

Abstract
Purpose
BRIP1 (FANCJ/BACH1), which encodes a DNA helicase that interacts with BRCA1, has been suggested to be a low penetrance breast cancer predisposing gene. We aimed to assess whether BRIP1 mutations contribute to breast cancer susceptibility in our population and, if so, to investigate the impact of such mutation(s) on BRIP1 function.
Experimental Design
A series of 49 breast/ovarian cancer families, devoid of a BRCA1/BRCA2 mutation, were screened for BRIP1 mutations. Functional analyses, including co-immunoprecipitation and stability assays, were employed to further characterize a previously unreported variant.
Results
Five sequence alterations were identified, of which four had been already described. Herein, we report a novel BRIP1 germ line mutation identified in a woman with early onset breast cancer. The mutation consists of a four-nucleotide deletion (c.2992-2995delAAGA) in BRIP1 exon 20 that causes a shift in the reading frame, disrupts the BRCA1-binding domain of BRIP1 and creates a premature stop codon. Functional analysis of the recombinant mutant protein in transfected cells showed that the truncation interferes with the stability of the protein and with its ability to interact with BRCA1. Loss of the wild type BRIP1 allele with retention of the mutated one was observed in the patient’s breast tumor tissue.
Conclusions
These results, by showing that the newly identified BRIP1 c.2992-2995delAAGA mutation is associated with instability and functional impairment of the encoded protein, provide further evidence of a breast cancer-related role for BRIP1.
Keywords: BRIP1, breast cancer susceptibility, BRCA1, tumor suppressor genes, Fanconi anaemia
INTRODUCTION
It is estimated that approximately 5-10% of all breast cancer cases result from an inherited predisposition to the disease (1). Thus far, two major high penetrance susceptibility genes have been identified: BRCA1 and BRCA2. Inheritance of a defective allele of either gene is sufficient to confer breast cancer predisposition with an estimated lifetime risk of ∼35-80% ((2-5) and reviewed in (6)). However, mutations in the BRCA genes fail to explain occurrence of the neoplasia in all breast cancer prone families (7). Other well-established cancer susceptibility genes, such as TP53, PTEN, and LKB1, contribute only to a small proportion of breast cancer in the general population, as their germ line mutations are very rare. The remainder breast cancer risk is, as yet, unexplained and likely due to cooperative defects in a larger number of low penetrance genes rather than to mutations in highly penetrant ones (7). In keeping with this notion, suitable candidates have been analyzed in search for functionally deficient variants that segregate with the cancer phenotype. Genes whose products are known to interact with BRCA1 and/or BRCA2 and to be involved in DNA repair or in the regulation of cell cycle progression are usually prioritized for such investigation (8). As a result, mutations of ATM, CHEK2, RAD50, Nibrin, PALB2 and BRIP1 have been found to associate with a modest (approximately two-fold) increase of breast cancer risk (9-14).
BRIP1 (BRCA1-interacting protein 1), also known as BACH1 (BRCA1-associated C-terminal helicase 1) was first identified as a 1249 amino acid (aa)-protein that interacts with the BRCA1 BRCTs in vivo (15). BRIP1 is ubiquitously expressed, it co-localizes with BRCA1 at sites of DNA damage, and contributes to its DNA repair function (15). Specifically, BRIP1-BRCA1 interaction, which has been shown to depend upon the cell cycle-regulated phosphorylation of BRIP1 at Serine 990 (16-18), is required for timely repair of DNA double strand breaks and for DNA damage-induced checkpoint control during the G2/M phase of the cell cycle (17).
Interestingly, BRIP1 maps to chromosome 17q22 near the BRCA1 locus (15). The frequent documentation of breast carcinomas that are wild-type for the BRCA genes but exhibit allelic losses encompassing the 17q21-q22 region, suggests that this chromosomal region may harbor an additional breast cancer susceptibility gene (19). Thus, both its functionally relevant interaction with BRCA1 and its chromosomal location render BRIP1 a candidate tumor suppressor gene.
In addition, the BRIP1 N-terminus shares substantial sequence homology to the catalytic and nucleotide-binding domains of known members of the DEAH helicase family. Indeed, BRIP1 was shown to be a bona fide ATP-dependent, 5′-3′ DNA helicase (20). Mutations in helicases (such as XPB, XPD, WRN, BLM, RecQL4) have been described in association with human genetic disorders (Xeroderma pigmentosum, Werner syndrome, Bloom syndrome, and Rothmund-Thomson syndrome, respectively), which are characterized by genome instability and predisposition to cancer (reviewed in (21-24)).
The demonstration that certain BRIP1 mutations, detected in women with early onset breast cancer, result in the synthesis of enzymatically defective proteins (20) provides biochemical support for the aformentioned notion that BRIP1 is a breast cancer gene. However, thus far, studies have failed to find any highly penetrant BRIP1 mutations, and most of the newly identified sequence alterations have not been resolved in terms of their biological significance (14, 25-28).
More recently, BRIP1 was shown to be bi-allelically inactivated in patients with Fanconi Anaemia (FA), a genetic disease (which can be autosomal recessive or X-linked) characterized by multiple congenital abnormalities, bone marrow failure, cellular hypersensitivity to interstrand DNA cross-linking agents and susceptibility to cancer (29). Specifically, BRIP1 mutations were found in patients with FA belonging to the complementation group J (30-33). Similarly, a bi-allelic inactivation of the breast cancer gene BRCA2 has been previously described in patients with FA complementation group D1 (34). More recently, bi-allelic defects in PALB2 have been demonstrated to cause FA subtype N (35, 36).
The aim of our study was to evaluate the contribution of alterations at the BRIP1 locus to breast cancer susceptibility in a cohort of 49 breast or breast/ovarian families in which no BRCA1/BRCA2 mutations were detected.
