

Introduction
Aldo-Keto Reductases (AKRs) are a rapidly growing protein superfamily that is highly conserved across prokaryotes and eukaroytes [1, 2]. These are generally monomeric NAD(P)H linked oxidoreductases, that are soluble, and consist of approximately 320 amino acids and have mol. wts = 34-37 kDa. These enzymes often convert carbonyl containing substrates to alcohols; aldehydes are converted to primary alcohols and ketones are converted to secondary alcohols. When the carbonyl functionality is found on a natural substrate (steroid hormone or prostaglandin) or a drug or xenobiotic, conversion to the corresponding alcohol functionalizes the product for conjugation and elimination. These enzymes thus play a central role in the metabolism of endogenous substrates, drugs, xenobiotics, and carcinogens and are likely to be as important as the CYP superfamily in dealing with toxic insults.
Currently, there are 114 members in the AKR superfamily distributed across 14 families. For a complete listing and nomenclature visit www.med.upenn.edu/akr. Proteins that belong to separate families have less than 40% sequence identity, and proteins that have greater than 60% similarity belong to the same subfamily. Each of the AKRs have common properties. They catalyze an ordered bi-bi reaction in which pyridine nucleotide binds first and leaves last. They are A-face specific dehydrogenases and catalyze the transfer of the 4-pro-R-hydride ion from the nicotinamide cofactor to the acceptor carbonyl [3]. NADP(H) binding is accompanined by the formation of a loose complex that isomerizes to a tight complex [4, 5]. The rate of release of NADP+ is a slow event and is controlled by the slow isomerization of the tight-complex to the loose-complex prior to release of cofactor.
There are 13 crystal structures of AKRs in the PDB. Each crystal structure has a common (α/β)8-barrel motif, in which an α-helix and a β-strand alternate 8-times [6-8]. The β-strands coalesce at the core of the structure to comprise the staves of a barrel, Fig. 1. In the available ternary complexes, the cofactor lies across the lip of the barrel and the substrate is orientated perpendicular to the cofactor. Three large loops at the back of the barrel define substrate specificity. At the base of the barrel a catalytic tetrad exists that is almost entirely conserved across the superfamily and consists of Tyr, Lys, His and Asp; where the catalytic tyrosine functions as the general acid-base in the proton relay to the substrate [9].
Fig. 1.

Structural motif of AKRs: showing the characteristic (α/β)8-barrel of the AKRs. After Hoog et al. (6).
Several AKRs have been implicated in carcinogen metabolism, these include the dihydrodiol dehydrogenases that oxidize polycyclic aromatic hydrocarbon trans-dihydrodiols to reactive and redox-active o-quinones (AKR1C1-AKR1C4 and AKR1A1) [10-14]; the NNK (4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone) carbonyl reductases that reduce tobacco-specific nitrosamino-ketones (NNK) to the corresponding alcohols (AKR1C1-AKR1C4)[15]; and the aflatoxin dialdehyde reductases (AKR7A subfamily) [16, 17]. This article will focus on the AKRs that display dihydrodiol dehydrogenase activity and their role in PAH activation.
Routes of Polycyclic Aromatic Hydrocarbon Activation (PAH)
PAH are major environment pollutants, they are prevalent in tobacco smoke and are suspect human lung carcinogens. PAH such as benzo[a]pyrene are metabolically activated to mediate their deleterious effects. Three major routes of PAH activation have been described, Fig. 2. In the first route, CYP peroxidase, in the presence of a peroxide substrate will catalyze peroxide bond cleavage and generate higher oxidation states of iron (compound I and compound II), which can be reduced in the presence of benzo[a]pyrene (BP) to yield a highly reactive BP radical cation which will alkylate DNA [18-20]. In the second route, BP is converted to BP-7,8-oxide (an arene oxide) by CYP1A1/CY1B1 which is then hydrolyzed by epoxide hydrolase to yield a trans-dihydrodiol (7,8-dihydroxy-,7,8-dihydro-benzo[a]pyrene or (-)-BP-7,8-diol). This route of metabolism is stereoselective since only the (-)-BP-7,8-diol is formed in vivo. (-)-BP-7,8-diol is then a substrate for the two remaining pathways of PAH activation. It can either undergo a second monoxygenation catalyzed by CYP1A1/CYP1B1 to yield (+)- anti-BPDE (7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene) [21, 22] or it can undergo NADP+-dependent oxidation catalyzed by AKRs to yield the corresponding reactive and redox-active o-quinone (benzo[a]pyrene-7,8-dione; BP-7,8-dione) [23, 24]. There is compelling evidence that (+)-anti-BPDE is an ultimate carcinogen derived from BP.
Fig. 2.

