

Protein prenylation involves the attachment of C15 (farnesyl) or C20 (geranylgeranyl) groups to proteins and is catalyzed by a class of enzymes known as prenyltransferases.1 The observation that inhibition of Ras farnesylation arrests the growth of tumor cells has been the motivating factor in developing inhibitors of prenyltransferases that can serve as anticancer drugs; currently several candidates are in Phase 3 clinical trials.2 Mechanistic analysis of enzymatic reactions can provide insights that are potentially useful in drug design. Since enzymes must bind to the transition state (TS) with greater affinity than the ground state, molecules that mimic the structure of the TS will have the highest possible affinity for the enzyme. Enzyme inhibitors based on such principles can manifest extraordinary affinity and selectivity;3 accordingly, we are interested in determining the TS structure for the reaction catalyzed by protein farnesyltransferase. Moreover, the detailed knowledge gained in these experiments should increase our understanding of how enzymes activate isoprenoid diphosphates for subsequent reaction. Such knowledge could be particularly useful for manipulating the reactivity of prenyltransferases and the closely related terpene cyclases for biotechnology purposes.
At present, the most reliable method to determine TS structure is through the use of computational methods in conjunction with experimentally measured kinetic isotope effect (KIE) measurements.4 While a large body of literature exists for KIE measurements performed on benzylic systems, reports for related allylic systems are sparse. Thus, as a prelude to enzymatic measurements, it was decided to investigate several model reactions, shown in Figure 1, first. Results from such experiments would provide KIE values for limiting associative (SN2) and dissociative (SN1) mechanisms and allow us to validate the computational methods that would be used in the subsequent determination of the enzymatic TS. For a model substrate, dimethylallyl chloride (1) was chosen. Solvolysis of 1 in benzyl alcohol (2) was used as a dissociative model while displacement with triphenylphosphine (5) was employed as an associative model.5 Kinetic analysis of these reactions revealed that the solvolysis reaction was first order in 1 and zero order in 2 whereas the other reaction was first order in both 1 and 5. To measure the 13C KIEs for the reaction, an NMR method was employed based on the work of Singleton6 and others; that approach involves the integration of 13C-NMR spectra obtained at natural abundance of reactant obtained prior to reaction and after substantial conversion. In the work reported here, the model reactions were performed, monitored by GC and terminated by vacuum distillation to recover the remaining starting material which was then derivatized by reaction with dimethylmalonate (7) and the resulting product (8) purified by flash chromatography. 13C-NMR spectra were obtained and integrated using C-4 as an internal standard. 13C KIEs were calculated from the ratios of peak areas determined from samples of the derivatized starting material (8) before and after reaction. A significant primary 13C KIE at C-1 was measured in the SN2 model reaction (1.040±0.003) whereas a value near unity (0.997±0.003) was observed in the SN1 model reaction. The reactants and TS structures for the SN1 and SN2 model reactions were determined at the density functional level of electronic structure theory with the 6–31+G(d) basis set.7 Analytic vibrational frequencies were computed for each stationary point and KIEs were predicted from the usual transition-state theory approach.8 The calculated primary 13C KIEs are 1.040 for the SN2 model reaction and 1.001 for the SN1 model reaction which are within experimental error of the measured values suggesting that the theoretical model should be useful for related reactions including those involving isoprenoid diphosphates.
Figure 1.
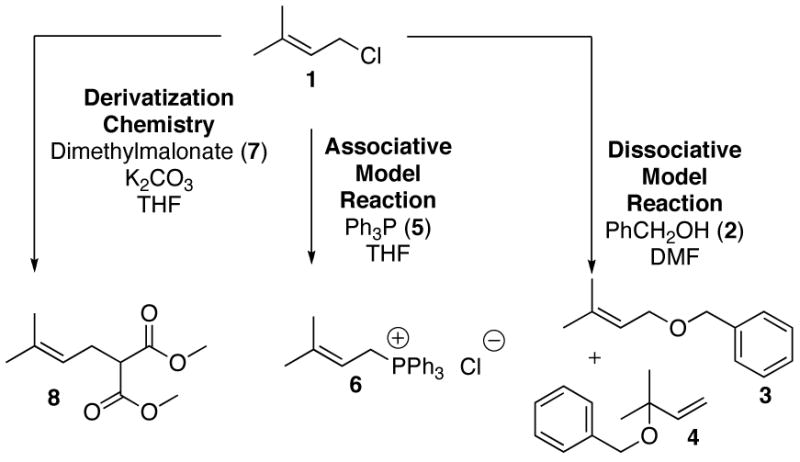
Model reactions and derivatization chemistry for SN1 and SN2 prenylation reactions.