MATERIALS and METHODS
Family recruitment
A total of forty nine breast or breast/ovarian cancer patients belonging to as many families mainly originating from Central-Northern Italy and referred to the University Hospital in Pisa (37), were defined as probands (index individuals) and were analyzed for BRIP1 germ line mutations. All probands displayed no detectable deleterious mutations upon full screening of BRCA1 and BRCA2 genes. In addition, when tested for the CHEK2 1100delC disease-linked mutation (11), they all resulted negative. Out of the 49 families, 40 (80%) were associated with breast cancer, 9 (20%) with both breast and ovarian cancer (Supplementary table 1). Most of the families included three or more breast/ovarian cancer cases. Thirty-five pedigrees showed also cases of other types of neoplasia. The average age at diagnosis of breast cancer among patients was 43.8 years (yrs), ranging between 21 and 73 yrs (Supplementary table 1). Family histories were obtained through detailed interviews and pedigrees were traced as far backward and laterally as possible. Families were classified as high-risk or moderate-risk cancer families depending on the criteria they met. Inclusion criteria for the 38 high risk-families were as follows: 1) three or more cases of breast and/or ovarian cancer in first-/second-degree relatives, or 2) two or more cases of breast and/or ovarian cancer in first-/second-degree relatives, of which at least one with bilateral or early onset (≤40 yrs) breast cancer or with breast and ovarian cancer. Each of the 4 moderate-risk families displayed two cases of breast/ovarian cancer in first-/second-degree relatives. In addition, 7 families with single cases of bilateral or early onset breast cancer or breast/ovarian cancer along with multiple (two or more) cases of other kinds of tumors in first-/second-degree relatives were included in the study. Written informed consent to collect pedigree data and a blood specimen for a study on cancer susceptibility was obtained from all probands. As controls, fifty individuals, from the same geographical region as the cancer cases, were anonymously recruited. They were all cancer-free at the time of blood donation and their age ranged between 21 and 87 yrs (average 48.5 yrs).
Mutation screening
A DNA sample of the index individual of each cancer family was analyzed. Genomic DNA was extracted from peripheral blood lymphocytes using a QIAamp DNA Mini kit (Qiagen). Twenty-two fragments of the BRIP1 gene, including both coding exons and flanking intronic sequences, were amplified by PCR using Taq Gold polymerase (Applied Biosystems). The size of PCR products ranged between 214 and 380 basepairs (bp). Screening for BRIP1 mutations was carried out by Single Strand Conformation Polymorphism (SSCP). Genomic DNA from cases showing an altered SSCP migration pattern was amplified and analyzed by bidirectional sequencing using a BigDye Terminator v3.1 Cycle Sequencing Ready Reaction kit (Applied Biosystems) on an ABI model 3100 capillary sequencer. Oligonucleotides for SSCP and sequencing were designed using the Primer3 Software based upon the sequence information obtained from the Genbank databases. Primer sequences and PCR conditions are available upon request.
Loss of heterozygosity analysis
Loss of heterozygosity (LOH) analysis was carried out on tumor tissue excised from the index individual of the Pisa#88 family. Laser-capture microdissection was performed to isolate pure populations of neoplastic and normal breast cells from 5 micron (μm) sections of the formalin-fixed paraffin-embedded breast carcinoma tissue block. The DNA was extracted from the dissected tumor and normal cells using a QIAamp DNA Mini Kit (Qiagen). Amplification was carried out by PCR using primers located in exon 20, and the resulting products were analysed by sequencing.
Plasmids
The full-length BRIP1 cDNA-containing pENTR/D-TOPO (wtBRIP1 pENTR/D-TOPO) vector (15), served as template to generate a 4 nucleotide (nt)-deletion (Δ) at the relevant site using the QuickChange mutagenesis kit (Stratagene). After sequencing to confirm the mutation, the ΔBRIP1-pENTR/D-TOPO vector was modified to insert an in frame Flag-encoding C terminal sequence (ΔBRIP1Flag-pENTR/D-TOPO). Next, both ΔBRIP1 and ΔBRIP1 Flag-encoding cDNAs were transferred, by LR recombinase-mediated reaction (Invitrogen), into a pcDNA-DEST47 destination vector (Invitrogen) for expression in mammalian cells. The ΔBRIP1-pcDNA-DEST47 vector was further used as template for PCR amplification. The PCR product was NotI-/ApaI-digested and subcloned into a pcDNA3.1 vector (Invitrogen) encoding a C-terminal Myc-6xHys tag (ΔBRIP1-pcDNA3.1/MycHis). All cloned cDNA constructs were fully sequenced prior to use. The mammalian expression vector pcDNA3.1 Myc-6xHis containing full length wtBRIP1 cDNA (wtBRIP1-pcDNA3.1/MycHis) was described previously (15). pEGFP-C2 (Clontech) and pRL-TK (Promega) plasmids were used as controls to ensure efficiency of transfection.
In vitro transcription and translation
Expression plasmids, generated as described above, and encoding the wt and the ΔBRIP1 protein products, were in vitro transcribed and translated using a TNT kit (Promega) according to the manufacturer’s specifications.
Establishment of a lymphoblastoid cell line
A lymphoblastoid cell line (LCL) was established by EBV-infection of Pisa#88 proband’s peripheral blood lymphocytes using standard protocols, and served as a source for DNA, RNA and proteins (detailed below).
DNA extraction and PCR amplification and analysis
Genomic DNA was extracted from the Pisa#88 LCL using a QIAamp DNA Mini Kit (Qiagen), and amplified with appropriate primer pairs depending on the applications. A number of cycles as low as 26 was used to avoid PCR artefacts which might be cause of preferential allelic amplification. Analysis of PCR products by bidirectional sequencing was carried out as described above. For electrophoretic analysis, PCR products were resolved on 4% Low Melting Point (LMP) agarose (Gibco) or 8% polyacrylamide denaturing gels, stained with Ethidium bromide and SYBR Gold (Invitrogen), dilution 1:10,000, respectively. The 4 bp length difference caused by the mutation allowed direct discrimination of the mutant and wt fragments (amplified with a pair of primers specifically designed to give rise to a small product encompassing the mutation site). Bands were visualized under UV light. The Image J software was utilized for quantitative analysis of the intensity of the bands.
Cell culture and transfection
HeLa, U2OS and 293T cells were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Gemini) and antibiotics in a humidified incubator at 37°C with an atmosphere containing 10% CO2. Grown to 50% confluence, cells were transfected with 5 to 10 micrograms of wtBRIP1 or ΔBRIP1-pcDNA3.1/MycHis constructs using FuGene 6 transfection reagent (Roche Diagnostic).
RNA extraction, cDNA synthesis and RT-PCR amplification and analysis
RNA was isolated from the Pisa#88 LCL or from GFP-, Myc-wtBRIP1- or Myc-ΔBRIP1-transfected 293T cells using the RNeasy mini kit (Qiagen). cDNA synthesis was carried out utilizing Superscript II RT Rnase H- reverse transcriptase (Invitrogen) and random hexamers (Invitrogen) according to the manufacturer’s instructions. RT-PCR to analyze the expression levels of endogenous transcripts in the Pisa#88 LCL was carried out with the same primer pair used for DNA, and the amplified fragments were discriminated on agarose and polyacrylamide gels, as previously detailed.