Metabolic routes of PAH activation:
Rat and Human AKRs with Dihydrodiol Dehydrogenase Activity
AKRs which convert PAH trans-dihydrodiols to PAH o-quinones do so because they have dihydrodiol dehydrogenase activity [25]. In this reaction the trans-dihydrodiol is oxidized in the presence of NADP+ to yield a ketol which spontaneously re-arranges to a catechol. The catechol is air-sensitive and undergoes two one electron autoxidations to produce an o-semiquinone anion radical which is then converted to the o-quinone. The o-quinones are reactive electrophiles which can undergo Michael addition reactions with macromolecules (protein, RNA and DNA) or react with cellular nucleophiles (e.g. GSH). Once formed the o-quinones can also be reduced back by two electrons to the catechol using cellular reducing equivalents e.g., NAD(P)H [25]. These o-quinones are not substrates for quinone reductase [26]. Alternatively, the o-quinones can undergo successive two 1 electron reduction reactions back to the catechol using NADPH cytochrome P450 reductases [26]. Each time the catechol is re-formed it is re-oxidized by molecular oxygen to form reactive oxygen species (superoxide anion , hydroxyl radical (OH·) and hydrogen peroxide (H2O2)) and the fully oxidized o-quinone. This establishes a futile redox-cycle that generates ROS multiple times. The ROS generated may then contribute to the initiation and promotional phases of PAH carcinogenesis.
The most extensively studied AKRs that have dihydrodiol dehydrogenase (DD) activity are as follows; AKR1C9 (rat 3α-hydroxysteroid/dihydrodiol dehydrogenase); AKR1C1 (human 3α(20α)-hydroxysteroid dehydrogenase (HSD) or DD1); AKR1C2 (human type 3 3α-HSD and bile-acid binding protein or DD2); AKR1C3 (human type 2 3α-HSD/type 5 17β-HSD and DDx); AKR1C4 (human type 1 3α-HSD/DD4); and human aldehyde reductase (AKR1A1), Table 1.
Table 1.
Relationship between rat and human AKR1C family members
| Nomenclature | Enzyme | Other Names | Percent Sequence Identity | Tissue Distribution |
|---|---|---|---|---|
| AKR1C9 | Rat dihydrodiol dehydrogenase | 3α-HSD | 67% | Liver |
| AKR1C1 | Human dihydrodiol dehydrogenase 1 | 20α(3α)HSD | 84.1% | Lung> Liver> Testis>Mammary Gland |
| AKR1C2 | Human dihydrodiol dehydrogenase 2 | Type 3 3α-HSD/bile acid binding protein | 99.4% | Lung & Liver > Prostate, Testis & Mammary Gland |
| AKR1C3 | Human dihydrodiol dehydrogenase X | Type 2 3α-HSD/Type 5 17β-HSD | 87.9% | Mammary gland > prostate> liver >lung |
| AKR1C4 | Human dihydrodiol dehydrogenase 4 | Type 1 3α-HSD Chlordecone reductase | 86% | Liver specific |
| AKR1A1 | Human dihydrodiol Dehydrogenase 3 | Aldehyde reductase | 46% | Ubiquitous |
Experimental Strategies
To study the role of AKRs in PAH activation requires a source of the recombinant enzymes, reliable assay methods, a source of PAH trans-dihydrodiol substrates and protocols for trapping the highly reactive PAH o-quinones. A source of PAH o-quinones is also required to study their chemical and biological properties. Biological properties of interest include their cytotoxicity, and genotoxicity (DNA-lesions and mutations).
Caution
All PAH are potentially hazaradous and should be handled in accordance with the “NIH Guidelines for the Laboratory Use of Chemical Carcinogens”
Cloning, Expression and Purification of Recombinant AKRs Cloning
Historically, AKR1C9 cDNA was cloned from a λgt11 expression library using a rabbit anti-rat AKR1C9 polyclonal antibody to yield a construct in pBluescriptSK+ [27]. Subsequent subcloning led to the construct of prokaryotic expression vectors (pKK-3α-HSD (AKR1C9) and pET16b-3α-HSD (AKR1C9) in which expression of the cDNA is driven by the hybrid trc promoter [28, 29].
By contrast the human enzymes (AKR1C1-AKR1C4 and AKR1A1) can be cloned by isoform specific RT-PCR [13]. In this approach first strand cDNA synthesis is conducted using human hepatoma (HepG2) cell polyA+-RNA as template, an oligo-dT primer and Superscript II RNAse H- reverse transcriptase (Gibco BRL). Aliquots of the first strand cDNA library are then mixed the 5′ and 3′ isoform-specific primers (Table 2), dNTPs and Vent-DNA polymerase (New England Biolabs). PCR is conducted for 30 cycles, and the PCR products (1.0 kB) purified by agarose gel electrophoresis. PCR amplified cDNAs are then ligated into the TA cloning vector pCRII (Invitrogen). The fidelity of positive inserts was established by dideoxysequencing. Coding regions were then excised using NcoI and BamH1 partial digestion and were inserted into the compatible NcoI and BamHI sites of the prokaryotic expression vector pET16b (Novagen). Utilization of the NcoI site in pET16b intentionally results in the loss of the histidine tag so that the expressed AKRs contain only their deduced amino acid sequence. His-tagged-fusion proteins are avoided since they generally require purification by Ni2+ chromatography and AKR1 family members are metal sensitive.
Table II.
Oligonucleotide Primers used for isoform-selective RT-PCR of human AKR1 CDNA's
| Isoform | Primer | Primer Sequence |
|---|---|---|
| AKR1C1 | 5′-primer | 5′-CCAGCCATGGATTCGAAATAT-3′ |
| 3′-primer | 3′-CCATGTTAATATTCATCAGAG-5′ | |
|
| ||
| AKR1C2 | 5′-primer | 5′-ACAGCCATGGATTCGAAATAC-3′ |
| 3′-primer | 3′-TCCATGTTAATATTCATCAGAA-5′ | |
|
| ||
| AKR1C3 | 5′-primer | 5′-CAGGCCATGGATTCCAAACAG-3′ |
| 3′-primer | 3′-TCCATGTTAATATTCATCTGAAT-5′ | |
|
| ||
| AKR1C4 | 5′-primer | 5′-CCATGGATCCCAAATATCAGC-3′ |
| 3′-primer | 3′-TCTATGCTAATATTCATCTGAA-5′ | |
|
| ||
| AKR1A1 | 5′-primer | 5′-GGGGGCCATGGCGGTTCCTG-3′ |
| 3′-primer | 3′-GGGAAATTACTGGGCARGACTCTG-5′ | |
NcoI-engineered start codons are in bold; stop-codons are underlined
Expression and Purification of Recombinant AKRs
pET-16b-AKR constructs are transformed into competent E. coli host expression strain C41 (DE3) [13, 14, 30]. This host strain is provided by Dr. J.E. Walker of the Medical Research Council Laboratory of Molecular Biology, Cambridge, U.K. Cultures are streaked on Luria-Bertania plates containing 100 μg/ml ampicillin for positive selection of single colonies. Single colonies are grown overnight in 5 mL starter cultures. Aliquots of each starter culture (2.5 mL) are used to inoculate 2 × 100 mL cultures which after 5 h are use to inoculate 2 × 2 liters of Luria-Bertania/ampicillin media. Once the absorbance at 600 nm has reached 0.6-0.8 OD units each 2 liter culture is induced with 2 mL 1M IPTG (isopropyl-thio-β-galactosidase) for overnight protein production.
Cells are harvested by centrifugation (8 × 250 mL bottles), washed twice in 10 mM Tris-HCl pH 8.6 containing 1 mM EDTA and 1 mM 2-mercaptoethanol and each pellet is suspended in 5 mL of the same buffer. AKR proteins are released from the cell pellets by sonication, and following centrifugation the sonicates are dialyzed into 10 mM Tris-HCl pH 8.6 containing 1 mM EDTA, 1 mM 2-mercaptoethanol and 20% glycerol. The dialyzed protein is then applied to DE-52 cellulose column equilibrated in the dialysis buffer and the AKR protein is eluted with a linear salt gradient of 10 –250 mM KCl, where the proteins elute at the mid-point in the gradient, (pI = 6.1). Following dialysis into 10 mM potassium phosphate pH 7.0 containing 1 mM EDTA, 1 mM 2-mercaptoethanol and 20% glycerol, the sample is purified by Sepharose-blue affinity column chromatography. After application, the sample is batch-eluted in a purified concentrated form in column buffer containing 1.0 M KCl. The purified AKRs are then dialyzed into protein storage buffer (20 mM potassium phosphate pH 7.0 containing 1 mM EDTA, 1 mM 2-mercaptoethanol and 30% glycerol).
Specific activities are monitored throughout the purification by measuring: the NAD+ dependent oxidation of 75 μM androsterone in 100 mM potassium phosphate pH 7.0 at 25 °C (AKR1C9 or AKR1C4), by measuring the oxidation of 1 mM 1-acenaphthenol in 100 mM potassium phosphate pH 7.0 at 25 °C (AKR1C1-AKR1C3) both in the presence of 2.3 mM NAD+, or by measuring the reduction of 4-nitrobenzaldehyde in 100 mM potassium phosphate buffer pH 7.0 at 25 °C in the presence of 200 μM NADPH (AKR1A1). The purity of peak chromatographic fractions is concurrently monitored by SDS-PAGE where expression of a 37 kDa protein is monitored. Specific activities for the homogeneous recombinant proteins are 1.6 (AKR1C9), 2.1 (AKR1C1), 2.5 (AKR1C2), 2.8 (AKR1C3) 0.21 (AKR1C4), and 6.0 (AKR1A1) μmoles substrate oxidized per min per mg where protein concentration is measured based on a Lowry determination relative to BSA. Typically 40 mg of homogeneous recombinant protein can be obtained from a 4 liter culture. Representative purification Tables are given for AKR1C1-AKR1C4 and AKR1A1, Table 3 [13, 30].
Table 3.
Purification Schemes for Five Recombinant Human Dihydrodiol Dehydrogenase (AKR) Isoforms a
| protein | purification step | volume (mL) | total protein (mg) | total activity (:mol min-1 mg-1) | specific activity (:mol min-1 mg-1) | purification factor (-fold) | yield (5%) |
|---|---|---|---|---|---|---|---|
| AKR1A1 | sonicate | 70 | 470 | 668 | 1.42 | 1 | 100 |
| DE52 cellulose | 12.5 | 88.5 | 233 | 2.63 | 1.85 | 34.7 | |
| Blue-Sepharose | 7.1 | 30.5 | 183 | 6.0 | 4.23 | 27.4 | |
| AKR1C1 | sonicate | 61 | 460 | 59 | 0.13 | ||
| DE52 cellulose | 16 | 23 | 46 | 2.0 | 15 | 78 | |
| Blue Sepharose | 8 | 20 | 42 | 2.1 | 16 | 71 | |
| AKR1C2 | sonicate | 77 | 510 | 42 | 0.08 | ||
| DE52 cellulose | 18 | 19 | 39 | 2.1 | 25 | 93 | |
| Blue-Sepharose | 11 | 15 | 37 | 2.5 | 31 | 88 | |
| AKR1C3 | sonicate | 51 | 310 | 65 | 0.21 | ||
| DE52 cellulose | 56 | 25 | 51 | 2.0 | 10 | 78 | |
| Blue-Sepharose | 5 | 12 | 34 | 2.8 | 13 | 52 | |
| AKR1C4 | sonicate | 44 | 270 | 4.6 | 0.02 | ||
| DE52 cellulose | 12 | 23 | 2.8 | 0.12 | 6 | 61 | |
| Blue-Sepharose | 8 | 13 | 2.7 | 0.21 | 11 | 59 |
Specific activities throughout the purifications were measured using 75 :M androsterone (AKR1C4) or 1 mM 1-acenaphthenol (AKR1C1-AKR1C3) as the substrate in reaction mixtures containing 100 mM KPO4 (pH 7.0) and 2.3 mM NAD+ at 25.0 °C.
Assay Methods for Dihydrodiol Dehydrogenase Activity
Continuous spectrophotometric assays based either on the NAD+ dependent oxidation of androsterone or 1-acenaphthenol can be used to monitor activity throughout the purification of the recombinant AKRs. However, these assays do not monitor the oxidation of PAH-trans-dihydrodiols directly; the following assays are recommended.
NADP+-dependent Oxidation of Benzenedihydrodiol
Both continuous and discontinuous assays have been developed to monitor the NADP+-dependent oxidation of the model trans-dihydrodiol (benzenedihydrodiol; 1,2-trans-dihydroxy-1,2-dihydro-cyclohexa-3,5-diene). Both assays require synthesis of the benzenedihydrodiol substrate [31, 32]. This is accomplished by the stoichiometric bromination of cyclohexadiene to yield 1,2-dibromo-cyclohexene, followed by epoxidation to yield 1,2-dibromo-4,5-epoxy-cyclohexane, and subsequent dehydrohalogenation to yield benzene oxide. Benzeneoxide is then subjected to alkali peroxidation to yield the peroxy intermediate which is reduced with NaBH4 to yield the final product which is purified by TLC, Fig. 3. The synthesis is low yielding because of the propensity of benezeneoxide to revert to phenol. Typically 100 mg of the product can be obtained if the synthesis is conducted on a 5 g scale.
Fig. 3.

