

Abstract
Neuroprotection can be induced by low doses of NMDA, which activate both synaptic and extrasynaptic NMDA receptors. This is in apparent contradiction with our recent findings that extrasynaptic NMDA receptor signaling exerts a dominant inhibitory effect on prosurvival signaling from synaptic NMDA receptors. Here we report that exposure to low preconditioning doses of NMDA results in preferential activation of synaptic NMDA receptors because of a dramatic increase in action potential firing. Both acute and long-lasting phases of neuroprotection in the face of apoptotic or excitotoxic insults are dependent on this firing enhancement. Key mediators of synaptic NMDA receptor-dependent neuroprotection, phosphatidylinositol 3 kinase-Akt (PI3 kinase-Akt) signaling to Forkhead box subgroup O (FOXO) export and glycogen synthase kinase 3β (GSK3β) inhibition and cAMP response element-binding protein-dependent (CREB-dependent) activation of brain-derived neurotrophic factor (BDNF), can be induced only by low doses of NMDA via this action potential-dependent route. In contrast, NMDA doses on the other side of the toxicity threshold do not favor synaptic NMDA receptor activation because they strongly suppress firing rates below baseline. The classic bell-shaped curve depicting neuronal fate in response to NMDA dose can be viewed as the net effect of two antagonizing (synaptic vs extrasynaptic) curves: via increased firing the synaptic signaling dominates at low doses, whereas firing becomes suppressed and extrasynaptic signaling dominates as the toxicity threshold is crossed.
Keywords: apoptosis, calcium [Ca], transcription, excitability, kinase, CREB, signal transduction, tolerance, NMDA receptor, neuroprotection, PI3 kinase
Introduction
Survival of many neuronal cell types is dependent on normal patterns of synaptic NMDA receptor activity (Ikonomidou and Turski, 2002; Hardingham and Bading, 2003). Elimination of NMDA receptor activity in vivo causes widespread apoptosis and exacerbates ongoing neurodegeneration in the developing and adult CNS (Ikonomidou et al., 1999, 2000; Monti and Contestabile, 2000; Olney et al., 2002; Adams et al., 2004). Blockade of NMDA receptor activity in vitro also exacerbates ongoing neurodegeneration, whereas enhancing synaptic NMDA receptor activity is neuroprotective (Papadia et al., 2005). Synaptic NMDA receptor activity promotes neuroprotection by activating pro-survival signal pathways, including cAMP response element-binding protein-dependent (CREB-dependent) gene expression (Lee et al., 2005; Papadia et al., 2005). CREB is a central mediator of neuroprotection (Lonze and Ginty, 2002; Dragunow, 2004) that controls a number of pro-survival genes (Mayr and Montminy, 2001; Impey et al., 2004; Zhang et al., 2005). In particular, expression of the CREB-dependent neurotrophin, brain-derived neurotrophic factor (BDNF) (Shieh et al., 1998; Tao et al., 1998), is regulated strongly by synaptic NMDA receptor activity in vitro (Hardingham et al., 2002), and lowered BDNF expression has been proposed to play a causal role in neurodegeneration that follows NMDA receptor blockade in vivo (Hansen et al., 2004).
We recently showed that, in contrast to synaptic NMDA receptor activation, extrasynaptic NMDA receptor activity promotes CREB inactivation and early excitotoxic events such as mitochondrial depolarization in hippocampal neurons (Hardingham and Bading, 2002; Hardingham et al., 2002). These findings have been recapitulated by others in cortical neurons (Leveill et al., 2005), raising the possibility that this is a general CNS phenomenon. CREB dephosphorylation by extrasynaptic NMDA receptor activity is dominant over CREB-activating signals (Sala et al., 2000; Hardingham et al., 2002). Moreover, activation of BDNF transcription by synaptic NMDA receptors is blocked by concurrent extrasynaptic NMDA receptor activation (Hardingham et al., 2002).
These observations are, however, at odds with the fact that low levels of bath-applied NMDA or glutamate are neuroprotective; these stimuli will activate both synaptic and extrasynaptic NMDA receptors, yet the pro-death extrasynaptic NMDA receptor-dependent signal is clearly not dominating. The anti-apoptotic effects of low doses of NMDA or glutamate have been demonstrated in several neuronal cell types in vitro, including cerebellar granule cells and hippocampal neurons (Balazs et al., 1988; Bambrick et al., 1995). Protection against excitotoxic trauma also can be induced by chronic exposure of neurons to a subtoxic dose of glutamate or NMDA (Chuang et al., 1992; Rocha et al., 1999; Raval et al., 2003) via a mechanism thought to involve CREB activation and BDNF synthesis (Monti et al., 2002; Jiang et al., 2005). In these scenarios pro-survival signaling is dominant and not suppressed by an extrasynaptic NMDA receptor-dependent CREB shut-off signal.
To address this apparent discrepancy, we analyzed the neuronal response to a range of NMDA doses on either side of the toxicity threshold. The reason for the transition from protecting to toxic doses of NMDA generally is assumed to be attributable to a quantitative increase in Ca2+ influx via NMDA receptors. However, we wanted to determine whether there were any qualitative differences in the neuronal response to NMDA as the dose rose from protective to toxic. In particular, we wanted to understand why pro-survival synaptic NMDA receptor signaling was able to dominate in neurons exposed to protecting doses of NMDA. We found that the patterns of NMDA receptor activity on either side of the toxicity threshold are very different; protecting doses enhance action potential firing rates, whereas toxic doses suppress it. Furthermore, this enhancement is essential for the resulting neuroprotection.