RT-PCR to compare mutant vs wt transcripts in transfected cells was carried out using specific primer pairs that allowed selective amplification of the exogenously expressed constructs. In both cases, mock RT-PCR reactions (from mRNA reverse-transcribed with no addition of reverse transcriptase) served as controls, and confirmed the absence of DNA contamination.
Immunoprecipitation, immunoblotting, and protein stability analysis
Forty-eight hours after transfection, cells were collected, washed with phosphate buffer saline (PBS) (pH 7.4), and lysed in NETN buffer (0.5% NP-40, 20 mM Tris (pH 8.0), 150 mM NaCl, and 1 mM EDTA) supplemented with 1 tablet/50 ml of Complete protease inhibitor cocktail (Roche). Lysates were cleared by centrifugation (16000g for 10 min) and incubated on ice for 1 hour with 1-2 μg of the relevant antibody (Ab). Next, 20 μl of slurry protein A or G Sepharose beads (Amersham Biosciences) were added to each lysate, and incubation continued with rotation for 40 min at 4°C. Beads were retrieved by centrifugation and washed three times in NETN buffer. Proteins bound to the beads were eluted by boiling in Laemmli sample buffer, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and electrotransferred to nitrocellulose membranes (Invitrogen). After blocking in 5% milk, western (W)-blot assays were carried out with the indicated primary Abs for 1 hour at room temperature, followed by incubation with horseradish peroxidase-conjugated secondary Abs for 1 hr at room temperature. Aliquots of lysates were also run and blotted for tubulin as a control for equal protein loading. Equal transfection efficiency among the samples was ensured by western blot detection of the co-transfected pEGFP-C2 plasmid. All blots were developed using ECL-plus western blotting detection system (Amersham Biosciences) and Biomax BLUE XB-1, MR or XAR films (Kodak). Cycloheximide-chase experiments were performed as described (38). The Image J software was used for densitometric analysis of the films.
To obtain whole cell lysates (WCEs) from EBV-immortalized lymphocytes, the cells were harvested, washed once in PBS, and lysed in NETN buffer, as previously described. WCEs were separated by SDS-PAGE and subjected to W-blotting as detailed above.
Antibodies
Two different rabbit polyclonal Abs, I-104 and I-82, were generated against BRIP1 in New Zealand White Rabbits. I-104 was produced against a C-terminal peptide of BRIP1 (N-KTHEIEIKNFKPSPSKNKGM-C). I-82 was generated against a recombinant full-length flag-tagged wild-type BRIP1 (rBRIP1) produced in High Five insect cells. I-82 was affinity-purified using rBRIP1 coupled to a solid support. The anti-BRCA1 rabbit antiserum was purchased from Upstate Biotechnology. The anti-BARD1 affinity-purified polyclonal antibody was purchased from Bethyl Laboratories Inc. The following monoclonal antibodies were used: anti-BRCA1 MS110 (39); anti-Myc, clones 4A6 (Upstate Biotechonology) and 9B11 (Cell Signaling Technology); anti-αtubulin, clone DM1A (Sigma); anti-GFP, clone C163 (Zymed Laboratories Inc). Secondary antibodies used for immunodetection were: HRP-conjugated sheep anti-mouse IgG and donkey anti-rabbit Ig (Amersham Biosciences) (dilution 1:5,000).
RESULTS
Mutational analysis of the BRIP1 gene performed in our cohort of 49 index individuals led to the identification of four germ line sequence alterations within the BRIP1 coding region together with one intronic variant (Table 1). Three sequence changes (c.2637G>A, c.2755C>T, c.3411C>T) had already been reported with comparable allelic frequencies (15, 25, 26, 28) and all were classified as neutral polymorphisms. The intronic variant IVS5-31 C>G in the region located between exons 5 and 6 was previously reported, as well (27). Neither of the two heterozygous missense mutations (c.139C>G, c.897G>A) described by Cantor et al. (2001) were found in our set of patients.
Table 1. BRIP1 sequence alterations.
| Exon/Intron | Nucleotide changea | Effect on protein | Allele frequency b (%) |
|---|---|---|---|
| Intron 5 | IVS5-31C>G | unknown | 1/98 (1%) |
| Exon 19 | 2637G>A | Glu879Glu | 32/98 (33%) |
| Exon 19 | 2755C>T | Pro919Ser | 48/98 (49%) |
| Exon 20 | 2992-2995delAAGA | Glu998fsX1057 | 1/98 (1%) |
| Exon 20 | 3411C>T | Tyr1137Tyr | 81/98 (83%) |
Numbering based on RefSeq NT_010783.13 (for the intronic variant) and NM_032043 (for all the variants within the coding region). For exonic variants, numbering starts at codon 1.
Frequency of second allele listed.
Interestingly, we identified a novel heterozygous sequence change in the last exon (exon 20) of BRIP1: a four nt-deletion starting at 2992 (c.2992-2995delAAGA) that causes a slippage of the open reading frame, leading to a premature stop at codon 1057 (Figure 1A and B). The sequence from the deletion to the stop bears no homology to wtBRIP1. Importantly, the aa change (aa 998-1057) caused by the newly identified mutation partially encompasses the BRCA1 binding domain (aa 979-1006) of BRIP1 (20) and might be predicted to affect the interaction between BRIP1 and BRCA1.
Figure 1. A novel BRIP1 sequence alteration identified in the family Pisa#88.