Synthesis of benzenedihydrodiol from cylcohexadiene.
The continuous assay measures the NADP+-dependent oxidation of benzenedihydrodiol in mixtures containing 1 mM benzenedihydrodiol, 2.3 mM NADP+ and 50 mM glycine pH 9.0 buffer at 25 °C. The discontinuous assay exploits the observation that benzenedihydrodiol is the only trans-dihydrodiol that is oxidized to stable hydroquinone (catechol) [33]. The assay links catechol production to catechol-O-methyl transferase using [3H]-S-adenosyl-L-methionine (SAM) as methyl donor to yield [3H]-guaiacol, which is isolated by extraction and counted. Assays are conducted in 0.1 mL systems containing 50 mM potassium phosphate, pH 7.8, 1.0 mM DTT, 1.0 mM MgCl2, 2.3 mM NADP+, 50 mM [3H]-SAM, 10 units COMT and 1.0 mM benzenedihydrodiol. Reactions are initiated by enzyme and quenched with 200 μL 1M HCl containing 2 μL/mL guaiacol as carrier. The reaction mixture is then extracted into 2 × 1.0 mL toluene. By counting the radioactive extracts and knowing the specific radioactivity of SAM it is possible to compute specific activities. Because the assay is radiometric it provides a 1000-5000 fold increase in sensitivity over the spectrophotometric assay, Fig. 4. Its advantage is that it can be used to measure dihydrodiol dehydrogenase activity in cell culture and in tissue extracts.
Fig. 4.
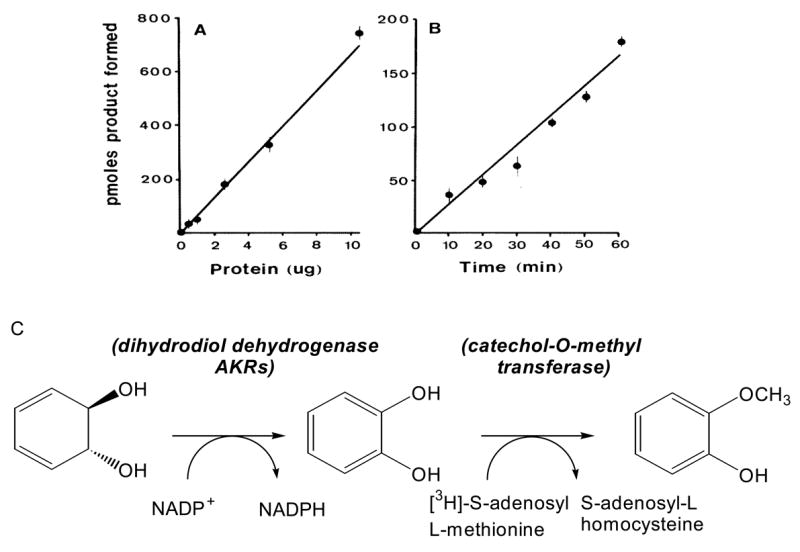
Coupled radiochemical assay for the detection of dihydrodiol dehydrogenase. Panel (A) shows linearity with protein where varying amounts of the 40-75% ammonium sulfate fraction of rat liver cytosol were incubated with 1 mM benezenedihydrodiol, 2.3 mM NADP+, 50 μM [3H]-S-adenosyl-L-methionine (0.10 μCi/nmol) and 10 units of COMT; panel (B) shows linearity with time in which the assay was initiated with 2.6 μg of protein; and panel (C) shows the bases of the assay in which the formation of catechol is linked to catechol-O-methyl transferase to yield [3H]-guaiacol. After, Ivins and Penning (33 ).
PAH Trans-Dihydrodiol Oxidation
To monitor the oxidation of PAH trans-dihydrodiols requires a source of substrates. The substrate of most relevance is (±)-trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene and is available in limited amounts from the NCI, Chemical Carcinogen Reference Standard Repository (MidWest Research Institute, Kansas City, MO 64110). Examination of a structural series of PAH trans-dihydrodiols is important from a structure-activity perspective because issues of regio-, stereo-, and conformational chemistry determine whether the trans-dihydrodiol is a proximate carcinogen or not. For example non-K region (-) R,R trans-dihydrodiols are potent proximate carcinogens (e.g,.(-) trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene) but other regio and stero-isomers are not (e.g., (±) trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene and (±) trans-4,5-dihydroxy-4,5-dihydrobenzo[a]pyrene) [34]. In systematic studies on trans-dihydrodiol oxidation, substrates containing increasing ring-number, bay-region methylation and fjord regions, need to be examined since the presence of these structural features often determines the carcinogenicity of the parent hydrocarbon. The structural features of interest are shown in Fig. 5 and PAH trans-dihydrodiols often examined as substrates are shown in Fig. 6. In this article there is not room to describe the synthesis of all the trans-dihydrodiol substrates. The synthesis of (±) trans-1,2-dihydroxy-1,2-dihydrophenanthrene is fairly typical. This is prepared from 1,2,3,4-tetrahydrophenanthrene-1-one (Aldrich) as follows: reduction of 1-keto-2,3,4-trihydro-phenanthrene with NaBH4 in methanol affords the corresponding alcohol, which is dehydrated with catalytic quantities of HCl in acetic acid to give the 3,4-dihydroarene [35]. Prevost reaction of the dihydroarene using silver benzoate and iodine yields the trans-dibenoxytetrahydroarene [36], which is reduced with DDQ to yield trans-dibenzoxydihydrophenanthrene [37]. Hydrolysis of the resultant dibenzoate with sodium methoxide yields the desired product, (±) trans-1,2-dihydroxy-1,2-dihydrophenanthrene.
Fig. 5.

Structural features that increase the carcinogencity in the parent PAH.
Fig. 6.

Structures of PAH trans-dihydrodiol substrates for AKRs. All trans-dihydrodiols shown are racemic; NP = naphthalene, Ph = phenanthrene; Ch = chrysene; 5Me-Ch = 5-methylchrysene; BP = benzo[a]pyrene; BA = benz[a]anthracene; 7MBA = 7-methylbenz[a]anthracene; 12MBA = 12-methylbenz[a]anthracene; and DMBA = 7,12-dimethylbenz[a]anthracene, DP =dibenz[a,l]pyrene; and BC = benzo[g]chrysene.
(±) trans-9,10-dihydroxy-9,10-dihydrophenanthrene is prepared by reducing phenanthrene-9,10-dione with lithium aluminium hydride [38, 39]. The remaining compounds can be synthesized by the routes cited (±) trans-1,2-dihydroxy-1,2-dihydrochrysene[40]; (±) trans-1,2-dihydroxy-1,2-dihydro-5-methyl-chrysene; (±) trans-7,8-dihydroxy-7.8-dihydro-5-methyl chrysene [41]; (±) trans-3,4-dihydroxy-3,4-dihydrobenz[a]anthracene [42]; (±) trans-3,4-dihydroxy-3,4-dihydro-7-methyl-benz[a]anthracene [43]; (±) trans-3,4-dihydroxy-3,4-dihydro-12-methyl-benz[a]anthracene ; (±) trans-3,4-dihydroxy-3,4-dihydro-7,12-dimethyl-benz[a]anthracene [44]; (±) trans-10,11-dihydroxy-10,11-dihydrobenz[g]chrysene [45]; and (±) trans-11,12-dihydroxy-11,12-dihydro-dibenzo[a,l]pyrene[46].
To determine whether these compounds are substrates for AKRs we have relied on two assays. First, we have monitored the oxidation of 20 μM (±)-trans-dihydrodiol in the presence of 2.3 mM NADP+ in AMPSO buffer at pH 9.0 by measuring changes in absorbance at 340 nm [23]. Under these conditions the reaction is pseudo-first-order where Km ≫ [S] so that the Michaelis-menton equation simplifies to v/[S] = Vmax/Km. This has allowed us to report the utilization rations for a large number of trans-dihydrodiol substrates for various AKRs [13, 23, 30, 47, 48]. In assigning utilization ratios it is important to consider that the reactions are performed in 8% DMSO and that this has an inhibitory effect on the initial velocity determinations which require correction. Because these absorbance assays give relatively small changes, the assay is usually validated by monitoring the disappearance of the trans-dihydrodiol substrate by RP-HPLC [13, 49].
RP-HPLC assays are conducted in 0.1 mL 100 mM potassium phosphate buffer pH 7.8 or 50 mM glycine (pH 9.0) containing 2.3 mM NADP+ and 20 μM trans-dihydrodiol. The substrate was dissolved in DMSO and the final organic solvent concentration was 8%. Reactions are initiated by the addition of homogeneous enzyme (2 -10 μg) and incubated for 2 h at 37 °C. Control incubations are performed in the absence of NADP+ or purified enzyme. Reaction mixtures are extracted into 0.2 mL EtOAC, pooled and evaporated to dryness and residues re-dissolved in methanol (50 μL) for RP-HPLC analysis.
RP-HPLC analysis were conducted by injecting aliquots (20 μL) onto a Zorbax Ultrasphere-ODS (10 um; 4.6 × 25 mm; Dupont Wilmington, DE) reverse-phase column. Compounds were separated using a 70 min linear gradient of 60-80% methanol-water (v/v). The solvent flow-rate was 0.5 mL/min and the chromatographic system was run at ambient temperature. The absorbance was monitored at 254 nm [13, 49]. Chromatography has been performed on a Perkin-Elmer LC480 diode-array HPLC, a Beckman-Sysem Gold HPLC and a Waters Alliance system. Comparison of specific activities yields a value of 5 – 6 nmoles of BP-7,8-diol oxidized/min/mg for the spectrophotometric assay, and 2.0 nmoles of BP-7,8-diol oxidized/min/mg based on the RP-HPLC assay. Typical progress curves for the consumption of BP-7,8-diol by AKR1C1-AKR1C4 based on RP-HPLC assays are shown, Fig. 7 [13].
Fig. 7.

Progress curves for the oxidation of BP-7,8-diol by AKR1C1-AKR1C4. Initial progress curves for the oxidation of the proximate carcinogen (±)-[1,3-3H]-BP-diol catalyzed by recombinant AKR1Cs. Following RP-HPLC analysis, the total cpm in the (±)-[1,3-3H]-BP-diol peak remaining at each time point was used to calculate the nmoles of BP-diol oxidized and was plotted versus time. The initial velocities (insets) were calculated from the linear portion of the data as shown: panel (A) AKR1C2; panel B AKR1C1; panel C AKR1C4; and panel D, AKR1C3.
To determine the sterochemical course of trans-dihydrodiol oxidation. RP-HPLC assays are performed until the reaction reached 50% completion. In some instances this is the end-point of the reaction [13, 23]. At this juncture the unreacted isomer(s) are extracted into ethyl-acetate, the remaining substrate is purified by TLC and the CD spectra recorded sufficient material for these assays requires an absorbance of 1.0 OD unit at 254 nm. The sign of the Cotton effect can be related to the stereochemical assignment for the unreacted isomer which has been established by NMR. AKR1C1-AKR1C4 oxidize both stereoisomers of racemic trans-dihydrodiols, however, pseudo-first order analysis of progress curves for oxidation shows that one diol is preferentially oxidized. CD spectra showed that for AKR1C1 and AKR1C2 the (+)-S,S-isomer was oxidized before the (-)-R,R-isomer see Fig. 8.
Fig. 8.