Materials and Methods
Hippocampal cultures, stimulation, and the induction of apoptosis/necrosis.
Hippocampal neurons from embryonic 21 d (E21) Sprague Dawley rats were cultured as described (Bading and Greenberg, 1991) except that the growth medium comprised Neurobasal A medium plus B27 (Invitrogen, Carlsbad, CA), 1% rat serum, and 1 mm glutamine. Experiments were performed after a culturing period of 10–12 d during which the hippocampal neurons developed a rich network of processes, expressed functional NMDA-type and AMPA/kainate-type glutamate receptors, and formed synaptic contacts (Hardingham et al., 2001, 2002). Neurons were treated with NMDA in the presence or absence of tetrodotoxin (TTX; 1 μm; Calbiochem, La Jolla, CA). Neurons were subjected to trophic deprivation (72 h) by transferring them from growth medium to a medium containing 10% MEM (Invitrogen) and 90% salt–glucose–glycine (SGG) medium [containing the following (in mm): 114 NaCl, 5.292 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 1 glycine, 30 glucose, 0.5 sodium pyruvate, plus 0.219% NaHCO3 and 0.1% phenol red; osmolarity, 325 mOsm/l] (Bading et al., 1993). “Trophic medium” contained added insulin–transferrin–selenite supplement (ITS; 7.5 μg insulin/ml; 7.5 μg transferrin/ml, and 7.5 ng sodium selenite/ml; Sigma, St. Louis, MO) in which neurons survived for >5 d without significant cell death and had high basal Akt activity because of the insulin supplementation. Staurosporine exposure (36 h) also was used to induce apoptosis (50 nm). Morphologically, trophically deprived and staurosporine-treated neurons show typical signs of apoptotic-like cell death (shrunken cell body and large round chromatin clumps). Examples of pictures of apoptotic nuclei of trophically deprived and staurosporine-treated neurons are shown in previous studies (Hardingham et al., 2002; Papadia et al., 2005). Neurons were fixed and subjected to 4′,6-diamidino-2-phenylindole (DAPI) staining, and cell death was quantified by the counting (blind) of the number of apoptotic nuclei as a percentage of the total. For the triggering of excitotoxic cell death, neurons were exposed to glutamate (40 μm) in our standard trophically deprived medium (90% SGG, 10% MEM) for 1 h, after which neurons were washed once and returned to fresh medium. Neurons that die in response to exposure to excitotoxic levels of glutamate exhibit swollen cell bodies and pyknotic nuclei with small irregular chromatin clumps, a characteristic of necrotic cell death as opposed to apoptotic-like death ((Fujikawa et al., 2000) (for example pictures, see Hardingham et al., 2002). Cell death was determined 24 h later by counting of the number of DAPI-stained pyknotic/necrotic nuclei as a percentage of the total.
Imaging and immunofluorescence studies.
All imaging was performed at 37°C on a Zeiss (Oberkochen, Germany) Axioplan2 microscope, using a laser-scanning confocal microscope driven by LSM510 software. Fluo-3 Ca2+ imaging and confocal laser-scanning microscopy were done as described (Hardingham et al., 1997). Ca2+ concentrations were expressed as a function of the Fluo-3 fluorescence (FF) [(F − Fmin)/(Fmax − F)]. For analyzing mitochondrial Ca2+, we loaded the neurons with 2 μm rhodamine-FF (Rhod-FF) for 30 min at room temperature, followed by extensive washing. Neurons loaded with Rhod-FF showed a punctate distribution of the indicator that very closely resembled that obtained with MitoTracker Red (data not shown), and the pattern was abolished by the mitochondrial uncoupler carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP) (see Fig. 2b, inset), confirming the mitochondrial localization of the dye. Mitochondrial membrane potential was analyzed as described (Hardingham et al., 2002), using rhodamine 123 (Invitrogen). Briefly, hippocampal neurons were loaded with rhodamine 123 (10 μg/ml or 26 μm) in SGG medium for 10 min, followed by extensive washing with SGG. Rhodamine 123 partitions into polarized mitochondria where it self-quenches at the concentration used. When mitochondria depolarize, rhodamine 123 leaks out of the mitochondria into the cytoplasm where it dequenches and fluoresces strongly. Maximum rhodamine 123 signal was obtained by eliminating the mitochondrial potential completely by exposing the neurons to the mitochondrial uncoupler FCCP (2.5 μm; Sigma). Immunofluorescence was performed as described (Hardingham et al., 2001). Pictures of Forkhead box subgroup O1-myelocytomatosis-expressing (FOXO1-myc-expressing) neurons were taken on a CCD camera driven by Openlab software, and the subcellular distribution was scored as exclusively nuclear, nuclear plus cytoplasmic (even distribution in nucleus and cytoplasm), or cytoplasmic (higher level in cytoplasm than in nucleus).
Figure 2.