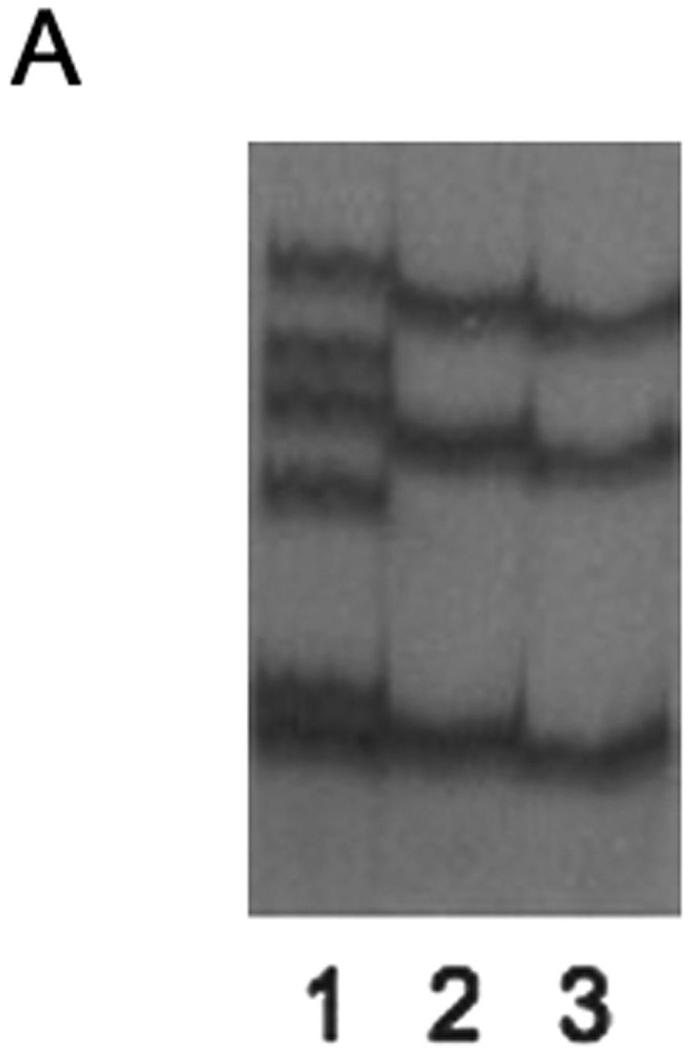

(A) Identification of a novel BRIP1 sequence variant. SSCP analysis after PCR amplification using primers located in BRIP1 exon 20 reveals an altered electrophoretic pattern in the sample corresponding to the proband of the family Pisa#88 (lane 1). Lanes 2 and 3, corresponding to the families Pisa#61 and Pisa#137, respectively, show a normal migration pattern, as ascertained in healthy control individuals (data not shown). (B) Characterization by sequence analysis of the BRIP1 gene alteration detected in the index case of the family Pisa#88. The chromatogram displays a 4 nucleotide-deletion, which starts at position 2992 (Genebank database sequence reference NM_032043, nucleotide numbering starting at codon 1), as indicated by an asterisk, and causes a shift of the open reading frame (ORF). The sequence of the wt and mutated alleles appear overlapped. The wt and mutated cDNA sequences are detailed (starting point marked by an arrowhead in the chromatogram). The corresponding amino acid sequences are reported, as well. The ORF shift (starting at codon 998) caused by the deletion results in a premature stop codon at position 1057. (C) Pedigree of the family Pisa#88, exhibiting the c.2992-2995delAAGA mutation. Black-filled symbols indicate individuals diagnosed with breast cancer (Br= breast cancer). Grey-filled symbols indicate individuals diagnosed with cancer other than breast (Co= colon cancer, Lu= lung cancer, Lym= lymphoma). The arrowhead indicates the index case.
The c.2992-2995delAAGA mutation was detected in a 49 year-old woman that had been diagnosed with breast cancer at the age of 40 and exhibited a family history of multiple cancers (Figure 1C). No breast cancer cases other than the proband and her mother (who was diagnosed at the age of 76) were known to have occurred in the family. However, the pedigree displayed two cases of colorectal cancer in the proband’s paternal aunt and grandmother. Moreover, both brothers of the paternal grandmother had been diagnosed with a neoplastic disease (lymphoma and lung cancer). Unfortunately, since DNA samples from any other suitable family member(s) were not available, co-segregation of the mutated allele with the cancer phenotype through the family could not be tested. Nevertheless, when 50 matched healthy control individuals were analyzed, they all tested negative for the c.2992-2995delAAGA mutation.
BRIP1 would conform to the classic paradigm for tumor suppressor genes if loss of the wild-type allele could be detected in tumor cells isolated from the affected individual. In order to address this possibility, we searched for LOH at the BRIP1 locus in breast carcinoma tissue from the patient carrier of the novel mutation. Remarkably, sequence analysis of the DNA extracted from the laser capture-microdissected specimen revealed that loss of the wild-type allele had occurred in the tumor cells, with retention of the mutant allele (Figure 2). This finding underscores the notion that BRIP1 has tumor-suppressive function and might participate in breast tumorigenesis.
Figure 2. Breast tumor tissue from Pisa#88 proband displays LOH at the BRIP1 locus.

Chromatograms of tumor and normal breast tissue DNA from Pisa#88 index individual. Loss of the wild type allele with retention of the mutant one is evident in the tumor sequence (upper panel). Normal tissue (lower panel) shows both mutated and wt alleles (an asterisk marks the beginning of the overlapping sequences).
We then sought to elucidate the possible biological consequences of the mutant BRIP1 allele by investigating whether the deletion had any effect on the function of the corresponding protein. Since the frameshift caused by the mutation was predicted to give rise to a premature translational stop, we decided to characterize the truncated protein product. First, we generated a number of BRIP1 constructs bearing the relevant mutation (ΔBRIP1), with or without a C-terminal tag (Flag or Myc). As a control, we utilized the wtBRIP1-pcDNA3.1/MycHis described previously (15). In vitro transcription and translation reactions were programmed with the wt and mutant plasmids. The obtained products were resolved by SDS-PAGE and analyzed by western blotting using Abs against BRIP1 or the Myc tag. As predicted, all the ΔBRIP1 constructs generated electrophoretically-distinguishable protein products that migrated approximately 20 kDa faster than the wtBRIP1 one, consistent with the expected size for ΔBRIP1 being ∼200 aa shorter than the wt protein (Figure 3A, lanes 3-5 vs 2). In addition, we ascertained that the mutant BRIP1 was not detected by an antibody generated against the extreme C-terminus of the protein whereas, under the same conditions, the full-length wtBRIP1 was easily recognized (Figure 3B). These results demonstrate that the BRIP1 c.2992-2995delAAGA mutation leads to the synthesis of an identifiable truncated protein that lacks the C-terminus.
Figure 3. BRIP1 2992-2995delAAGA gives rise to a truncated protein that is not detectable in vivo by western blot analysis.