CD Spectroscopy to determine the stereochemical preference of BP-7,8-diol oxidation. AKR1C1-AKR1C4 oxidize both stereoisomers of (+)-BP-7,8-diol. Pseudo-first order plots of BP-7,8-diol oxidation catalyzed by AKR1C1 and AKR1C2 indicated that decay curves for BP-7,8-diol were bi-phasic indicating a preference for one isomer. RP-HPLC assays were used to monitor the progress curves of 10 mL reactions containing 50 μM BP-7,8-diol, 2.3 mM NADP+ in 50 mM glycine buffer pH 9.0 and 8% DMSO. When 50% of the diol was consumed the reaction was quenched with ethyl actetate and unoxidized dihydrodiol substrates remaining were isolated by TLC and their CD-spectra recorded. The CD spectrum of the racemic (+)-BP-7,8-diol was subtracted from the enatiomerically enriched substrates to determine the sign of the Cotton effect. (A) CD spectra of the unreacted BP-7,8-diol recovered from the AKR1C2 reaction; and (B) CD spectra of the unreacted BP-7,8-diol recovered from the AKR1C1 reaction. After Burczynski et al ( ).
PAH-Trans-Dihydrodiol Specificity of AKRs
Based on the assays described a fairly complete picture of the trans-dihydrodiol specificity of AKRs has emerged. (1) PAH trans-dihydrodiols of increasing ring size are substrates; (2) K-region trans-dihydrodiols e.g., trans-4,5-dihydroxy-4,5-dihydrobenzo[a]pyrene are not substrates; (3) AKR1C1-AKR1C4 do not exhibit stereospecificity in that they will oxidize both the (+)- and the (-) stereoisomers of racemic BP-7,8-diol and DMBA-3,4-diol; (4) AKR1A1 is stereoselective in that it will only oxidize the (-)-BP-7,8-diol; (5) depending on the ring arrangement different AKRs can be involved in the metabolism of the PAH trans-dihydrodiol [13, 23, 30, 48, 50].
Characterization of PAH o-Quinones as Products of PAH trans-dihydrodiol Oxidation
PAH o-quinones produced by the AKR-dependent oxidation of PAH trans-dihydrodiols are highly reactive and readily form buffer conjugates making their isolation difficult. They can be isolated from in vitro reactions if these are performed in the presence of a potent nucleophile as a trapping agent (e.g., 2-mercaptoethanol). The trapped product is a thio-ether conjugate that arises from 1,4-Michael addition to the PAH –o-quinone. Validation of the structure of the product requires LC/MS characterization of the thio-ether conjugate and its comparison to an authentic synthetic standard [24].
Preparation of the 2-Mercaptoethanol Conjugates of PAH o-Quinones
Reactions (10 mL) containing 50 mM potassium phosphate buffer pH 7.0, 5 mM 2-mercaptoethanol and 20 μM PAH o-quinone in 8% DMSO are incubated for 18 h at 37 °C. The reaction was terminated by extraction with ethyl-acetate ( 2.0 ml). Organic extracts are combined, dried over sodium sulfate, and the organic solvent removed under reduced pressure. The resulting residue was chromatographed on a 20 × 20 cem semipreparative silica TLC plate using chloroform : methanol (4: 1) as mobile phase. For the mercaptoethanol BP-7,8-dione conjugate, a single purple spot Rf = 0.77 was observed which can be eluted from the plate and analyzed by EIMS, LC/MS and [ 1H]-NMR [24].
Characterization of the Product of AKR Oxidation of BP-7,8-diol by trapping with 2-mercaptoethanol
Incubations (50 mL) containing 20 mM BP-7,8-diol, 2.3 mM NADP+, 5 mM 2-mercaptoethanol and 8% DMSO are reacted with purified recombinant AKR ( 1 mg) for 20 h at 37 °C. Reactions are terminated by extraction with ethyl-acetate and the product is isolated and characterized as before.
EIMS spectra for BP-7,8-dione thio-ether conjugate yield a m/z = 358 M+; and fragment ions that correspond to the successive loss of 2 carbonyls m/z 358 → 330, and 298 → 270 which is diagnostic of the structure [24]. LC/MS gives m/z = 359 MH+; and m/z =331 (MH+ - CO) [30]. [1H]-NMR shows the diagnostic loss of the C9- vinyl proton d = 6.6 ppm and its replacement by a singlet due to substitution at C10 [24].
LC/MS data were acquired on a Finnigan LCQ ion trap mass spectrometer (ThermoQuest, San Jose CA) equipped with a Finnigan atmospheric pressure chemical (APCI) source. The on-line chromatography was performed using a Waters Alliance 2690 HPLC system (Waters Corp., Milford, MA). A YMC C18 ODS-AQ column was used at a flow rate of 0.9 ml/min. Solvent A was 5 mM ammonium acetate in water containing 0.01% trifluoroacetic acid and solvent B was 5 mM ammonium acetate in methanol containing 0.01% trifluoroacetic acid with the gradient run as follows; 30% solvent B at 0 min 30%, solvent B at 5min, 100% solvent B at 16 min, 100% solvent B at 24 min and 30% solvent B at 26 min [30].
The identification of PAH-o-quinone-thio ether conjugates in enzymatic reactions with AKRs suggests the following sequence of events: oxidation of the trans-dihydrodiol would yield a ketol which tautomerizes to yield the air-sensitive catechol, the catechol undergoes autoxidation to yield the PAH o-quinone, reaction with 2-mercaptoethanol yields a ketol, which rearranges to the catechol which air oxidizes to the o-quinone-conjugate, Fig. 9.
Fig. 9.