Protein farnesyltransferase (PFTase) catalyzes the transfer of a prenyl group from farnesyl diphosphate (FPP, 9) to proteins including Ras that terminate in CVIA and related sequences. Since the enzyme recognizes only the four C-terminal residues, tetrapeptides are efficient substrates for the enzyme with kinetic constants similar to full-length protein substrates.9 To measure Vmax/KM (V/K) KIEs under steady state conditions, the substrate, FPP, must manifest a low commitment factor (cf).10 Isotope trapping experiments, using FPP as a substrate, to measure the cf suggest significant commitment to catalysis.11 In contrast, similar experiments using the related, weaker binding isoprenoid, geranyl diphosphate (GPP, 10) gave a low value of cf (cf = 0.057, data not shown) indicating that KIEs measured using GPP would not be masked by excessive commitment. Based on the work described above with the model substitution reactions, it was noted that the primary 13C KIE was a particularly sensitive reporter of the extent of bonding between the electrophilic carbon center and incoming nucleophile. Thus it was decided to measure the primary 13C KIE using [1-13C]-GPP as a substrate. The labeled substrate was prepared in four steps using commercially available [1-13C]-triethylphosphonoacetate. For the KIE measurement, a mixture of [1-13C]-GPP and unlabeled GPP was incubated with the peptide N-Dansyl-GCVIA (11) in the presence of PFTase. Reactions were performed either with limiting peptide or limiting GPP. After enzymatic incorporation, the farnesylated peptide product, 12, was purified by reversed-phase HPLC and analyzed by ESI-MS (ion trap instrument) operating in the zoom scan mode. The mass spectra of the samples were deconvoluted into ratios of labeled and unlabeled species using spectra obtained from pure labeled and unlabeled products; the V/K KIE using [1-13C]-GPP as a substrate calculated from that data was found to be 1.039±0.003. A similar approach was used to measure the α-secondary 2H KIE, using [1-2H2]-GPP (prepared from [1-2H2]-geraniol)12 as a substrate, which was found to be 1.068±0.003. To determine the structural significance of these KIE values, a TS structure for the nonenzymatic reaction between ethane thiolate and GPP via an associative mechanism was computationally determined; a density functional level of electronic structure theory using the mPW1N functional in combination with the 6-31+G(d) basis set was employed for those calculations. An associative model was chosen for the starting point for these calculations since a significant primary 13C KIE was observed only in the SN2 model reaction described above. For the nonenzymatic reaction between GPP and ethane thiolate, the primary 13C KIE was calculated to be 1.067. Interestingly, this value is significantly higher than the experimentally measured value of 1.039 determined for the enzymatic reaction indicating that the TS structure for the protein-catalyzed process must be somewhat different. Iterative cycles of altering the C-O (leaving group) and C-S (nucleophile) bond lengths and calculation of the resulting primary 13C KIE was used to generate a plot of the variation of the 13C KIE with C-O and C-S bond lengths; a similar plot was created for the α-secondary 2H KIE. Using the two KIE reported here enabled us to computationally identify a set of C-O (1.69 Å) and C-S (3.70 Å) TS bond lengths that are consistent with both measurements; a proposed structure for the TS is shown in Figure 3. Interestingly, the C-O bond lengths in the model SN2 TS and enzymatic TS are essentially the same. However, the C-S bond lengths are substantially different with the enzymatic TS (3.70 Å) manifesting less bonding to C-1 of GPP than is present in the model system (2.80 Å). This suggests lower electron density at C-1 than is typified by SN2 reactions and provides a rationale for why substrates containing electron withdrawing substituents decrease the enzymatic reaction rate.13 It should be noted that the KIEs reported here for yeast PFTase are similar, but not identical, to KIEs recently reported by Pais et al for a mammalian PFTase.14 These variations may reflect differences in the TS structures between the yeast and mammalian enzymes that could be exploited for drug design.