(A) Electrophoretic mobility of endogenous (lane 1) and in vitro transcribed and translated BRIP1 (IVT-BRIP1) proteins (lanes 2-5). IVT-ΔBRIP1 proteins (lanes 3-5) exhibited faster electrophoretic mobility compared to the IVT-wtBRIP1 (lane 2), consistent with the smaller size of the mutated protein products. Flag- and Myc-tagged IVT-ΔBRIP1 proteins (lanes 4-5) migrated slightly slower than IVT-ΔBRIP1 with no tag (lane 3). (B) The I82 Ab, generated against full-length rBRIP1, recognized both IVT-wtBRIP1 and IVT-ΔBRIP1 proteins (upper blot). The I104 Ab, raised against BRIP1 extreme C-terminus, failed to recognize BRIP1 while still being able to react with wtBRIP1 (lower blot, lane 2 vs 1). (C) Western-blot analysis of EBV-immortalized lymphocytes from the Pisa#88 proband revealed absence of a truncated BRIP1 protein (lane 5). IVT-wt/-ΔBRIP1 proteins (lanes 1-3), 293T cell extract (lane 4) and EBV-immortalized lymphocyte lysates from unrelated individuals (lanes 6-8) were run as controls. Molecular mass, in Kilodaltons (kDa), is indicated on the left of each blot.
Next, we aimed to determine the expression of the ΔBRIP1 protein in vivo. For this purpose, a lymphoblastoid cell line was established, by EBV-infection, from the peripheral blood lymphocytes of the heterozygous patient, and analysis of the endogenous BRIP1 protein was performed by W-blotting. Only a wtBRIP1 product was observed when either a polyclonal antibody (Figure 3C, lane 5) or a pool of monoclonal antibodies (data not shown) were used. This observation indicates that ΔBRIP1, if expressed, is significantly less abundant than wtBRIP1 in EBV-immortalized lymphocytes.
Since mRNAs bearing a premature termination codon (PTC) are known to be targeted for degradation via a surveillance mechanism named nonsense-mediated mRNA decay (NMD) (reviewed in (40, 41)), we searched for an imbalanced expression of the transcripts (mutant vs wt) that could explain the absence of a detectable mutant protein. As a result, direct sequencing and polyacrylamide gel electrophoresis analysis of RT-PCR products revealed the presence of both mutant and wt alleles in the Pisa#88 LCL (Supplementary figure 1A and B). Furthermore, the expression levels of both transcripts appeared comparable, as confirmed after quantitation of the band intensities (Supplementary figure 1B and C, and data not shown). This finding is consistent with the notion that PTCs residing within the last exon do not trigger NMD (42-44) and argues against preferential allelelic degradation as an explanation for the inability to detect the mutant protein.
We, therefore, reasoned that the lack of expression of ΔBRIP1 in the patient’s immortalized lymphocytes could be due to the inherent instability of the mutant protein. To test this possibility, we set out to characterize an exogenously over-expressed ΔBRIP1 protein in cultured cells. HeLa cells were transiently transfected with Myc-wtBRIP1 or Myc-ΔBRIP1 encoding constructs. Using anti-Myc or anti-BRIP1 antibodies, we were unable to detect a mutant protein product of the predicted size (data not shown). A low expression of the truncated protein was observed after transfection of the relevant constructs in U2OS cells (data not shown). We then exploited the 293T cell line, which expresses SV40 large T antigen, to allow T antigen-dependent amplification of the SV40 origin-containing plasmids. Even in this setting, the expression of the ΔBRIP1 protein was lower than the expression of the wt counterpart (Figure 4A). Co-transfection with GFP- or Luciferase-encoding plasmids allowed us to confirm equal efficiency of the transfection for both wt and ΔBRIP1 constructs (Figure 4A and data not shown). Furthermore, we ruled out that the mutation could affect the protein by rendering ΔBRIP1 insoluble: indeed, the mutated protein was easily extractable in cell lysis buffer from transfected cells with no significant residual amount detected in the pellet (data not shown). Moreover, a fairly equal level of expression for both Myc-wtBRIP1 and Myc-ΔBRIP1 transcripts was demonstrated by RT-PCR carried out with primers specifically designed to amplify the exogenous mRNAs (Supplementary figure 2). These observations suggest that the lower level of ΔBRIP1 detected after transfection might be due to instability of the truncated protein caused by the mutation.
Figure 4. The recombinant ΔBRIP1 is unstable when expressed in 293T cells.


(A) Lysates from 293T cells transfected with a GFP-encoding plasmid alone (lane 1) or in combination with either Myc-wtBRIP1- (lane 2) or Myc-ΔBRIP1-encoding constructs (lanes 3-5) were resolved by SDS-PAGE and probed with the indicated Abs. A significantly reduced abundance of ΔBRIP1 compared to wtBRIP1 was evidenced with both anti-Myc (lane 3 vs lane 2) and anti-BRIP1 (lanes 3-5 vs lane 2) Abs. (B) Cycloheximide-chase analysis of 293T cells expressing Myc-tagged wt or ΔBRIP1 proteins. Equal amounts of lysates, prepared at the indicated time points, were analyzed by western blot with anti-Myc and anti-tubulin Abs. A significant decrease of the ΔBRIP1 signal intensity was detectable already after 1 hour, as opposed to the steady level of wtBRIP1 signal. The experiment was repeated three times with consistent results. (C) Densitometric analysis of a representative cycloheximide chase experiment. Films were scanned and analyzed using the Image J software. The intensity of each band, after normalization to tubulin, which served as loading control, was plotted to determine the half-lives of the wt and mutant BRIP1 proteins.
We sought to address this possibility by utilizing a cycloheximide-chase method (38) to determine the half-lives of both mutant and wt proteins. Transient transfection of 293T cells using equal amounts of the aforementioned Myc-wtBRIP1- and Myc-ΔBRIP1-encoding constructs, was performed followed by cycloheximide treatment to block new protein synthesis. When cell lysates obtained at different time points were blotted with an anti-Myc specific Ab, ΔBRIP1 appeared to be, indeed, significantly less stable than wtBRIP1 (Figure 4B). An estimated half-life of ∼1 hour was observed for the mutant protein, whereas wtBRIP1 was shown to be significantly more stable, having a half-life of ∼5 hours (Figure 4C), consistent with previous findings (15). These results indicate that the c.2992-2995delAAGA mutation impairs the stability of the corresponding truncated BRIP1 protein.
