rt with constant rocking, and then centrifuged at max speed in a microcentrifuge for 15 min. The supernatant was then transferred to 50 mL conical tube and 4 volumes of cold acetone were added. Acetone precipitated protein pellets were washed twice with 80% acetone in water. Pellets were dried under N2 and prepared for fractionation using the ZOOM IEF fractionator according to manufacturer’s instructions. After IEF fractionation, samples from the different pH ranges were acetone precipitated as described above. Samples were then dissolved in 45 μL Laemmli sample buffer (0.06 M Tris-HCl pH 6.8, 0.01% bromophenol blue, 1.5% SDS, 10% glycerol) and analyzed by SDS-PAGE using 4–20% gradient gel. Gels were stained with Coomassie and subjected to 32P-phosphorimaging at rt. Radiolabeled protein bands were excised using pipette tip (3 spots from each band). For tryptic digestion of proteins, samples were reduced and alkylated using 10 mM dithiothreitol and 100 mM iodoacetamide in water, then digested with 100 ng of trypsin in 25mM NH4HCO3.
Mass Spectral Analysis of Labeled Proteins
Samples from tryptic digestion were spotted onto a MALDI target and eluted with 70% CH3CN, 0.2% formic acid containing 5 mg/mL MALDI matrix (α-Cyano-4-hydroxycinnamic acid). 0.5 μl was then spotted onto the MALDI target. Analysis was performed using an Applied Biosystems 4700 Proteomics Analyzer with TOF/TOF Optics. MALDI-MS data was acquired in reflector mode from a mass range of 700 – 4000 Daltons and 1250 laser shots were averaged for each mass spectrum. Each sample was internally calibrated using the trypsin autolysis products (m/z of 842.51 and 2211.10) as internal standards. The eight most intense ions from the MS spectrum were then subjected to MS/MS analysis. The mass range was 70 to precursor ion with a precursor window of −1 to 3 Daltons with an average 5000 laser shots for each spectrum. A peak list was created by GPS Explorer software (Applied Biosystems) from the raw data based on signal to noise filtering and included de-isotoping. The resulting file was then searched by MASCOT (Matrix Science) using user specified databases. A tolerance of 20 ppm was used if the sample was internally calibrated and 200 ppm tolerance if the default calibration was applied. Protein identification was validated by the following criteria: greater than 20 ppm mass accuracy on all MS ions and all ions in at least two MS/MS spectra, which were not modified, had to be accounted for.
Supplementary Material
Image of the SDS-PAGE used for the first mass spectral analysis of the photolysis reaction of Hevea Brasiliensis washed rubber particles with [32P]3b. General methods and materials. Copies of 1H-NMR, 31P-NMR, and mass spectral data for 3b as well as the intermediates in its synthesis. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research is supported by the National Institutes of Health Grant GM58442 and National Institutes of Health Predoctoral Training Grant T32-GM08700 (to A.J.D.)
References
- 1.Liscum L. New Comp Biochem. 2002;36:409–431. [Google Scholar]