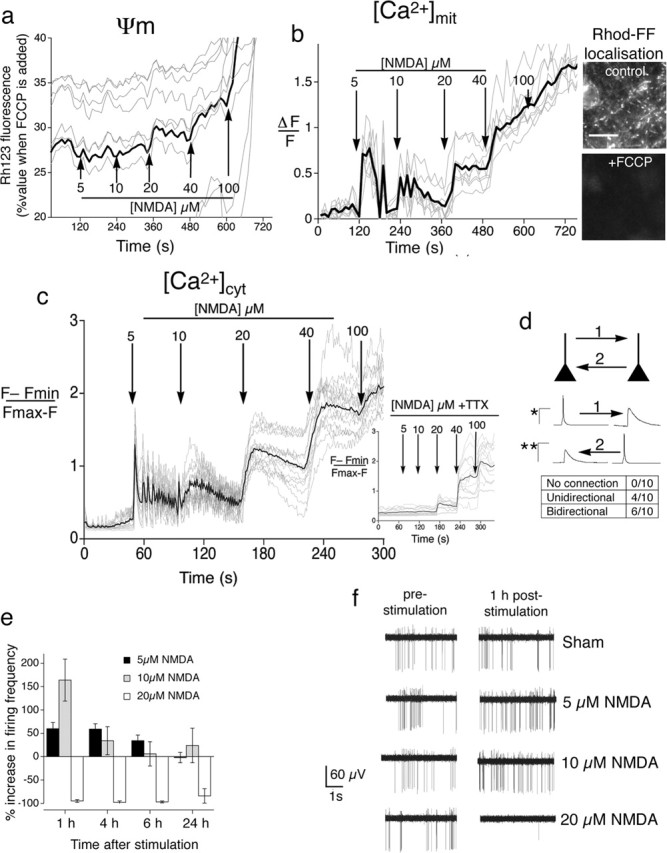
Qualitative differences in the cellular response to protecting versus toxic doses of NMDA. a, Only toxic doses of NMDA perturb the mitochondrial membrane potential. NMDA was added to neurons at regular intervals at increasing concentrations. Loss of mitochondrial membrane potential is indicated by an increase in fluorescence (Keelan et al., 1999) and is expressed as a percentage of the signal obtained when mitochondria are depolarized completely by FCCP treatment at the end of the experiment. Gray lines indicate the traces of individual neurons; the black line indicates the mean. b, Toxic doses of NMDA induce sustained elevated mitochondrial Ca2+, unlike protecting doses. Neurons were subjected to Rhod-FF imaging and exposed to a stepwise increase in NMDA. Gray lines indicate the traces of individual neurons; the black line indicates the mean. Inset, Punctate Rhod-FF localization is abolished by the mitochondrial uncoupler FCCP. Scale bar, 20 μm. c, Protecting doses of NMDA induce AP-dependent intracellular Ca2+ transients. Gray lines indicate the traces of individual neurons; the black line indicates the mean. Neurons were subjected to Fluo-3 imaging in the absence or presence (inset) of TTX. d, Determining the degree of synaptic connectivity between pairs of nearby neurons (≤ 250 μm apart). Example is shown of the AP and resulting EPSP in a pair of neurons synaptically connected in both directions (“1” and “2”). ∗Calibration applies to induced AP (50 mV/50 ms); ∗∗calibration applies to EPSP triggered in postsynaptic neuron (5 mV/50 ms). e, Protecting doses of NMDA strongly enhance neuronal network activity. Shown is an MEA recording of neurons exposed to the indicated levels of NMDA and monitored over time. Mean ± SEM is shown of eight individual arrays for each condition. f, Examples of MEA recording traces.
Paired whole-cell and microelectrode array recordings.
Paired whole-cell recordings were made of nearby 10 d in vitro (10 DIV) neurons (within a single field of view, approximately ≤250 μm apart). The cultures were maintained in a submersion chamber continually perfused (2–3 ml/min) with artificial CSF containing the following (in mm): 119 NaCl, 3.5 KCl, 1 NaH2PO4, 2 CaCl2, 1 MgSO4, 26 NaHCO3 and 10 glucose. The solution was bubbled with 5% CO2/95% O2, and cultures were recorded at room temperature (21–24°C). The intracellular electrodes were 3–5 MΩ in resistance and were filled with the following (in mm): 110 K-gluconate, 10 KCl, 2 MgCl2, 2 Na2ATP, 0.03 Na2GTP, 10 HEPES, corrected to pH 7.3. Synaptic connections were tested by injecting sufficient current into one of the neurons to produce an action potential (AP) and analyzing the membrane potential of the other neuron in the short time window after this AP. Connections were tested in both directions. EPSPs were typically >1 mV, consistent with previous studies (Malinow, 1991).
Microelectrode array (MEA) recording was performed as described (Arnold et al., 2005). Briefly, neurons were plated onto MEA dishes containing a grid of 60 planar electrodes (Multi Channel Systems, Reutlingen, Germany) at a density of ∼400 cells/mm2 (neurons and glia). The distance between the electrodes on the MEA dishes was 200 μm, and the electrode diameter was 30 μm. Recordings were acquired with an MEA-60 amplifier board (from 10 Hz to 35 kHz; gain, 1200; sampling frequency, 20 kHz; Multi Channel Systems). Recordings were done in SGG plus 10% MEM plus ITS supplement (Sigma) after a culturing period of 10–12 d. Before the application of NMDA the basic network activity was recorded for 5 min. NMDA was added to the cultures in concentrations of 5, 10, or 20 μm. Another 5 min recording was performed immediately (30 s) after the NMDA application. Cultures were put back into the incubator, and 5 min recordings were repeated at 1, 4, 6, and 24 h after the addition of NMDA. After 24 h 1 μm TTX (Sigma) was added to the medium before a final recording to verify the activity dependence of the observed signal. Spikes were detected with the integrated spike detector of the MC Rack software (Multi Channel Systems). Frequency analysis was done with NeuroExplorer (Nex Technologies, Littleton, MA). All results are given as the mean ± SEM.