Although these studies show that the mutant BRIP1 protein is unstable, we were intrigued by the fact that the c.2992-2995delAAGA mutation disrupts the previously described BRCA1-binding domain (15, 20) and wondered whether ΔBRIP1, even in low concentration, could still interact with BRCA1. To this end, we attempted to assess, by co-immunoprecipitation (Co-IP) analysis, the potential impact of the novel BRIP1 sequence change on the BRIP1-BRCA1 interaction in vivo. 293T cells were transfected with GFP and either Myc-wtBRIP1- or Myc-ΔBRIP1-encoding constructs, and IP with an anti-Myc monospecific antibody was carried out, followed by W-blotting for BRCA1. Co-immunoprecipitated BRCA1 was always nearly absent in the IP from ΔBRIP1-transfected cells, whereas a clearly detectable BRCA1 signal was observed even when serial dilutions of the Myc-IP from wtBRIP1-transfected cells were loaded onto the gel (Figure 5). These results indicate that ΔBRIP1, which bears a significantly altered BRCA1-binding domain, is impaired in its ability to interact with BRCA1. In addition, because BRIP1 and BARD1 have been reported to indirectly interact with one another, BRCA1 being the mediator of such interaction (15), we asked whether the defective binding of ΔBRIP1 to BRCA1 translated into a defective interaction with BARD1, as well. Indeed, in Myc-ΔBRIP1 complexes the amount of co-immunoprecipitated BARD1 was significantly decreased compared to what observed in Myc-wtBRIP1 complexes (Figure 5). Furthermore, results from reciprocal IPs (i.e. BRCA1 IP followed by western blotting for Myc) consistently showed a significantly impaired interaction between BRCA1 and the mutant BRIP1 protein (Supplementary figure 3).
Figure 5. ΔBRIP1 exhibits defective ability to interact with BRCA1.

293T cells were transfected with Myc-wtBRIP1-, Myc-ΔBRIP1- or GFP-encoding plasmids. Lysates (lanes 1-3) and corresponding Myc-IPs (lanes 4-8) were analyzed by W-blotting with the indicated antibodies. BRCA1 and BARD1 were detected in Myc-wtBRIP1 complexes even when the IP was diluted up to 10 fold (lanes 4-6), whereas they were both barely detectable in undiluted Myc-ΔBRIP1 complexes (lane 7). IP from GFP-transfected cells (lane 8) served as control.
Taken together these results indicate that the c.2992-2995delAAGA mutation affects BRIP1 function and impairs its ability to interact with BRCA1.
DISCUSSION
Our goal was to evaluate whether and to what extent defects in the BRIP1 gene contribute to familial breast cancer in selected BRCA1-/BRCA2-negative breast/ovarian cancer families. The mutational screening we carried out led to the identification of five BRIP1 sequence changes in a series of 49 affected individuals. Four alterations (IVS5-31C>G, c.2637G>A, c.2755C>T, c.3411C>T) had already been reported (15, 25-27), of which only one (c.2755C>T) causes a non synonymous aa substitution and introduces a Ser (Pro919Ser) in the BRCA1 binding domain thus generating a potential phosphorylation site. An association between such polymorphism and an increased breast cancer risk by the age of 50 years has been suggested (45) but it is, as yet, not confirmed (28, 46).
The fifth sequence variant we characterized (c.2992-2995delAAGA) is a unique mutation that causes a change of 9 out of 28 aa within the BRCA1-binding domain (979-1006) of BRIP1 and introduces a premature stop codon. The mutation was identified in a woman diagnosed with early onset breast cancer who lacked a strong family history for the disease. Importantly, we found LOH at the BRIP1 locus in breast cancer tissue from the same patient.
To our knowledge, this is the first report of a heterozygous germ line mutation in the BRIP1 gene, which gives rise to a truncated protein product, predicted to be impaired in its biological role, and is associated with loss of the wtBRIP1 allele in the corresponding breast tumor tissue. The latter result is consistent with the classic Knudson’s two hit hypothesis for loss of tumor suppressor function (47) and supports the speculation that the novel mutation we describe herein might have contributed to the development of breast cancer in the patient. Notably, although most of the BRIP1 sequence alterations described so far associated with Fanconi Anaemia have a truncating effect (31, 32), the c.2992-2995delAAGA mutation was not previously reported in FA-J patients.
Even though analysis of BRIP1 transcripts evidenced a fairly equal expression of both wt and mutant alleles (thus suggesting that the c.2992-2995delAAGA mutation, which falls in BRIP1 last exon, is not subject to NMD (43)), W-blotting on lysate from Pisa#88 LCL revealed undetectable levels of endogenous mutant protein. Consistent with this observation, the recombinant ΔBRIP1 protein was inefficiently expressed in transfected cells because it is unstable. Indeed, instability of mutated BRIP1 proteins has already been reported in studies where cells from the heterozygous carriers could be directly tested (15). Furthermore, truncated BRIP1 species have not been detected, so far, in FA-J cells (31-33).
Given the instability of the truncated protein, the interaction with BRCA1 being preserved or disrupted would likely represent an irrelevant point in vivo. However, one cannot rule out the possibility that ΔBRIP1 is still expressed at low levels in tissues other than lymphoid. When co-immunoprecipitation experiments were performed in transfected cells, we observed that the described c.2992-2995delAAGA mutation affects BRIP1 interaction with BRCA1. In fact, even though both Ser990 and Phe993 (recently suggested to play a major role in anchoring BRIP1 binding to the first and second BRCT, respectively (16)) are preserved in BRIP1, it is conceivable that the altered aa sequence starting at 998 might have an impact on the interaction.
Interestingly, disease-linked mutations occurring in the BRCT domains are sufficient to abrogate BRCA1-BRIP1 interaction in vivo (48). Furthermore, mutations within the helicase domains of BRIP1 were shown to interfere with double-strand break repair in a BRCA1-binding dependent manner (20). One may, therefore, hypothesize that impairment or loss of such interaction, due to BRIP1 mutations that perturb the stability of the protein and/or its ability to bind to the partner, may be a critical cancer predisposing event. In addition, more recent studies have reported BRCA1-independent functions of BRIP1 (49), and our observations by protein chromatography (R. Drapkin, unpublished) suggest that the majority of BRIP1 exists in a native complex without BRCA1. In such a scenario, one can imagine that in the patient’s breast tumor tissue, which, as shown here, has lost the wt allele, the instability of ΔBRIP1 combined with possible functional defects of the residual mutant protein might render the tumor cells virtually BRIP1-deficient, thus having a dramatic impact on biological processes in which BRIP1 is involved.
Taken together, our data suggest a causative role for BRIP1 in breast cancer disease reinforcing the possibility that BRIP1 mutations may account for a small percentage of inherited breast cancer cases. A recent model of breast cancer susceptibility suggests that a sizeable fraction of the still unaccounted familial aggregation (50) maybe best explained by a large number of low penetrance genes whereby their individual small effects combine multiplicatively (7, 51). Of note, Seal et al. have recently reported that BRIP1 truncating mutations act as low penetrance breast cancer alleles in the British population (14). Interestingly, BRIP1 truncating mutations were found to be associated with a high relative risk for carriers aged less than 50, as opposed to BRIP1 missense variants which were described, instead, to associate with a much lower risk (14).