Conversion of PAH trans-dihydrodiols to PAH o-quinone thio-ether conjugates
Oxygen Metabolism During AKR Mediated Trans-Dihydrodiol Oxidation
The conversion of trans-dihydrodiols to fully-oxidized PAH o-quinones catalyzed by AKRs suggests that this reaction sequence will be accompanied by concomitant changes in reactive oxygen species (ROS, , OH· and H2O2). Enzymatic trans-dihydrodiol oxidation occurs concomitantly with the depletion of molecular oxygen, the formation of superoxide anion, and the generation of H2O2 [51].
The consumption of molecular oxygen during the enzymatic oxidation of trans-dihydrodiols can be monitored using Clark-style oxygen electrodes in 600 μL chambers containing the reaction components trans-dihydrodiol oxidation, Fig. 10 The reaction is initiated by the addition of enzyme. The output from the electrode was connected through an amplifier (Instech) which allows the simultaneous display of oxygen concentration and the rate of oxygen uptake as a function of time. The data acquisition software allows the recording of 1-1,000 data points/min. All oxygen uptake experiments are performed at 25 °C assuming that the amount of dissolved oxygen is 0.137 μmoles in 0.6 mL at 25 °C at 760 mm Hg.
Fig. 10.
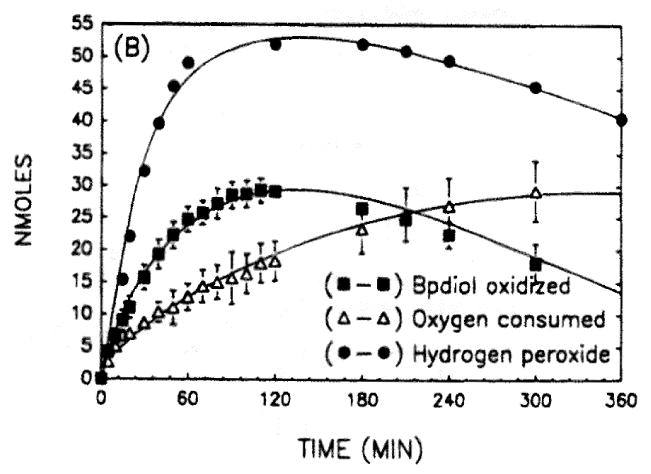
Oxygen uptake and hydrogen peroxide formation during the enzymatic oxidation of BP-7,8-diol. Reactions contained 20 μM BP-7,8-diol in 8% DMSO, 2.3 mM NADP+ and 50 mM glycine buffer pH 9.0. Oxygen uptake was measured in 600 μL chambers with a micro-oxygen electrode. Nmoles of BP-diol oxidized was monitored spectrophotometrically at 340 nM and nmoles of hydrogen peroxide formed were measured discontinuously using horse-radish peroxidase and tetramethylbenzidine as co-reductant.
The concurrent formation of H2O2 during the enzymatic oxidation of PAH –trans-dihydrodiols is measured by linking H2O2 production to the horseradish peroxidase catalyzed oxidation of 3,3,5,5-tetramethylbenzidine E = 30,680 M-1 cm-1 [52].
The concurrent formation of superoxide anion during the enzymatic oxidation of PAH trans-dihydrodiol is measured by monitoring the rate of acetylated cytochrome c readuction that is inihibited by superoxide dismutase at 550 nm (E = 19,600 M-1 cm-1. )[53]. Typical traces of oxygen metabolism during the enzymatic oxidation of BP-7,8-diol are shown, in Fig. 10. Inspection of these traces shows that during the autoxidation of the intermediate catechol, H2O2 is produced before O2 consumption.
Direct measurement of ROS produced in the reaction can be monitored with spin-trapping agents such as DMPO in the presence of DMSO cosolvent. Reaction systems (200 μL) containe 45 μM BP-7,8-diol, 2.3 mM NADP+, 8% DMSO and 50 mM glycine buffer pH 9.0 plus 50 mM DMPO. Reactions were initiated by the addition of enzyme 50 μg. EPR spectra were recorded on an Varian E109 EPR spectrometer operating in the X-band (9.25 GHz) employing 0.1M modulation amplitude and 200 mW microwave power with a receiver gain set at 5 × 104 [51].
A six-line hyperfine splitting pattern can be observed during the AKR mediated oxidation of BP-7,8-diol, Fig. 11. This splitting pattern can be assigned to DMPO-CH3, and is abolished by the presence of superoxide dismutase and catalase suggesting that it is both and H2O2 dependent. An identical DMPO spin-adduct can be observed with DMPO and DMSO, using xanthine and xanthine oxidase to produce superoxide anion. In this sequence the spin-adduct is only destroyed by catalase but is unaffected by SOD, which will produce H202. Thus the difference between the two spin-adducts is that the one formed in the enzymatic reaction has a dependency on , suggesting that this radical may be an initiating radical while the spin adduct produced in the model system is entirely dependent upon H2O2 [51].
Fig. 11.
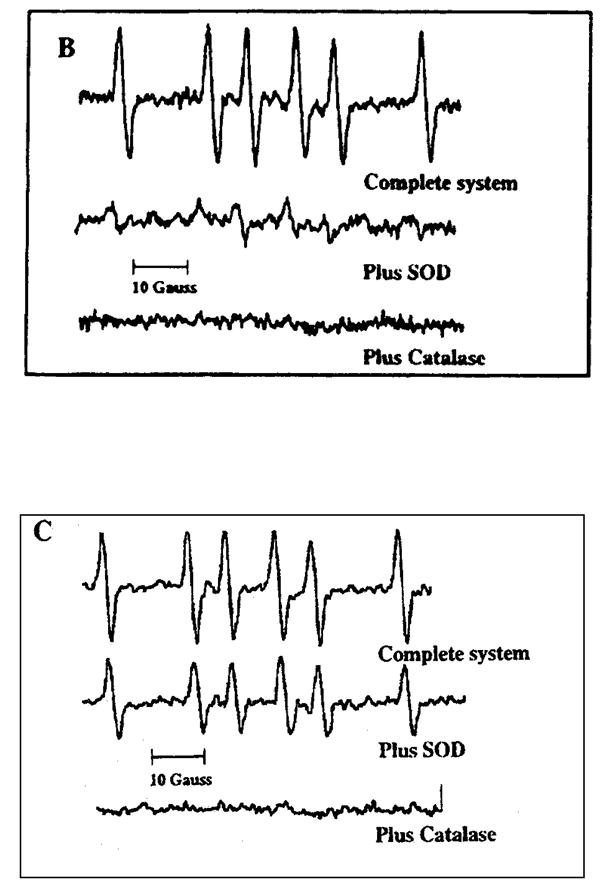
EPR spectra obtained during the enzymatic oxidation of (±)-BP-7,8-diol using DMPO as the spin-trapping agent. Panel A; complete system for the enzymatic oxidation of BP-diol plus 50 mM DMPO taken at 78 min (upper spectrum); complete system for the enzymatic oxidation of BP-diol plus 50 mM DMPO and SOD taken at 60 min (middle spectrum); complete system for the enzymatic oxidation of BP-diol plus 50 mM DMPO and catalase taken at 71 min (lower spectrum). Where the complete system contained: 45 μM BP-diol, 2.3 mM NADP+, 8% DMSO 50 mM glycine buffer pH 9.0 and 50 mM DMPO in 200 μL. Panel B; superoxide generating system (hypoxanthine/xanthine oxidase) plus DMSO as co-solvent and DMPO as spinning trapping agent, complete system at 57 min (upper spectrum); complete system plus SOD at 38 min; and complete system plus catalase at 56 min (lower spectrum). Where the complete system contained: 800 μM hypoxanthine, 8%DMSO, 100 mM potassium phosphate buffer pH 8.0 plus 50 mM DMPO and 30 mU xanthine oxidase.
Formation of the DMPO-CH3 spin adduct can be best explained by the following scheme:
(where MSOH is methanesulfinic acid)
Based on the temporal relationship that H2O2 is produced before O2 is consumed, and that superoxide anion is the initiating radical the following mechanism for autooxidation is proposed. Superoxide anion first acts as base to remove a proton from the catechol to form a hydroperoxy radical. The hydroperoxy radical then acts as oxidant on the catecholate to yield an o-semiquinone anion radical and hydrogen peroxide. The o-semiquinone radical is then oxidized by molecular oxygen to produce the fully oxidized PAH o-quinone. Trace metal ions are believed to cause the initiating Fenton-chemistry, Fig. 12.
Fig. 12.

Mechanism of catechol autooxidation
Detection of the AKR Pathway in Cell Culture
Detection of AKR expression in human cells can be achieved using isoform specific RT-PCR (for transcripts); immunoblot analysis using rabbit anti-rat AKR1C9 antisera (which cross-reacts with AKR1C1-AKR1C4) and by direct functional enzymatic assays (see earlier). Using this approach a number of human cells lines that represent a null-environment for AKR expression have been identified and are available for stable transfection: MCF-7 (mammary carcinoma) and H358, and H-441 (bronchoalveolar cells) [54]. By contrast the same assays show that HepG2 and the human lung adenocarcinoma cell line A549 are rich sources of AKRs [13, 14].
The AKR dependent production of PAH o-quinones has been detected in rat hepatocytes, MCF7-cells stably overexpressing AKR1C9 and in human lung cell lysates A549 [14, 54, 55]. In rat hepatocytes BP-7,8-dione formation was characterized by EIMS and abolished by the AKR inhibitor indomethacin [55]. In the MCF-7 AKR1C9 transfectants, BP-7,8-dione was detected by co-chromatography versus synthetic standards and by diode-array spectrometry[54]. The quinone was not formed in the mock-transfected controls. In A549 cells the conversion of DMBA-3,4-diol to DMBA-3,4-dione was monitored by trapping the quinone as its mono- and bis-thioether conjugates which can be characterized by LC/MS [14].
Properties of PAH o-Quinones Produced by AKRs
In order to study the properties PAH o-quinones produced by AKRs, it is necessary to synthesize these quinones in sufficient quantities. For each trans-dihydrodiol substrate for AKRs the corresponding o-quinone product is desired, Fig. 13. The quinones synthesized include: naphthalene-1,2-dione (NP-1,2-dione) [56], phenanthrene-1,2-dione [57]. The remainder: chrysene-1,2-dione, 5-methylchrysene-1,2-dione, 5-methylchrysene-7,8-dione, benzo[a]pyrene-7,8-dione (BP-7,8-dione), benz[a]anthracene-3,4-dione, 7-methyl-benz[a]anthracene-3,4-dione (BA-3,4-dione), 7,12-dimethylbenz[a]anthracene-3,4-dione (DMBA-3,4-dione) and benzo[g]chrysene-11,12-dione can besynthesized by somewhat similar routes [58, 59]. The synthetic sequence to BP-7,8-dione, involves reduction of 7-keto-8,9,10-trihydrobenzo[a]pyrene, to yield the 7-hydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene. Following dehydration, epoxidation to the 7,8-oxide provides a route to the enol-acetate which is then oxidized in the presence of Fremy's salt with a phase-transfer catalyst to yield the BP-7,8-dione, Fig [58, 59].
Fig. 13.