Antibodies and Western blotting.
Total cell lysates were boiled at 100°C for 5 min in 1.5× sample buffer (1.5 m Tris, pH 6.8, 15% glycerol, 3% SDS, 7.5% β-mercaptoethanol, 0.0375% bromophenol blue). Gel electrophoresis and Western blotting were performed via the XCell SureLock system (Invitrogen), using precast gradient gels (4–20%) according to the manufacturer's instructions. The gels were blotted onto polyvinylidene difluoride membranes, which then were blocked for 1 h at room temperature with 5% (w/v) nonfat-dried milk in TBS with 0.1% Tween 20. Next the membranes were incubated at 4°C overnight with the primary antibodies diluted in blocking solution; anti-phospho Ser473 Akt (1:1000), anti-Akt (1:1000), anti-phospho extracellular signal-regulated kinases 1/2 (ERK1/2; 1:2000), and anti-phospho glycogen synthase kinase 3β (GSK3β; 1:500) antibodies were obtained from Cell Signaling Technology (Beverly, MA). The phospho-tyrosine kinase A (TrkA) antibody that also recognizes TrkB phosphorylated on tyrosine 515 was from Cell Signaling Technology (1:1000) as well. Note that hippocampal neurons do not express TrkA (Ip et al., 1993; Friedman, 2000), making the detection of phospho-TrkB unambiguous. Anti-TrkB (1:1000) and anti-myc (for immunofluorescence; 1:200) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). For the visualization of Western blots HRP-based secondary antibodies were used, followed by chemiluminescent detection on Kodak X-OMAT film.
Transfections and gene expression analysis.
Neurons were transfected with cAMP response element-luciferase (CRE-Luc) and thymidine kinase promoter–Renilla luciferase (pTK-RL) (Promega, Madison, WI) or FOXO-Luc at 8 DIV, using Lipofectamine 2000 (Invitrogen) and stimulated 24 h after transfection (for 6 h). Luciferase assays were performed by using the Dual Glo assay kit (Promega) with firefly luciferase-based CRE reporter gene activity normalized to the Renilla control (pTK-RL plasmid) in all cases. The mean ± SEM of four experiments is shown. RNA was isolated via the Qiagen (Hilden, Germany) RNeasy isolation reagents after passage of the cells through a QiaShredder column. Reverse transcription-PCR (RT-PCR) was performed by using the Pro-Star one step RT-PCR kit from Stratagene (La Jolla, CA). For each gene the cycle time was optimized such that reactions were terminated within the linear amplification range. The sequences of the primers used included 5′-TCATCCAGTTCCACCAGG-3′ and 5′-CCCTCATAGACATGT-TTGC-3′ for BDNF plus 5′-TGTCGTGGAGTCTACTGG-3′ and 5′-CAGCATCAAAGGTGGAGG-3′ for glyceraldehyde phosphate dehydrogenase (GAPDH).
Results
Subtoxic doses of NMDA protect against apoptotic and excitotoxic insults
To investigate neuroprotection afforded by subtoxic preconditioning levels of NMDA (NMDApre), we first ascertained the highest tolerated dose of NMDA that did not affect neuronal health or viability adversely (Fig. 1a). Analysis of nuclear pyknosis in neurons exposed to NMDA for 24 h revealed that 10 μm NMDA is tolerated consistently by 10 DIV hippocampal neurons, in contrast to higher concentrations. Consistent with previous studies, tolerated doses of NMDA were neuroprotective. Apoptosis induced either by trophic deprivation or by exposure to staurosporine was reduced in neurons exposed to 5 or 10 μm NMDA (Fig. 1b). Our previous work had shown that neuroprotection evoked by an episode of synaptic activity lasted for a long time after that activity had ceased (Papadia et al., 2005), so we decided to see whether NMDApre also evoked long-lasting neuroprotection (Fig. 1c). Neurons were exposed to NMDApre before, as opposed to during, trophic deprivation or staurosporine exposure. This previous exposure also triggered significant neuroprotection for at least 48 h after NMDApre exposure was terminated, showing that NMDApre-induced neuroprotection also had a long-lasting component. We also assayed the ability of NMDApre to protect neurons against excitotoxic cell death induced by toxic levels of glutamate (Fig. 1d). NMDApre afforded significant protection against a toxic dose of glutamate (40 μm), reducing the amount of death by >50%.
Figure 1.