In conclusion, the screening we carried out revealed a previously unreported constitutive, functionally deleterious BRIP1 mutation in a woman with early onset breast cancer and, importantly, LOH at the BRIP1 locus in the corresponding breast tumor tissue. The scarce number of breast cancer cases in the Pisa#88 pedigree is consistent with the low penetrance suggested for BRIP1 mutations whereby some of the unaffected individuals might be potential carriers. Unfortunately, the inability to check for segregation of the mutation in other individuals of the same family did not allow us to explore the possible link between BRIP1 mutations and neoplasia in tissues other than breast.
It would be interesting to know whether heterozygous BRIP1 mutation carriers in FA families have an excess risk of developing breast cancer or other malignancies. So far, unlike BRCA2, whose mono-allelic mutations confer high risk of breast cancer, and whose bi-allelic mutations predispose to childhood solid and haematological malignancies (52), BRIP1 seems to be linked only to a modest risk of breast cancer. Nevertheless, recent studies suggest that BRIP1 may also be inactivated via an epigenetic mechanism in certain head and neck cancers (53). Future investigation will certainly help to gain insights into the role of BRIP1 in the FA pathway and to shed light on how that relates to its tumor suppression function.
ACKNOWLEDGEMENTS
We thank the patients and their family members for cooperation. We are deeply grateful to Prof. D.M. Livingston (Dana-Farber Cancer Institute, Boston, MA) for considerable advice and insights, Dr. V. Joukov (Dana-Farber Cancer Institute, Boston, MA) for helpful discussions and constructive criticism during the preparation of the manuscript, and Prof. S. Cantor (University of Massachusetts Medical School, Worcester, MA) for providing us with reagents and for giving us useful suggestions. We would also like to thank Dr. P. Aretini (MGM Biotecnologie SRL, Pisa, Italy) for invaluable help with artwork, Dr. J. Garber (Dana-Farber Cancer Institute, Boston, MA), Dr. R. Kennedy (Dana-Farber Cancer Institute, Boston, MA), Dr. M. Loda (Dana-Farber Cancer Institute, Boston, MA) and Dr. P. Vermeer (University of Iowa, Iowa City, IA) for reading the manuscript, Dr. C. Colin, Dr. P. O’Donovan, Dr. D. Silver for help with techniques and reagents, and all other members of the Livingston and Drapkin laboratories.
Funding: this work was supported by an AIRC (Associazione Italiana per la Ricerca sul Cancro) regional grant to M.A.C. A.D.N. is a recipient of a postdoctoral fellowship by the Susan G. Komen Breast Cancer Foundation. M.T. was funded by the Fondazione Comel at the time of this study. B.S. is a recipient of a doctoral fellowship by the Austrian Academy of Science. R.D. is supported by the NCI (K08 CA108748; Ovarian Cancer SPORE P50CA105009), the Robert and Debra First Fund, the Fannie Rippel Foundation, the Randi and Joel Cutler Ovarian Cancer Research Fund, the Phi Beta Psi Sorority Charity Trust Fund, and the Ovarian Cancer Research Fund (OCRF).
Footnotes
ELECTRONIC DATABASE INFORMATION
The URLs and accession numbers for data presented herein are as follows: GenBank, http://ncbi.nlm.nih.gov/GenBank/ for BRIP1 genomic (accession number NT_010783) and cDNA (accession number NM_032043) sequences. Primer3 Software, http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi.
REFERENCES
- 1.Rahman N, Stratton MR. The genetics of breast cancer susceptibility. Annu Rev Genet. 1998;32:95–121. doi: 10.1146/annurev.genet.32.1.95. [DOI] [PubMed] [Google Scholar]
- 2.Anglian Breast Cancer Study Group Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br J Cancer. 2000;83:1301–8. doi: 10.1054/bjoc.2000.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ford D, Easton DF, Stratton M, et al. The Breast Cancer Linkage Consortium Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am J Hum Genet. 1998;62:676–89. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336:1401–8. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- 5.Thorlacius S, Struewing JP, Hartge P, et al. Population-based study of risk of breast cancer in carriers of BRCA2 mutation. Lancet. 1998;352:1337–9. doi: 10.1016/s0140-6736(98)03300-5. [DOI] [PubMed] [Google Scholar]
- 6.Fackenthal JD, Olopade OI. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat Rev Cancer. 2007;7:937–48. doi: 10.1038/nrc2054. [DOI] [PubMed] [Google Scholar]
- 7.Antoniou AC, Easton DF. Models of genetic susceptibility to breast cancer. Oncogene. 2006;25:5898–905. doi: 10.1038/sj.onc.1209879. [DOI] [PubMed] [Google Scholar]
- 8.Walsh T, King MC. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–5. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 9.Erkko H, Xia B, Nikkila J, et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007;446:316–9. doi: 10.1038/nature05609. [DOI] [PubMed] [Google Scholar]
- 10.Heikkinen K, Rapakko K, Karppinen SM, et al. RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis. 2006;27:1593–9. doi: 10.1093/carcin/bgi360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meijers-Heijboer H, van den Ouweland A, Klijn J, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31:55–9. doi: 10.1038/ng879. [DOI] [PubMed] [Google Scholar]
- 12.Rahman N, Seal S, Thompson D, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39:165–7. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Renwick A, Thompson D, Seal S, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38:873–5. doi: 10.1038/ng1837. [DOI] [PubMed] [Google Scholar]
- 14.Seal S, Thompson D, Renwick A, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–41. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- 15.Cantor SB, Bell DW, Ganesan S, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–60. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- 16.Shiozaki EN, Gu L, Yan N, Shi Y. Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: implications for signaling. Mol Cell. 2004;14:405–12. doi: 10.1016/s1097-2765(04)00238-2. [DOI] [PubMed] [Google Scholar]
- 17.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–42. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 18.Williams RS, Lee MS, Hau DD, Glover JN. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat Struct Mol Biol. 2004;11:519–25. doi: 10.1038/nsmb776. [DOI] [PubMed] [Google Scholar]
- 19.Callahan R. Somatic mutations that contribute to breast cancer. Biochem Soc Symp. 1998;63:211–21. [PubMed] [Google Scholar]