Figure 3.
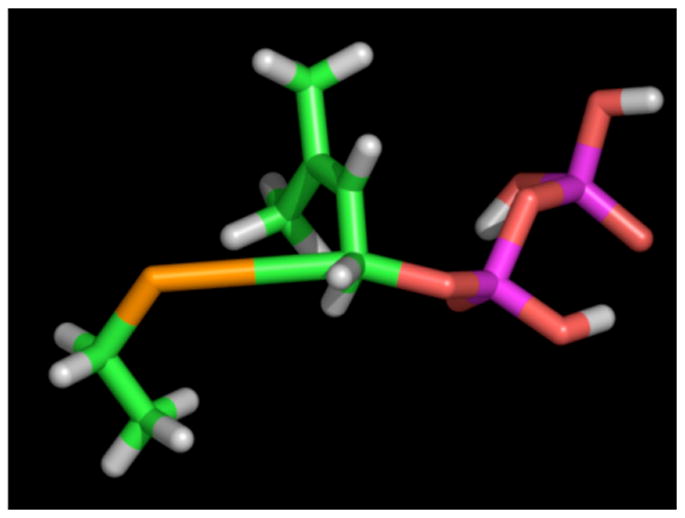
Proposed TS structure for PFTase catalyzed reaction based on calculations using GPP and ethane thiolate. The second isoprene unit is omitted for clarity. Colors: S (orange), C (green) and O (red).
In summary, we present here, a TS structure for the reaction catalyzed by PFTase that complements X-ray crystallographic studies15 and provides a clear structural framework for understanding the results of previous mechanistic investigations of this enzyme including stereochemical16,17 and kinetic analyses.14,18 The experiments reported here provide powerful insights into an important class of biological reactions through a combination of model chemistry, computation and kinetic analysis. Finally, it should be noted that the KIE analysis described here was accomplished through experiments performed with stable isotopes and did not require radiolabeled compounds. It is likely that the role of ESI-MS and NMR in KIE analyses of biological processes will continue to grow in the future.
Supplementary Material
Details for the synthesis of [1-13C]-GPP, cf determination, KIE experiments, TS calculations and data analysis are included. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 2.
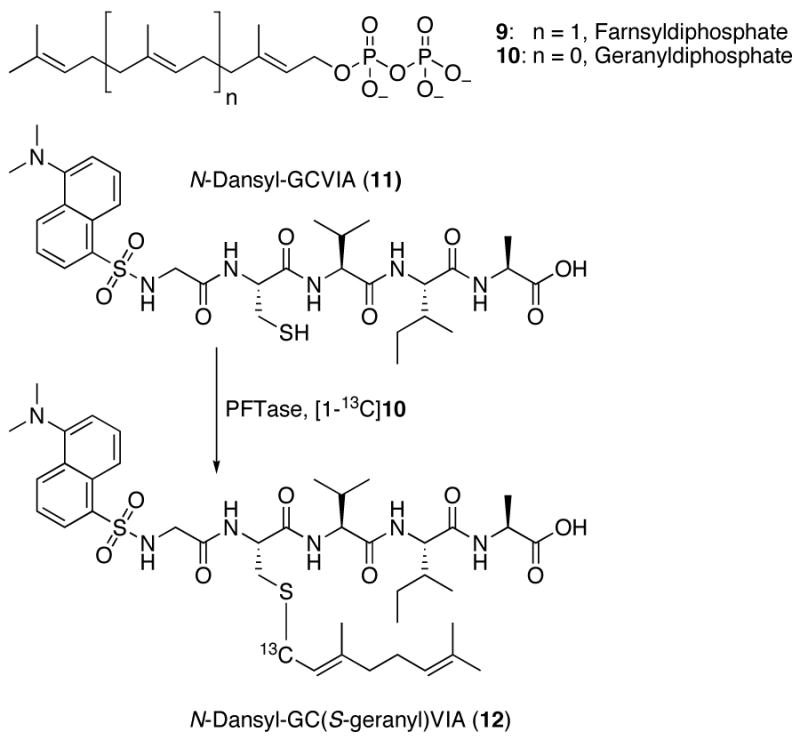
Isoprenoid substrates and prenylation reaction catalyzed by PFTase studied here.