Structures of PAH o-Quinones: products of AKR reactions. NP = naphthalene, Ph = phenanthrene; Ch = chrysene; 5Me-Ch = 5-methylchrysene; BP = benzo[a]pyrene; BA = benz[a]anthracene; 7MBA = 7-methylbenz[a]anthracene; 12MBA = 12-methylbenz[a]anthracene; and DMBA = 7,12-dimethylbenz[a]anthracene, after Sukumaran and Harvey ( ).
Three Classes of PAH o-Quinones Produced by AKRs
PAH o-quinones produced by AKRs differ in their reactivity and redox-activity. Reactivity is determined by measuring the bimolecular rate constants for the addition of thiols (2-mercaptoeethanol, N-acetyl-L-cysteine and glutathione) based on RP-HPLC analyses [60]. Bimolecular rate constants for the addition of N-acetyl-L-cysteine with either (+)-anti-BPDE or BP-7,8-dione show that they equi-reactive[60, 61]. Redox activity is determined by measuring the rate of NADPH oxidation and the rate of consumption of molecular oxygen during redox-cycling in the absence and presence of subcellular fractions (e.g., mitochondria and microsomes) [62, 63]. These properties predict the cytotoxicity of the o-quinones and the mechanisms responsible for cell death.
Class I o-quinones (e.g. NP-1,2-dione and DMBA-3,4-dione) are highly electophilic and redox active. They are potent cytotoxins affecting hepatoma cell viability (acute affect) and cell survival (chronic effect). They produce the most superoxide anion and semiquinone radical. Cell death is associated with a change in redox-state decreased NADPH / NADP and NAD, GSH/GSSH, see Table [62, 63].
Class II o-quinones (e.g. 5-methlychrysene-1,2-dione and BA-3,4-dione) are less electrophilic but are not redox active. They are potent cytoxins affecting cell survival only. They produce mainly o-semiquinone anion radicals and cell death is likely due to macromolecule damage via the o-semiquinone radical [62, 63].
Class III o-quinones (e.g. BP-7,8-dione) are as electophilic as Class II o-quinones and are redox active but produce mainly superoxide anion suggesting that the o-semiquinone radical may be transient. They are potent cytotoxins affecting cell viability only. Cell death is likely due to GSH depletion, which occurs without a change in redox state [62, 63].
Detection of o-Semiquinone Anion Radicals
Reduction of representative PAH o-quinones (NP-1,2-dione and BP-7,8-dione) in the presence of NADPH coupled with EPR leads to the detection of the o-semiquinone radicals. O-Semiquinone anion radicals may differ in their stability. Naphthalene-1,2-o-semquinone radical gives a 12 line hyperfine splitting pattern indicating that the free radical can be stabilized throughout the ring system. By contrast benzo[a]pyrene-7,8-o-semiquinone radical gives a 4 line hyperfine splitting pattern showing that the free radical is stabilized only over the terminal benzo-ring and may be shorter-lived this would be consistent with the existing classification of the o-quinones, Fig. 15. [64].
Fig. 15.
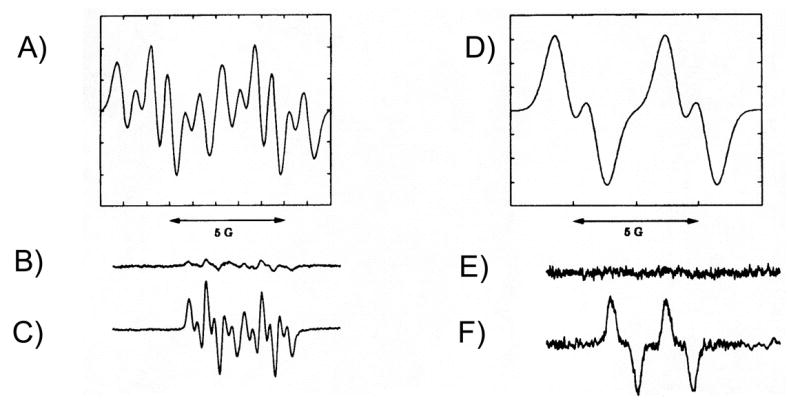
EPR spectra of o-semiquinone (SQ) radicals of NP-1,2-dione and BP-7,8-dione. Panel A, is a simulated spectrum of the SQ radical of NP-1,2-dione; panel B, is the spectrum of NP-1,2-dione (0.78 mM) taken in 8%DMSO; panel C is the spectrum of NP-,12-dione plus 10 mM NADPH taken anaerobically; panel D is a simulated spectrum of the SQ radical of BP-7,8-dione; panel E is the spectrum of BP-7,8-dione (50 uM) taken in 8%DMSO; and panel F is the spectrum of BP-7,8-dione plus 10 mM NADPH taken anaerobically.
PAH o-quinone Reactivity with Amino Acids and Proteins
PAH o-quinones can also undergo 1,4-Michael addition with the side chains of reactive amino acids to form conjugates. These reactions are likely to modify cellular proteins leading to a change in protein function. Many transcription factors (e.g., AP-1, Nf-κB) contain reactive thiols and it is speculated that their modification by PAH o-quinones may cause a change in cell signaling. Model reactions have been performed with NP-1,2-dione and amino acids. Reactions with N-acetyl-L-cysteine and cysteine are informative since they show that after the initial addition reaction subsequent rearrangements can occur leading to more complex products e.g. p-iminoquinones, Fig. 16. Michael addition products have also been characterized for the addition of N-t-boc-L-aspartic acid α-benzoyl ester, N-α-acety-L-lysine methyl ester and N-α-benzyl-L-histidine methyl ester to show that reactive amino acid side chains can form discrete amino acid conjugates [65].
Fig. 16.

PAH o-quinone amino acid conjugates. N-acetyl-L-cysteine (N-acetyl-S-(3,4-dihydro-3,4-dioxo-1-naphthyl)-L-cysteine) conjugate 1; L-cysteine (L-cysteine (N-acetyl-S-(3,4-dihydro-3,4-dioxo-1-naphthyl)-L-cysteine) conjugate 2; p-iminoquinone-L-cysteine (N-3-hydrox-4-oxo-1-naphthyl)imino-L-cysteine) conjugate 3; protected L-aspartyl (O-(3,4-dihydroxy-1-naphthyl)-N-t-Boc-L-aspartyl benzyl ester) conjugate 4; protected L-lysine ( 1-N-(3,4-dihydro-3,4-dioxo-1-naphthyl)-Nα-acetyl-L-lysine methyl ester) conjugate 5; and protected L-histidyl (1-N-(3,4-dihydro-3,4-dioxo-1-naphthyl)-Nα-benzyl-L-histidyl methyl ester 6;
DNA-Lesions Caused by PAH o-Quinones
PAH o-quinones will cause both covalent and oxidative modification of DNA. Interest exists in characterizing these reactions because they may lead to mutation of critical protoncogenes and tumor suppressor genes, increase the mutational load, and contribute to the causation of human lung cancer. The most common mutations seen in human lung cancer are G to T transversions in K-ras (12th codon) and G to T transversions in p53 (codons, 245, 248 and 249) [66-68]. DNA-lesions observed with PAH o-quinones should provide routes to these mutations if they are to a play role in lung carcinogenesis.
Covalent Adducts with DNA
PAH o-quinones have the capacity to form stable N2-deoxyguanosine and N6-deoxyadenosine adducts. Detection of stable BP-7,8-dione DNA-adducts can be achieved by reacting of [3H]-BP-7,8-dione (Chemsyn, Lenexa, Ks) with calf-thymus DNA, followed by enzymatic digestion to the constituent deoxyribonucleosides. This leads to the isolation of a single major adduct that coelutes on RP-HPLC with the adduct formed by addition of the o-quinone to oligo-p(dG)10, followed by digestion [69]. By contrast no adducts were detected when [3H]-BP-7,8-dione was reacted with poly-dC, poly-dT or poly-dA. No formal structure exists for the stable N2-deoxyguanosine adduct due to the difficulty in synthesizing authenticated standards.
Current routes to these standards include two approaches. In the first route their is activation of the o-quinone by bromination followed by attack with the O-TBDMS protected deoxyribonucleoside. In the second approach the amino-quinone is formed followed by attack from the halogenated deoxyribonucleoside [70]. Bulky stable deoxyribonucleoside adducts could give rise to G to T transversions depending on whether there is error-free or error-prone trans-lesional synthesis by replicative DNA polymerases.
PAH o-quinones can also form N7-guanine and N7-adenine depurinating adducts leaving behind an abasic site. N7-guanyl depurinating adducts of NP-1,2-dione, phenanthrene-1,2-dione and BP-7,8-dione can be synthesized by conducting reactions with the o-quinone and deoxyribonucloside in the presence of acetic acid [71].
Typical reactions involve reacting a PAH o-quinone (10 mg) in acetonitrile/DMSO (2 mL) to a stirred solution of a 5-fold molar exces of dGuo in a 1 : 1 mixture of acetic acid : water (4 mL). The reaction mixture is purified by solid-phase extraction on a C18-Sep-Pak cartridge which is washed successfully with 1% acetic acid/water (30 mL and water (3 mL). The product is then eluted with 75% methanol : water, and following removal of the organic solvent, the final product is obtained by lyophilization and analyzed by RP-HPLC.
LC/ESI/MS analysis of the purified 7-(benzo[a]pyrene-7,8-dion-10-yl) guanine adduct gave a protonated molecular ion at m/z = 432 consistent with the N7 depurinating adduct. CID of the m/z = 432 ion generated adduct derived fragment ions at m/z = 281 (MH+ -guanine) and 253 (MH+-guanine-CO). Additional product ions consistent with the proposed structure were observed at m/z = 410 (MH+-NH3), 404 (MH+ -CO), 389 (MH+-HNCO), 362 (MH+-CO, NH2CN) and 308 (MH+ -CO, NH2CN, 2HCN), Fig. 18. These adducts have also been detected in o-quinone reactions with calf-thymus DNA. In terms of mutagenesis, abasic sites created by depurination provide a route to G to T transversions. DNA polymerases often introduce and A opposite the abasic site so that upon replication a G to T ransversion occurs [72].
Fig. 18.

Collision-induced dissociation spectrum of the m/z 432 (MH+) ion of the benzo[a]pyrene-7,8-dione-guanine adduct, obtained on a Finnigan LCQ ion trap.
Oxidative Damage of DNA
Different forms of oxidative DNA damage are possible as a result of PAH o-quinone redox-cycling, Fig. 17. The ROS produced could cause DNA strand breaks (single and double strand breaks); formation of oxidatively damaged bases (e.g., 8-oxo-2-deoxyguanosine and thymine glycol); the formation of base-propenals from 4′OH radical attack of deoxyribose leading to malondialdehyde-dG adducts; and the indirect formation of adducts that may arise from lipid peroxidation breakdown products. Many of these lesions can be seen with PAH o-quinones [64].
Fig. 17.