- 20.Cantor S, Drapkin R, Zhang F, et al. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci U S A. 2004;101:2357–62. doi: 10.1073/pnas.0308717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellis NA. DNA helicases in inherited human disorders. Curr Opin Genet Dev. 1997;7:354–63. doi: 10.1016/s0959-437x(97)80149-9. [DOI] [PubMed] [Google Scholar]
- 22.Hickson ID. RecQ helicases: caretakers of the genome. Nat Rev Cancer. 2003;3:169–78. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 23.Karow JK, Wu L, Hickson ID. RecQ family helicases: roles in cancer and aging. Curr Opin Genet Dev. 2000;10:32–8. doi: 10.1016/s0959-437x(99)00039-8. [DOI] [PubMed] [Google Scholar]
- 24.van Brabant AJ, Stan R, Ellis NA. DNA helicases, genomic instability, and human genetic disease. Annu Rev Genomics Hum Genet. 2000;1:409–59. doi: 10.1146/annurev.genom.1.1.409. [DOI] [PubMed] [Google Scholar]
- 25.Luo L, Lei H, Du Q, et al. No mutations in the BACH1 gene in BRCA1 and BRCA2 negative breast-cancer families linked to 17q22. Int J Cancer. 2002;98:638–9. doi: 10.1002/ijc.10214. [DOI] [PubMed] [Google Scholar]
- 26.Karppinen SM, Vuosku J, Heikkinen K, Allinen M, Winqvist R. No evidence of involvement of germline BACH1 mutations in Finnish breast and ovarian cancer families. Eur J Cancer. 2003;39:366–71. doi: 10.1016/s0959-8049(02)00498-7. [DOI] [PubMed] [Google Scholar]
- 27.Rutter JL, Smith AM, Davila MR, et al. Mutational analysis of the BRCA1-interacting genes ZNF350/ZBRK1 and BRIP1/BACH1 among BRCA1 and BRCA2-negative probands from breast-ovarian cancer families and among early-onset breast cancer cases and reference individuals. Hum Mutat. 2003;22:121–8. doi: 10.1002/humu.10238. [DOI] [PubMed] [Google Scholar]
- 28.Vahteristo P, Yliannala K, Tamminen A, et al. BACH1 Ser919Pro variant and breast cancer risk. BMC Cancer. 2006;6:19. doi: 10.1186/1471-2407-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taniguchi T, D’Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006;107:4223–33. doi: 10.1182/blood-2005-10-4240. [DOI] [PubMed] [Google Scholar]
- 30.Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet. 2005;37:953–7. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- 31.Levitus M, Waisfisz Q, Godthelp BC, et al. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J. Nat Genet. 2005;37:934–5. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- 32.Levran O, Attwooll C, Henry RT, et al. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–3. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- 33.Litman R, Peng M, Jin Z, et al. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–65. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 34.Howlett NG, Taniguchi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–9. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 35.Reid S, Schindler D, Hanenberg H, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–4. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 36.Xia B, Dorsman JC, Ameziane N, et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet. 2007;39:159–61. doi: 10.1038/ng1942. [DOI] [PubMed] [Google Scholar]
- 37.Aretini P, D’Andrea E, Pasini B, et al. Different expressivity of BRCA1 and BRCA2: analysis of 179 Italian pedigrees with identified mutation. Breast Cancer Res Treat. 2003;81:71–9. doi: 10.1023/a:1025428807472. [DOI] [PubMed] [Google Scholar]
- 38.Maki CG, Howley PM. Ubiquitination of p53 and p21 is differentially affected by ionizing and UV radiation. Mol Cell Biol. 1997;17:355–63. doi: 10.1128/mcb.17.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scully R, Ganesan S, Brown M, et al. Location of BRCA1 in human breast and ovarian cancer cells. Science. 1996;272:123–6. doi: 10.1126/science.272.5258.123. [DOI] [PubMed] [Google Scholar]
- 40.Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- 41.Kuzmiak HA, Maquat LE. Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med. 2006;12:306–16. doi: 10.1016/j.molmed.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 42.Cheng J, Belgrader P, Zhou X, Maquat LE. Introns are cis effectors of the nonsense-codon-mediated reduction in nuclear mRNA abundance. Mol Cell Biol. 1994;14:6317–25. doi: 10.1128/mcb.14.9.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998;23:198–9. doi: 10.1016/s0968-0004(98)01208-0. [DOI] [PubMed] [Google Scholar]
- 44.Perrin-Vidoz L, Sinilnikova OM, Stoppa-Lyonnet D, Lenoir GM, Mazoyer S. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum Mol Genet. 2002;11:2805–14. doi: 10.1093/hmg/11.23.2805. [DOI] [PubMed] [Google Scholar]
- 45.Sigurdson AJ, Hauptmann M, Chatterjee N, et al. Kin-cohort estimates for familial breast cancer risk in relation to variants in DNA base excision repair, BRCA1 interacting and growth factor genes. BMC Cancer. 2004;4:9. doi: 10.1186/1471-2407-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Closas M, Egan KM, Newcomb PA, et al. Polymorphisms in DNA double-strand break repair genes and risk of breast cancer: two population-based studies in USA and Poland, and meta-analyses. Hum Genet. 2006;119:376–88. doi: 10.1007/s00439-006-0135-z. [DOI] [PubMed] [Google Scholar]
- 47.Knudson AG., Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Clapperton JA, Manke IA, Lowery DM, et al. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat Struct Mol Biol. 2004;11:512–8. doi: 10.1038/nsmb775. [DOI] [PubMed] [Google Scholar]
- 49.Peng M, Litman R, Xie J, et al. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. Embo J. 2007;26:3238–49. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Antoniou AC, Pharoah PD, McMullan G, et al. Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population-based study. Genet Epidemiol. 2001;21:1–18. doi: 10.1002/gepi.1014. [DOI] [PubMed] [Google Scholar]
- 51.Ponder BA, Antoniou A, Dunning A, Easton DF, Pharoah PD. Polygenic inherited predisposition to breast cancer. Cold Spring Harb Symp Quant Biol. 2005;70:35–41. doi: 10.1101/sqb.2005.70.029. [DOI] [PubMed] [Google Scholar]
- 52.Reid S, Renwick A, Seal S, et al. Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet. 2005;42:147–51. doi: 10.1136/jmg.2004.022673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wreesmann VB, Estilo C, Eisele DW, Singh B, Wang SJ. Downregulation of fanconi anemia genes in sporadic head and neck squamous cell carcinoma. ORL J Otorhinolaryngol Relat Spec. 2007;69:218–25. doi: 10.1159/000101542. [DOI] [PubMed] [Google Scholar]