Exposure to subtoxic doses of NMDA induces long-lasting neuroprotection. a, Determining the toxicity threshold of NMDA in 10 DIV hippocampal neurons. Neurons were exposed to the indicated levels of NMDA for 24 h before fixation and assessment of nuclear pyknosis. b, Subtoxic doses of NMDA protect neurons against apoptosis induced by trophic deprivation or staurosporine. Neurons were subjected to trophic deprivation (72 h) or staurosporine (36 h) in the presence of the indicated concentrations of NMDA. c, Subtoxic doses of NMDA induce long-lasting neuroprotection. Neurons were exposed to the indicated concentrations of NMDA for 12 h, after which all activity and NMDA receptors were blocked by TTX plus MK-801. Neurons then were exposed to the indicated apoptotic stimuli. d, Subtoxic doses of NMDA protect neurons against excitotoxicity. Neurons were exposed to the indicated levels of NMDA for 12 h before the toxic insult (Glutamate; 40 μm) for 1 h. Death was assessed 24 h later. In all panels, the mean ± SEM is shown of at least three independent experiments. An asterisk indicates significant neuroprotection as compared with control neurons not exposed to NMDA; p < 0.05; paired two-tailed t test.
Mitochondrial responses to NMDA alter dramatically as the toxicity threshold is crossed
To ascertain whether there are any qualitative differences between the cellular response to NMDApre versus toxic doses of NMDA, we performed a series of imaging experiments. The bath application of toxic levels of NMDA and glutamate induced sustained elevated levels of intracellular Ca2+ that were particularly efficient at promoting mitochondrial Ca2+ uptake and loss of membrane potential, which preceded and predicted cell death (White and Reynolds, 1996; Peng and Greenamyre, 1998).
Mitochondrial membrane potential (Ψm) was assayed semi-quantitatively, using the indicator rhodamine 123 (Keelan et al., 1999; Hardingham et al., 2002). We found that NMDApre (at 5 and 10 μm) had no effect on Ψm (Fig. 2a), consistent with the fact that these doses are tolerated (and indeed are neuroprotective). NMDA (20 μm) causes cell death in a small percentage of neurons (Fig. 1a), and, consistent with this marginal toxicity, we observed a modest decrease in Ψm (Fig. 2a). However, going beyond the toxicity threshold (> 20 μm) immediately caused a large decrease in Ψm (Fig. 2a). Accumulation of Ca2+ into mitochondria through the uniporter is a major cause of mitochondrial depolarization and a central mediator of excitotoxicity (Stout et al., 1998; Nicholls, 2004). Rhodamine-based Ca2+ indicators are a useful tool for measuring mitochondrial Ca2+ (Peng et al., 1998; Collins et al., 2001; Kovacs et al., 2005), because they partition into polarized mitochondria caused by their positive charge. Neurons loaded with Rhod-FF showed a punctate distribution of the indicator that very closely resembled that obtained with MitoTracker Red (data not shown), and the pattern was abolished by the mitochondrial uncoupler FCCP (Fig. 2b, inset), confirming the mitochondrial localization of the dye. Rhod-FF imaging of mitochondrial Ca2+ in neurons exposed to rising concentrations of NMDA revealed that, although NMDApre did cause increases in mitochondrial Ca2+, they were transient (Fig. 2b). In contrast, toxic levels of NMDA (NMDAtox) clearly showed a sustained accumulation of mitochondrial Ca2+. Thus mitochondria appear to respond in a qualitatively different manner to NMDAtox as compared with NMDApre. However, the reason for this simply could be quantitative differences in NMDA receptor activity.
Neuroprotective doses of NMDA enhance firing; toxic doses suppress it
To investigate this, we decided to characterize the cytoplasmic Ca2+ transients that were causing these differing responses from the mitochondria. We performed Fluo-3-based Ca2+ imaging on neurons exposed to rising NMDA (Fig. 2c). Strikingly, we found that NMDApre (5 and 10 μm) triggered Ca2+ transients that were highly oscillatory in nature. In sharp contrast, as soon as the threshold into toxicity was crossed (≥ 20 μm), the Ca2+ levels became sustained, elevated, and non-oscillatory in nature. The oscillatory nature of the Ca2+ transients induced by NMDApre strongly indicated that a large component of the Ca2+ influx was attributable to AP firing. In support of this, the Ca2+ spikes were abolished with the sodium channel blocker TTX (Fig. 2c, inset). To test whether these oscillations were being regulated by synaptic activity, we examined an expanded view of the Ca2+ oscillations in Figure 2c (also see supplemental Fig. S1, available at www.jneurosci.org as supplemental material). This revealed that the individual neurons were oscillating synchronously, strongly indicating that the network was coupled synaptically. To quantify the degree of synaptic connectivity in our cultures, we performed a series of paired recordings of nearby neurons. Of 10 pairs of neurons tested within two separate cultures, all were connected synaptically; four exhibited unidirectional synaptic connectivity, whereas six were coupled bidirectionally (Fig. 2d).
To monitor directly the impact of NMDApre on neuronal firing, we grew hippocampal neurons on a MEA and recorded their change in firing rate after treatment with NMDA. Both 5 and 10 μm NMDA induced a sustained increase in firing, with firing rates significantly increased even after 1 h of exposure (Fig. 2e,f, examples). Network activity levels were increased more strongly by 10 μm than by 5 μm NMDA; however, activity levels returned to baseline more quickly. In contrast to the activity-promoting effects of NMDApre, 20 μm NMDA caused no increase in spiking; indeed even spontaneous firing was blocked (Fig. 2e,f, examples), strongly indicating that the neurons were chronically depolarized.