Covalent and oxidative DNA lesions caused by PAH o-quinones.
DNA strand scission can be detected using RF φX174DNA and DNA fragmentation can be detected using poly dG: poly dC by agarose gel electrophoresis following incubation with sub-micromolar concentrations of PAH o-quinones under redox-cycling conditions. Redox-cycling conditions are maximized by the presence of NADPH and CuCl2, whereas DNA strand scission or fragmentation is abolished by enzymatic scavengers of ROS (SOD and catalase) and chemical scavengers of ROS (Tiron and mannitol). Under anaerobic conditions in which only the o-semiquinone anion radical is formed no DNA strand scission was observed [64]. The DNA strand scission that is seen in vitro may lead to cytotoxic rather than mutagenic consequences.
PAH o-quinones in the presence of NADPH produce sufficient ROS to form 8-oxo-deoxyguanosine (8-oxo-dGuo). Interest in this lesion exists since the C8 oxygen causes base mispairing with adenine if the lesion is unrepaired [73, 74]. Like depurination these lesions would also provide a straightforward route to G to T transversions. Thus both depurination and 8-oxo-dGuo formation could provide routes to the G to T transversions that dominate in K-ras and p53.
Sensitive detection of 8-oxo-dGuo is required to detect this lesion and the method of choice has been EC-HPLC which provides sensitivity in the fmole range, where the amount of 8-oxo-dGuo is normalized to dGuo. Pronounced increases in the amount of 8-oxo-dGuo can be seen when salmon testis DNA is treated with NP-1,2-dione under redox cycling conditions (20 μM NP-1,2-dione, 200 μM NADPH), Table 4. Levels of 8-oxo-dGuo are decreased by the addition of the S9 fraction from rat liver due to protein sequestration but elevated with SOD due to the increased production of H2O2. Measurements of 8-oxo-dGuo and the precautions necessary to prevent adventitious oxidation of dG are described elsewhere in this series [75].
Table 4.
Detection of 8-oxo-dGuo in salmon testis DNA exposed to NP-1,2-dione under redox cycling conditions
| Treatment | Peak Area | Ratio | |
|---|---|---|---|
| dGuo | 8-oxo-dGuo | 8-oxo-dGuo/dGuo | |
| DNA w/Dig Enz | 235 μC | 8.2 nC | 2.4 per 105 |
| DNA w/DMSO, S9, NADPH, Dig. Enz | 230 μC | 8.3 nC | 2.5 per 105 |
| DNA w/NP-1,2-dione, S9, NADPH, Dig Enz | 228 μC | 38 nC | 23.0 per 105 |
| DNA w/NP-1,2-dione, NADPH, Dig Enz | 229 μC | 525 nC | 239 per 105 |
| DNA w/NP-1,2-dione, NADPH, SOD, Dig. Enz | 253 μC | 2230 nC | 892 per 105 |
PAH o-Quinones and p53 Mutagenesis
We have adapted a yeast reporter gene assay that allows us to detect mutation of p53 mediated by PAH o-quinones produced by AKRs [76, 77]. In this assay, plasmid p53 is treated with PAH o-quinones in the absence and presence of redox-cycling conditions (at concentrations of o-quinone that insufficient to cause significant strand scission). The resulting mixture of wt p53 and lesioned DNA is mixed with a gap-repair plasmid and used to transform a host yeast strain containing a p53 reporter gene. The gapped-repair plasmid contains sites for homologous recombination. The transformed host produces wt and mutated p53 from a ADH promoter 5′to the sites of homologous recombination. The host also has integrated into its chromosome an adenine reporter gene driven by the p21 promoter. Wt p53 will bind to the p21 promoter and in the presence of limiting adenine the yeast colonies will synthesize adenine and turn white. By contrast mutated p53 will be unable to bind to the p21 promoter and in the presence of limiting adenine the yeast colonies will not synthesize adenine and colonies will turn red. This positive selection allows us to score change-in-function mutations in p53 that eliminate its transcriptional competency. Using this assay we have shown that BP-7,8-dione is mutagenic at sub-micromolar concentrations, provided a redox-cycling system is present (Table 5), that the mutation rate (% red colonies / white colonies) is abolished by ROS scavengers, and that a significant number of G to T transversions were observed, implicating 8-oxo-dG in their formation.
Table 5.
NP-1,2-dione and BP-7,8-dione mutate p53 under redoc-cycling conditions
| Treatment | NP-1,2-dione | BP-7,8-dione | ||||
|---|---|---|---|---|---|---|
| Colonies | Colonies | |||||
| total | red | % red | total | red | % red | |
| aquinone alone | 922 | 11 | 1.2 | 3380 | 32 | 0.9 |
| quinone + NADPH | 896 | 13 | 1.5 | 3160 | 31 | 1.0 |
|
| ||||||
|
bμM quinone +
NADPH + CuCl2 |
||||||
|
| ||||||
| 0 μM | 798 | 16 | 2.0 | 3290 | 34 | 1.0 |
| 0.031 μM | 812 | 31 | 3.8 | 3284 | 41 | 1.2 |
| 0.0625 μM | 1232 | 54 | 4.4 | 3080 | 51 | 1.7 |
| 0.125 μM | 756 | 46 | 6.1 | 2536 | 69 | 2.1 |
| 0.25 μM | 484 | 45 | 9.3 | 2200 | 113 | 5.1 |
p53 mutagenesis was determined in the presence of 20 μM PAH o-quinone alone
p53 mutagenesis was determined in the presence of increasing PAH o-quinone concentration in the presence of 1 mM NADPH and 100 μM CuCl2
AKR Stable Transfection Systems To Measure DNA-Lesions and p53 Mutations
Strong evidence exists for the formation of stable and depurinating covalent adducts of PAH o-quinones and for the oxidative lesions based on in vitro measurements, however, the DNA lesions have not been detected within a cellular context. Stable overxpression of human AKRs into heterologous systems will address this concern. Several systems currently exist, these include the stable overexpression of AKR1A1 (aldehyde reductase) in human lung bronchoalevolar cells (H-358 and H-441) and the observation that A549 cells endogenously overexpress AKR1C1-AKR1C3. Treatment of cells with PAH trans-dihydrodiol substrates would be predicted to cause the DNA-lesions described, and should be attenuated by AKR1A1 inhibitors (alrestat and tolerstat) and AKR1C inhibitors (indomethacin and 6-medroxyprogesterone acetate).
MCF7-AKR1A1 and A549-AKR1C1-AKR1C3 overexpressing cells will also allows us to determine whether exposure of these cells to PAH trans-dihydrodiols will cause change in function mutations in p53. MCF7 cells and A549 cells contain wt p53, following treatment of the cells with PAH trans-dihydrodiols, p53 can be obtained by RT-PCR and substituted into the yeast reporter gene assay to detect p53 mutations. Increased p53 mutation should be attenuated by AKR inhibitors. Such an approach will provide a proof-of-principle concept that AKRs generate reactive and redox-active o-quinones and cause change-in-function mutations in p53.
Fig. 14.

Synthesis of BP-7,8-dione.
Fig. 19.