- 2.Fraser PD, Bramley PM. Prog Lipid Res. 2004;43:228–265. doi: 10.1016/j.plipres.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 3.Christianson DW. Chem Rev (Washington, DC, U S) 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 4.Zhang FL, Casey PJ. Annu Rev Biochem. 1996;65:241–69. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 5.Puskas JE, Gautriaud E, Deffieux A, Kennedy JP. Prog Pol Sci. 2006;31:533–548. [Google Scholar]
- 6.Brunner TB, Hahn SM, Gupta AK, Muschel RJ, McKenna WG, Bernhard EJ. Cancer Res. 2003;63:5656–68. [PubMed] [Google Scholar]
- 7.Crul M, de Klerk GJ, Beijnen JH, Schellens JH. Anti-cancer Drugs. 2001;12:163–84. doi: 10.1097/00001813-200103000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Ohkanda J, Blaskovich MA, Sebti SM, Hamilton AD. Prog Cell Cycle Res. 2003;5:211–7. [PubMed] [Google Scholar]
- 9.Sebti SM, Hamilton AD. Oncogene. 2000;19:6584–93. doi: 10.1038/sj.onc.1204146. [DOI] [PubMed] [Google Scholar]
- 10.Kato K, Cox AD, Hisaka MM, Graham SM, Buss JE, Der CJ. Proc Natl Acad Sci U S A. 1992;89:6403–7. doi: 10.1073/pnas.89.14.6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaon I, Turek TC, Distefano MD. Tetrahedron Lett. 1996;37:8833–8836. [Google Scholar]
- 12.Omer CA, Kral AM, Diehl RE, Prendergast GC, Powers S, Allen CM, Gibbs JB, Kohl NE. Biochemistry. 1993;32:5167–76. doi: 10.1021/bi00070a028. [DOI] [PubMed] [Google Scholar]
- 13.Turek TC, Gaon I, Distefano MD, Strickland CL. J Org Chem. 2001;66:3253–64. doi: 10.1021/jo991130x. [DOI] [PubMed] [Google Scholar]
- 14.Yokoyama K, McGeady P, Gelb MH. Biochemistry. 1995;34:1344–54. doi: 10.1021/bi00004a029. [DOI] [PubMed] [Google Scholar]
- 15.Dorman G, Prestwich GD. Biochemistry. 1994;33:5661–73. doi: 10.1021/bi00185a001. [DOI] [PubMed] [Google Scholar]
- 16.Gaon I, Turek TC, Weller VA, Edelstein RL, Singh SK, Distefano MD. J Org Chem. 1996;61:7738–7745. doi: 10.1021/jo9602736. [DOI] [PubMed] [Google Scholar]
- 17.Marecak DM, Horiuchi Y, Arai H, Shimonaga M, Maki Y, Koyama T, Ogura K, Prestwich GD. Bioorg Med Chem Lett. 1997;7:1973–1978. [Google Scholar]
- 18.Turek TC, Gaon I, Gamache D, Distefano MD. Bioorg Med Chem Lett. 1997;7:2125–2130. [Google Scholar]
- 19.Zhang YW, Koyama T, Marecak DM, Prestwich GD, Maki Y, Ogura K. Biochemistry. 1998;37:13411–20. doi: 10.1021/bi972926y. [DOI] [PubMed] [Google Scholar]
- 20.Turek-Etienne TC, Strickland CL, Distefano MD. Biochemistry. 2003;42:3716–24. doi: 10.1021/bi0266838. [DOI] [PubMed] [Google Scholar]
- 21.Webb Y, Zhou X, Ngo L, Cornish V, Stahl J, Erdjument-Bromage H, Tempst P, Rifkind RA, Marks PA, Breslow R, Richon VM. J Biol Chem. 1999;274:14280–7. doi: 10.1074/jbc.274.20.14280. [DOI] [PubMed] [Google Scholar]
- 22.Zhang CX, Chang PV, Lippard SJ. J Am Chem Soc. 2004;126:6536–7. doi: 10.1021/ja049533o. [DOI] [PubMed] [Google Scholar]
- 23.Blau NFWTS, Buess CM. J Chem Eng Data. 1970;15:206–208. [Google Scholar]
- 24.Popjak G, Hadley C. J Lipid Res. 1985;26:1151–9. [PubMed] [Google Scholar]
- 25.Zgani I, Menut C, Seman M, Gallois V, Laffont V, Liautard J, Liautard JP, Criton M, Montero JL. J Med Chem. 2004;47:4600–4612. doi: 10.1021/jm049861z. [DOI] [PubMed] [Google Scholar]
- 26.Bartlett DL, King CHR, Poulter CD. Anal Biochem. 1985;149:507–15. doi: 10.1016/0003-2697(85)90606-2. [DOI] [PubMed] [Google Scholar]