Acknowledgments
This work was supported by the National Institutes of Health (GM58442) and the National Science Foundation (CHE-0610183).
References
- 1.Zhang FL, Casey PJ. Ann Rev Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 2.Doll RJ, Kirschmeier P, Bishop WR. Curr Opin Drug Disc Dev. 2004;7:478–486. [PubMed] [Google Scholar]
- 3.Taylor Ringia EA, Tyler PC, Evans GB, Furneaux RH, Murkin AS, Schramm VL. J Am Chem Soc. 2006;128:7126–7127. doi: 10.1021/ja061403n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schramm VL. Ann Rev Biochem. 1998;67:693–720. doi: 10.1146/annurev.biochem.67.1.693. [DOI] [PubMed] [Google Scholar]
- 5.O’Brien PJ, Herschlag D. Biochemistry. 2002;41:3207–3225. doi: 10.1021/bi012166y. [DOI] [PubMed] [Google Scholar]
- 6.Singleton DA, Thomas AA. J Am Chem Soc. 1995;117:9357–8. [Google Scholar]
- 7.Hehre WJ, Radom L, Schleyer PvR, Pople JA. Ab Initio Molecular Orbital Theory. 2. Wiley; New York: 1986. [Google Scholar]
- 8.Cramer CJ. Essentials of Computational Chemistry: Theories and Models. 2. John Wiley & Sons; Chichester: 2004. [Google Scholar]
- 9.Moores SL, Schaber MD, Mosser SD, Rands E, O’Hara MB, Garsky VM, Marshall MS, Pompliano DL, Gibbbs JB. J Biol Chem. 1991;266:14603–14610. [PubMed] [Google Scholar]
- 10.Northrop DB. Biochemistry. 1975;14:2644–2651. doi: 10.1021/bi00683a013. [DOI] [PubMed] [Google Scholar]
- 11.Furfine ES, Leban JJ, Landavazo A, Moomaw JF, Casey PJ. Biochemistry. 1995;34:6857–6862. doi: 10.1021/bi00020a032. [DOI] [PubMed] [Google Scholar]
- 12.Clausen VA, Edelstein RL, Distefano MD. Biochemistry. 2001;40:3920–3930. doi: 10.1021/bi002011a. [DOI] [PubMed] [Google Scholar]
- 13.Dolence JM, Poulter CD. Proc Natl Acad Sci USA. 1995;92:5008–5011. doi: 10.1073/pnas.92.11.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pais JE, Bowers KE, Fierke CA. J Am Chem Soc. 2006;128:15086–15087. doi: 10.1021/ja065838m. [DOI] [PubMed] [Google Scholar]
- 15.Reid TS, Terry KL, Casey PJ, Beese LS. J Mol Biol. 2004;545:417–433. doi: 10.1016/j.jmb.2004.08.056. [DOI] [PubMed] [Google Scholar]
- 16.Mu YQ, Omer CA, Gibbs RA. J Am Chem Soc. 1996;118:117–123. [Google Scholar]
- 17.Edelstein RL, Weller VA, Distefano MD, Tung JS. J Org Chem. 1998;63:5298–5299. [Google Scholar]
- 18.Huang C-c, Hightower KE, Fierke CA. Biochemistry. 2000;39:2593–2602. doi: 10.1021/bi992356x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details for the synthesis of [1-13C]-GPP, cf determination, KIE experiments, TS calculations and data analysis are included. This material is available free of charge via the Internet at http://pubs.acs.org.