We know that AP bursting in our cultures triggers Ca2+ spikes that are mainly dependent on influx via synaptic NMDA receptors, augmented by release from internal stores (Hardingham et al., 2001, 2002). Thus we conclude that NMDApre is inducing Ca2+ spikes by promoting AP firing, which in turn triggers Ca2+ influx via synaptic NMDA receptors. However, the Ca2+ oscillations also may contain components that are not synaptic in origin, arising from the AP firing its self.
NMDApre neuroprotection via phosphatidylinositol-3 kinase-Akt signaling relies on enhanced firing
Figure 2 demonstrates that the cellular response to a low neuroprotective dose of NMDA is qualitatively different from that elicited by a toxic dose. Neuroprotective doses enhance firing, whereas toxic doses suppress it. This is intriguing, given that we recently showed that AP bursting triggered by network disinhibition is strongly neuroprotective because of the resulting enhancement of synaptic NMDA receptor activity (Papadia et al., 2005). Continuous synaptic NMDA receptor activity induced by network disinhibition promotes neuroprotection against apoptotic insults via activation of the phosphatidylinositol-3 kinase-Akt (PI3K-Akt) pathway (Papadia et al., 2005). We therefore tested the ability of NMDApre to activate this neuroprotective pathway and the role of AP firing in its induction.
Western blot analysis revealed that NMDApre (at 10 μm) activated Akt (Fig. 3a) (for normalized quantitation of three independent blots, see Fig. 3b), consistent with previous studies (Lafon-Cazal et al., 2002; Sutton and Chandler, 2002). Strikingly, NMDApre induction of Akt was mainly dependent on AP firing; TTX dramatically impaired the induction of Akt by NMDApre (Fig. 3a) (for quantitation, see Fig. 3b). Signaling to other pathways by NMDApre was similarly sensitive to TTX; activation of ERK1/2 by NMDApre also was blocked by TTX (Fig. 3a).
Figure 3.
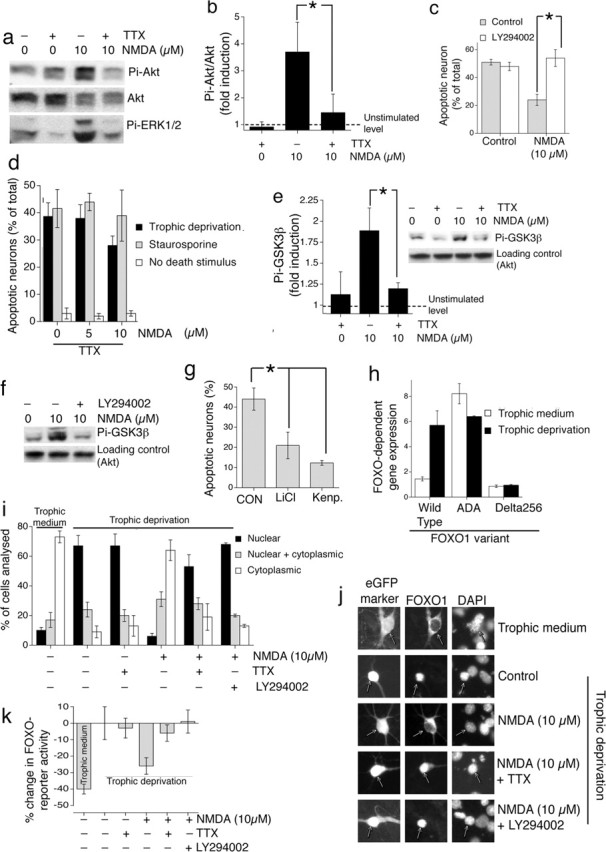
NMDApre neuroprotective signaling via Akt-mediated GSK3β inhibition and FOXO export and reliance on enhanced firing. a, Activation of Akt and ERK1/2 signaling by NMDApre is activity-dependent. Neurons were stimulated as indicated for 1 h, followed by Western blot analysis of phospho-Ser473 Akt (Pi-Akt) and phospho-ERK1/2 (Pi-ERK1/2). Example shown is typical of three similar experiments. b, Analysis of Pi-Akt Western band intensity normalized to levels of Akt. c, The PI3K inhibitor LY294002 blocks synaptic NMDA receptor-dependent neuroprotection. Neurons were placed in trophically deprived medium and treated with LY294002 (30 μm) 1 h before treatment with 10 μm NMDA. Death was assessed after 72 h. d, The anti-apoptotic effect of NMDApre is dependent on synaptic activity. Neurons were subjected to trophic deprivation (72 h) or staurosporine (36 h) in the presence of the indicated concentrations of NMDA in the presence of TTX (1 μm). Compare the results with Figure 1b (NMDA treatment in the absence of TTX). e, f, NMDApre-induced GSK3β phosphorylation is AP dependent and PI3K dependent. Neurons were stimulated as indicated for 1 h, followed by Western blot analysis of phospho-Ser9 GSK3β levels. g, Inhibition of GSK3β is sufficient to confer neuroprotection in the absence of NMDApre-induced Akt signaling. Neurons were placed in trophically deprived medium and treated with either kenpaullone (Kenp.; 2 μm) or LiCl (10 mm; adjusting the [NaCl] to keep overall osmolarity constant). Death was assessed after 72 h. h, Neurons subjected to trophic deprivation support high FOXO activity. Neurons were transfected with FOXO-Luc containing six tandem arrays of the FOXO consensus (TTGTTTAC) plus an expression construct for FOXO1 [wild type, Akt phosphorylation site mutant (ADA), and Δ256, which lacks the transcriptional activation domain]. Neurons either were subjected to trophic deprivation for 48 h or were left in trophic medium and then assayed for firefly luciferase activity (normalized to Renilla transfection control). i, NMDApre-induced Akt activity promotes FOXO export. Neurons were transfected with an expression construct for FOXO1-myc and placed either in trophic medium or trophically deprived medium for 36 h. Where used, LY294002 (30 μm) was preincubated for 1 h before stimulation. Neurons were then stimulated for 1 h as indicated, followed by fixation and processing for immunofluorescence with an anti-myc (9E10) antibody. For each treatment, >150 cells were analyzed across three independent experiments. j, Examples of pictures analyzed in i. k, NMDApre-induced Akt activity reduces endogenous FOXO activity. Neurons were transfected with FOXO-Luc and stimulated overnight as indicated and then assayed for firefly luciferase activity (normalized to Renilla transfection control). In applicable panels, ∗p < 0.05; paired two-tailed t test (n = 3–5); mean ± SEM.