- 27.Sagami H, Morita Y, Ogura K. J Biol Chem. 1994;269:20561–6. [PubMed] [Google Scholar]
- 28.Coates RM, Ley DA, Cavender PL. J Org Chem. 1978;43:4915–22. [Google Scholar]
- 29.Umbreit MA, Sharpless KB. J Am Chem Soc. 1977;99:5526–8. [Google Scholar]
- 30.Hammerschmidt F. Monatsh Chem. 1991;122:389–398. [Google Scholar]
- 31.Freeman GA, Rideout JL, Miller WH, Reardon JE. J Med Chem. 1992;35:3192–6. doi: 10.1021/jm00095a014. [DOI] [PubMed] [Google Scholar]
- 32.Strickland CL, Weber PC, Windsor WT, Wu Z, Le HV, Albanese MM, Alvarez CS, Cesarz D, del Rosario J, Deskus J, Mallams AK, Njoroge FG, Piwinski JJ, Remiszewski S, Rossman RR, Taveras AG, Vibulbhan B, Doll RJ, Girijavallabhan VM, Ganguly AK. J Med Chem. 1999;42:2125–35. doi: 10.1021/jm990030g. [DOI] [PubMed] [Google Scholar]
- 33.Strickland CL, Windsor WT, Syto R, Wang L, Bond R, Wu Z, Schwartz J, Le HV, Beese LS, Weber PC. Biochemistry. 1998;37:16601–11. doi: 10.1021/bi981197z. [DOI] [PubMed] [Google Scholar]
- 34.Bartlett DL, King CH, Poulter CD. Methods Enzymol. 1985;110:171–84. doi: 10.1016/s0076-6879(85)10073-x. [DOI] [PubMed] [Google Scholar]
- 35.Turek TC, Gaon I, Distefano MD. J Labeled Compds Radiopharm. 1997;39:139–146. [Google Scholar]
- 36.McMullen AI, McSweeney GP. Biochem J. 1966;101:42–47. doi: 10.1042/bj1010042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Czuppon AB, Chen Z, Rennert S, Engelke T, Meyer HE, Heber M, Baur X. J Allergy Clin Immunol. 1993;92:690–7. doi: 10.1016/0091-6749(93)90012-5. [DOI] [PubMed] [Google Scholar]
- 38.Yeang HY, Cheong KF, Sunderasan E, Hamzah S, Chew NP, Hamid S, Hamilton RG, Cardosa MJ. J Allergy Clin Immunol. 1996;98:628–39. doi: 10.1016/s0091-6749(96)70097-0. [DOI] [PubMed] [Google Scholar]
- 39.Dennis MS, Light DR. J Biol Chem. 1989;264:18608–17. [PubMed] [Google Scholar]
- 40.Durauer A, Csaszar E, Mechtler K, Jungbauer A, Schmid E. J Chromatogr, A. 2000;890:145–58. doi: 10.1016/s0021-9673(00)00241-7. [DOI] [PubMed] [Google Scholar]
- 41.Mayer MP, Prestwich GD, Dolence JM, Bond PD, Wu HY, Poulter CD. Gene. 1993;132:41–7. doi: 10.1016/0378-1119(93)90512-2. [DOI] [PubMed] [Google Scholar]
- 42.Stirtan WG, Poulter CD. Arch Biochem Biophys. 1995;321:182–90. doi: 10.1006/abbi.1995.1384. [DOI] [PubMed] [Google Scholar]
- 43.Zhang FL, Diehl RE, Kohl NE, Gibbs JB, Giros B, Casey PJ, Omer CA. J Biol Chem. 1994;269:3175–80. [PubMed] [Google Scholar]
- 44.Cassidy PB, Dolence JM, Poulter CD. Methods Enzymol. 1995;250:30–43. doi: 10.1016/0076-6879(95)50060-x. [DOI] [PubMed] [Google Scholar]
- 45.Pompliano DL, Gomez RP, Anthony NJ. J Am Chem Soc. 1992;114:7945–6. [Google Scholar]
- 46.Krishnakumar R, Cornish K, Jacob J. J Rubber Res (Kuala Lumpur, Malaysia) 2001;4:131–139. [Google Scholar]
- 47.Cornish KSDJ, Grosjean O-K, Goodman N. J Nat Rubb Res. 1993;8:275–285. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Image of the SDS-PAGE used for the first mass spectral analysis of the photolysis reaction of Hevea Brasiliensis washed rubber particles with [32P]3b. General methods and materials. Copies of 1H-NMR, 31P-NMR, and mass spectral data for 3b as well as the intermediates in its synthesis. This material is available free of charge via the Internet at http://pubs.acs.org.