Yeast reporter assay for p53 mutation.
Acknowledgments
The methods and techniques described could not have been developed without the contribution of many talented colleagues including: Drs. Thomas E. Smithgall, Lynn Flowers, Varanasi Murty, Kirsten McCoull, Elizabeth Glaze, Michael E. Burczynski, Nisha Palackal and Sridhar Gopishetty. Synthetic approaches were developed in collaboration with Dr. Ronald G. Harvey at the Ben May Institute for Cancer Research, and mass-spectrometry was performed in the laboratory of Dr. Ian Blair, at the Center for Cancer Pharmacology, University of Pennsylvania School of Medicine. Many PAH-metabolites were made available from the National Cancer Institute Chemical Carcinogen Standard Reference Repository and the work was supported by grants R01 CA39505 and P01 CA092537 awarded to T.M.P.
References
- 1.Jez JM, Flynn TG, Penning TM. Biochem Pharmacol. 1997;54:639–647. doi: 10.1016/s0006-2952(97)84253-0. [DOI] [PubMed] [Google Scholar]
- 2.Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Biochem J. 1997;326:625–636. doi: 10.1042/bj3260625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Askonas LJ, Ricigliano JW, Penning TM. Biochem J. 1991;278:835–841. doi: 10.1042/bj2780835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimshaw CE, Bohren KM, Lai CJ, Gabbay KH. Biochemistry. 1995;34:14356–14365. doi: 10.1021/bi00044a012. [DOI] [PubMed] [Google Scholar]
- 5.Ratnam K, Ma H, Penning TM. Biochemistry. 1999;38:7856–7864. doi: 10.1021/bi982838t. [DOI] [PubMed] [Google Scholar]
- 6.Hoog SS, Pawlowski JE, Alzari PM, Penning TM, Lewis M. Proc Natl Acad Sci USA. 1994;91:2517–2521. doi: 10.1073/pnas.91.7.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett MJ, Schlegel BP, Jez JM, Penning TM, Lewis M. Biochemistry. 1996;35:10702–10711. doi: 10.1021/bi9604688. [DOI] [PubMed] [Google Scholar]
- 8.Bennett MJ, Albert RH, Jez JM, Ma H, Penning TM, Lewis M. Structure. 1997;5:799–812. doi: 10.1016/s0969-2126(97)00234-7. [DOI] [PubMed] [Google Scholar]
- 9.Schlegel BP, Jez JM, Penning TM. Biochemistry. 1998;37:3538–3548. doi: 10.1021/bi9723055. [DOI] [PubMed] [Google Scholar]
- 10.Hara A, Taniguchi H, Nakayama T, Sawada H. J Biochem. 1990;108:250–254. doi: 10.1093/oxfordjournals.jbchem.a123189. [DOI] [PubMed] [Google Scholar]
- 11.Hara A, Matsurra K, Tamada Y, Sato K, Miyabe Y, Deyashiki Y, Ishida T. Biochem J. 1996;313:373–376. doi: 10.1042/bj3130373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deyashiki Y, Taniguchi H, Amano T, Nakayama T, Hara A, Sawada H. Biochem J. 1992;282:741–746. doi: 10.1042/bj2820741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burczynski ME, Harvey RG, Penning TM. Biochemistry. 1998;37:6781–6790. doi: 10.1021/bi972725u. [DOI] [PubMed] [Google Scholar]
- 14.Palackal NT, Lee SH, Harvey RG, Blair IA, Penning TM. J Biol Chem. 2002;277:24799–24808. doi: 10.1074/jbc.M112424200. [DOI] [PubMed] [Google Scholar]
- 15.Atalla A, M E. Chem Biol Inter. 2001;130-132:737–748. doi: 10.1016/s0009-2797(00)00304-5. [DOI] [PubMed] [Google Scholar]
- 16.Ellis EM, Judah DJ, Neal GE, Hayes JD. Proc Natl Acad Sci USA. 1993;90:10350–10354. doi: 10.1073/pnas.90.21.10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knight LP, Primiano T, Groopman JD, Kensler TW, Sutter TR. Carcinogenesis. 1999;20:1215–1223. doi: 10.1093/carcin/20.7.1215. [DOI] [PubMed] [Google Scholar]
- 18.Cavalieri EL, Rogan EG. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]
- 19.Devanesan PD, RamaKrishna NVS, Todorovic R, Rogan EG, Cavalieri EL, Jeong H, Jankowiak R, Small GJ. Chem Res Toxicol. 1992;5:302–309. doi: 10.1021/tx00026a024. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Devanesan PD, Higginbotham S, Ariese F, Jankowiak R, Small GJ, Rogan EG, Cavalieri E. Chem Res Toxicol. 1996;9:897–903. doi: 10.1021/tx960004a. [DOI] [PubMed] [Google Scholar]
- 21.Gelboin HV. Physiol Rev. 1980;60:1107–1166. doi: 10.1152/physrev.1980.60.4.1107. [DOI] [PubMed] [Google Scholar]
- 22.Conney AH. Cancer Res. 1982;42:4875–4917. [PubMed] [Google Scholar]
- 23.Smithgall TE, Harvey RG, Penning TM. J Biol Chem. 1986;261:6184–6191. [PubMed] [Google Scholar]
- 24.Smithgall TE, Harvey RG, Penning TM. J Biol Chem. 1988;263:1814–1820. [PubMed] [Google Scholar]
- 25.Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS. Chem Res Toxicol. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- 26.Flowers-Geary L, Harvey RG, Penning TM. 1992;11:49–58. [Google Scholar]
- 27.Pawlowski JE, Huizinga M, Penning TM. J Biol Chem. 1991;266:8820–8825. [PubMed] [Google Scholar]
- 28.Pawlowski JE, Penning TM. J Biol Chem. 1994;269:13502–13510. [PubMed] [Google Scholar]
- 29.Ma H, Penning TM. Proc Natl Acad Sci USA. 1999;96:11161–11166. doi: 10.1073/pnas.96.20.11161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palackal NT, Burczynski ME, Harvey RG, Penning TM. Biochemistry. 2001;40:10901–10910. doi: 10.1021/bi010872t. [DOI] [PubMed] [Google Scholar]
- 31.Vogel E, Gunther H. Angew Chem Int Ed Eng. 1967;6:385–401. [Google Scholar]
- 32.Jeffrey AM, Yeh HJ, Jerina DM, Demarinis RM, Foster CH, Piccolo DE, Berchtold GA. J Amer Chem Soc. 1974;96:6929–6937. [Google Scholar]
- 33.Ivins JK, Penning TM. Cancer Res. 1987;47:680–684. [PubMed] [Google Scholar]
- 34.Dipple A, Harvey RG, editors. American Chemical Society; Washington, D.C.: 1984. pp. 1–17. [Google Scholar]
- 35.Yagi H, J DM. J Amerc Chem Soc. 1975;97:3185–3192. [Google Scholar]
- 36.Lehr RE, Schaefer-Ridder M, Jerina DM. J Org Chem. 1977;42:736–744. [Google Scholar]
- 37.Fu PP, H RG. Tetrahedron Lett. 1977;24:2059–2062. [Google Scholar]
- 38.Booth J, Boyland E, Turner EE. J Chem Soc. 1950:1188–1190. [Google Scholar]
- 39.Cortez C, H RG. Org Synth. 1978;58:12–16. [Google Scholar]
- 40.Fu PP, H RG. J Org Chem. 1979;44:3778–3784. [Google Scholar]
- 41.Pataki J, Lee H, Harvey RG. Carcinogenesis. 1983;4:399–402. doi: 10.1093/carcin/4.4.399. [DOI] [PubMed] [Google Scholar]
- 42.Harvey RG, Sukumaran KB. Tetrahedron Lett. 1977:2387–2390. [Google Scholar]
- 43.Lee HH, Harvey RG. J Org Chem. 1979;44:4948–4953. [Google Scholar]
- 44.Lee HH, Harvey RG. J Org Chem. 1986;51:3502–3507. [Google Scholar]
- 45.Harvey RG. Polycyclic aromatic hydrocarbons. Academic Press; New York: 1997. pp. 129–188. [Google Scholar]
- 46.Gill HS, Kole PL, Wiley JC, Li KM, Higginbotham S, Rogan EG, Cavalieri EL. Carcinogenesis. 1994;15:2455–2460. doi: 10.1093/carcin/15.11.2455. [DOI] [PubMed] [Google Scholar]
- 47.Smithgall TE, Harvey RG, Penning TM. Cancer Res. 1988;48:1227–1232. [PubMed] [Google Scholar]
- 48.Palackal NT, Lee SH, Harvey RG, Blair IA, Penning TM. J Biol, Chem. 277:24799–24808. doi: 10.1074/jbc.M112424200. [DOI] [PubMed] [Google Scholar]
- 49.Flowers-Geary L, Harvey RG, Penning TM. Chem Res Toxicol. 1992;5:576–583. doi: 10.1021/tx00028a018. [DOI] [PubMed] [Google Scholar]
- 50.Burczynski ME, Palackal NT, Harvey RG, Penning TM. Polycyclic Aromatic Compunds. 1999;215:205–209. [Google Scholar]
- 51.Penning TM, Ohnishi ST, Ohnishi T, Harvey RG. Chem Res Toxicol. 1996;9:84–92. doi: 10.1021/tx950055s. [DOI] [PubMed] [Google Scholar]
- 52.Liem HH, Cradenas F, Tavassoli M, Poh-Fitzpatrick MB, Muller-Eberhard U. Anal Biochem. 1979;98:388–393. doi: 10.1016/0003-2697(79)90157-x. [DOI] [PubMed] [Google Scholar]
- 53.Azzi A, Montecucco C, Richter C. Biochem Biophys Res Commun. 1975;65:597–603. doi: 10.1016/s0006-291x(75)80188-4. [DOI] [PubMed] [Google Scholar]
- 54.Tsuruda L, Hou Yt, Penning TM. Chem Res Toxicol. 2001;14:856–862. doi: 10.1021/tx0100035. [DOI] [PubMed] [Google Scholar]
- 55.Flowers-Geary L, Harvey RG, Penning TM. Carcinogenesis. 1996;16:2707–2715. doi: 10.1093/carcin/16.11.2707. [DOI] [PubMed] [Google Scholar]
- 56.Fieser LF, Blatt AH, editors. Wiley; New York: 1943. p. 430. [Google Scholar]
- 57.Fieser LF. J Amer Chem Soc. 1929;51:1896–1906. [Google Scholar]
- 58.Fu PP, Cortez C, Sukumaran KB, Harvey RG. J Org Chem. 1979;44:4265–4271. [Google Scholar]
- 59.Sukamaran KB, Harvey RG. J Org Chem. 1980;45:4407–4413. [Google Scholar]
- 60.Murty VS, Penning TM. Chem-Biol Inter. 1992a;84:169–188. doi: 10.1016/0009-2797(92)90077-x. [DOI] [PubMed] [Google Scholar]
- 61.Islam NB, Gupta SC, Yagi H, Jerina DM, Whalen DL. J Amer Chem Soc. 1990;112:6363–6369. [Google Scholar]
- 62.Flowers-Geary L, Harvey RG, Penning TM. Chem Res Toxicol. 1993;6:252–260. doi: 10.1021/tx00033a002. [DOI] [PubMed] [Google Scholar]
- 63.Flowers-Geary L, Bleczinski W, Harvey RG, Penning TM. Chemico-Biol Inter. 1996;99:55–72. doi: 10.1016/0009-2797(95)03660-1. [DOI] [PubMed] [Google Scholar]
- 64.Flowers L, Ohnisni ST, Penning TM. Biochemistry. 1997;36:8640–8648. doi: 10.1021/bi970367p. [DOI] [PubMed] [Google Scholar]
- 65.Sridhar G, Murty VS, Lee SH, Blair IA, Penning TM. Tetrahedron Lett. 2001;57:407–412. [Google Scholar]
- 66.Rodenhius S. Cancer Biology. 1992;3:241–247. [PubMed] [Google Scholar]
- 67.Soussi T. The p53 Database. 2001 http://p53.curie.fr/p53%20site%20version%202.0/database/p53_database.html.
- 68.Hainaut P, Pfeifer GP. Carcinogenesis. 2001;22:367–374. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 69.Shou M, Harvey RG, Penning TM. Carcinogenesis. 1993;14:475–482. doi: 10.1093/carcin/14.3.475. [DOI] [PubMed] [Google Scholar]
- 70.Aldo-Keto reductases and toxicant metabolism. In: Gopishetty SR, Harvey RG, Lee SH, Balir IA, Penning TM, editors; Penning TM, Petrash J Mark, editors. ACS Symposium Series. Oxford University Press; 2003. In Press. [Google Scholar]
- 71.McCoull KD, Rindgen D, Blair IA, Penning TM. Chem Res Toxicol. 1999;12:237–346. doi: 10.1021/tx980182z. [DOI] [PubMed] [Google Scholar]
- 72.Sagher D, Strauss B. Biochemistry. 1983;22:4518–4526. doi: 10.1021/bi00288a026. [DOI] [PubMed] [Google Scholar]
- 73.Breen AP, Murphy JP. Free Radical Biol & Med. 1995;18:1033–1077. doi: 10.1016/0891-5849(94)00209-3. [DOI] [PubMed] [Google Scholar]
- 74.Wang D, Kreutzer DA, Essigmann JM. Mutation Res. 1998;400:99–115. doi: 10.1016/s0027-5107(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 75.Shigenaga MK, Park JW, Cundy KC, Gimeno CJ, Ames BN. Methods Enz. 1990;186:521–530. doi: 10.1016/0076-6879(90)86146-m. [DOI] [PubMed] [Google Scholar]
- 76.Yu D, Penning TM, Field JM. Polycyclic Aromatic Compounds. 2002;3-4:881–891. [Google Scholar]
- 77.Yu D, Berlin JA, Penning TM, Field JM. Chem Res Toxicol. 2002;15:832–842. doi: 10.1021/tx010177m. [DOI] [PubMed] [Google Scholar]