We found that neuroprotection induced by NMDApre is indeed dependent on the PI3K-Akt pathway; protection was abolished by the PI3K inhibitor 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) (Fig. 3c), as well as by the structurally distinct PI3K inhibitor wortmannin and by an Akt inhibitor, the 3′-modified phosphatidyl inositol analog HIMO (1l-6-hydroxymethyl-chiro-inositol 2-(R)-2-O-methyl-3-O-octadecylcarbonate (Hu et al., 2000) (data not shown).
We therefore predicted that PI3K-Akt-dependent neuroprotection induced by continued exposure to NMDApre was mediated by AP firing. We compared the ability of NMDApre (at 5 and 10 μm) to evoke neuroprotection in the presence or absence of TTX. We found that the anti-apoptotic effect of NMDApre (in the face of trophic deprivation or staurosporine treatment) was abolished in the presence of TTX (compare Figs. 3d, 1b). We conclude that NMDApre-dependent anti-apoptotic signaling, reliant on Akt signaling, is induced by the dramatic increase in firing rate triggered by NMDApre. Chronic low-level activation of NMDA receptors (by NMDApre in the presence of TTX) is unable to induce Akt-dependent neuroprotection.
An important target of Akt signaling is GSK3β, which promotes mitochondrial Bax translocation during neuronal apoptosis (Linseman et al., 2004). Akt is able to inhibit GSK3β activity via phosphorylation on serine 9 (Ser9) and prevent neuronal apoptosis (Crowder and Freeman, 2000; Hetman et al., 2000). Western blot analysis revealed that NMDApre induced robust phosphorylation of GSK3β on Ser9, which was blocked by TTX treatment (Fig. 3e, inset). This phosphorylation also was blocked by the PI3K-Akt inhibitor LY294002 (Fig. 3f), confirming its dependence on the PI3K-Akt signaling cassette. We next investigated whether GSK3β inhibition promotes neuroprotection in our experimental system. Recent kinase inhibitor specificity studies led the authors to recommend the parallel use of LiCl and kenpaullone for identifying the role of GSK3β in physiological processes (Bain et al., 2003). We inhibited GSK3β with either LiCl or kenpaullone (Fig. 3g), and both conferred significant neuroprotection in the absence of NMDApre. Kenpaullone does have one other known target in cyclin-dependent kinase 2 (cdk2). However, treatment of neurons with a distinct cdk2 inhibitor (roscovitine) had no neuroprotective effect (data not shown). Although we cannot rule out that the observed neuroprotective effects of LiCl and kenpaullone are attributable to unknown targets of these agents, it appears that GSK3β inhibition by Akt-mediated phosphorylation may be an important neuroprotective mechanism triggered by NMDApre via the dramatic increase in firing rate.
Another key effector of pro-survival signaling by Akt is the FOXO family of transcription factors, which regulate the transcription of several pro-death genes, including Fas ligand (Brunet et al., 2001; Arden and Biggs, 2002). Akt phosphorylates and triggers nuclear export of FOXO members, thereby inhibiting FOXO-dependent gene expression (Brunet et al., 1999; Kops et al., 1999; Nakae et al., 2000). We therefore investigated whether FOXOs were a target for NMDApre-induced Akt activity in our model of trophic deprivation. We found that, in trophically deprived neurons, FOXO activity was high; expression of wild-type FOXO1 resulted in high FOXO-luciferase reporter activity under conditions of trophic deprivation as compared with neurons supplied with a medium containing good trophic support (Fig. 3h). This high activity is likely to be attributable to low FOXO phosphorylation; in support of this, activity of a mutant FOXO (ADA) (Nakae et al., 2000) with its Akt phosphorylation sites mutated revealed high activity in both trophically deprived and enriched conditions (Fig. 3h).
Because FOXO activity is regulated at the level of subcellular localization, we looked at the spatial distribution of myc-tagged FOXO1 (FKHR). We found that, in neurons supplied with a medium containing good trophic support, FOXO1 was predominantly cytoplasmic. However, in neurons placed in trophically deprived medium, FOXO1 is almost exclusively nuclear (Fig. 3j, compare top two rows) (for quantitation, see Fig. 3i). In trophically deprived neurons NMDApre induces nuclear export of FOXO1, which is inhibited by the PI3K-Akt inhibitor LY294002 (Fig. 3i,j). Akt dependence of the export process is underlined additionally by our observation that NMDApre fails to induce the export of FOXO-ADA-myc (data not shown). Consistent with the TTX sensitivity of Akt activation, TTX also reduces the NMDApre-induced export of FOXO1 (Fig. 3i,j).
We next quantified the degree to which NMDA (±TTX) was able to reduce FOXO reporter activity, compared with the control condition (Fig. 3k). For comparison, the level of FOXO reporter activity in neurons placed in trophic medium also is shown to give an idea of the “full scale deflection” of the experiment, because in trophic medium FOXO is almost entirely cytoplasmic. The high level of endogenous FOXO activity in trophically deprived neurons was reduced by NMDApre in a TTX- and LY294002-sensitive manner (Fig. 3k), corroborating the localization studies.
Thus, although NMDApre induces strong Akt-dependent prosurvival signaling and targets GSK3β and FOXO inhibition, this is reliant on the enhancement of AP firing induced by NMDApre. In the absence of this firing (TTX-treated), the direct, chronic low-level NMDA receptor activation induced by NMDApre is insufficient to support robust FOXO export or GSK3β inhibition.
The long-lasting CREB-dependent phase of neuroprotection also relies on enhanced firing
The bath applications of high doses of NMDA or glutamate are very poor inducers of CRE-dependent gene expression (Bading et al., 1993; Ghosh et al., 1994; Hardingham et al., 1999) because of extrasynaptic NMDA receptor signaling to CREB dephosphorylation (Hardingham and Bading, 2002; Hardingham et al., 2002). However, low levels of NMDA (NMDApre) can induce the long-lasting phase of anti-apoptotic neuroprotection (Fig. 1c), which we have shown is CREB-dependent (Papadia et al., 2005). We therefore decided to study the activation of CREB family-dependent gene expression by NMDApre. Neurons were transfected with a CRE-luciferase reporter gene and stimulated with 10 μm NMDA in the presence or absence of TTX (Fig. 4a). TTX dramatically reduced the induction of CRE-mediated gene expression. The reliance on AP firing rate enhancement on NMDApre-induced CREB activation also applied to the CREB-dependent long-lasting phase of neuroprotection, in which the apoptosis-inducing stimulus was applied after NMDApre exposure was terminated. TTX treatment rendered NMDApre unable to promote long-lasting neuroprotection (Fig. 4b). Because synaptic NMDA receptor-dependent neuroprotection against excitotoxic insults is also CREB-dependent (Lee et al., 2005), we predicted a similar effect of TTX on NMDApre-induced protection against an excitotoxic insult. This indeed was observed; TTX treatment rendered NMDApre unable to promote protection against a toxic concentration of glutamate (Fig. 4c). Thus for all death paradigms the neuroprotection induced by NMDApre is mediated indirectly via the enhancement of firing rates. In the absence of firing, the chronic low-level activation of NMDA receptors is clearly insufficient to induce protection. Given that doses of NMDA higher than 10 μm are toxic, our data indicate that, in the absence of firing activity, NMDA is a very poor inducer of neuroprotection in hippocampal neurons.
Figure 4.
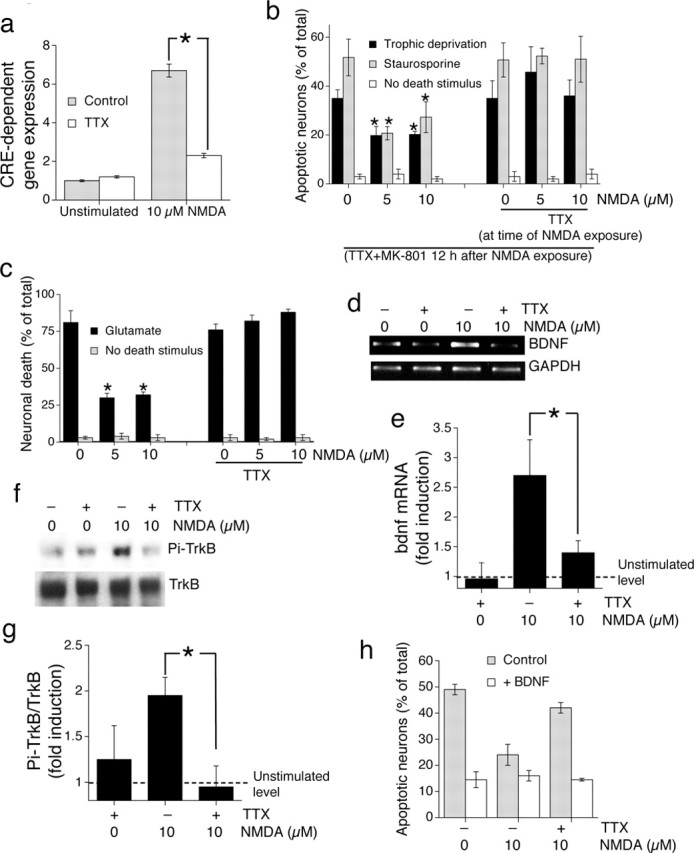
NMDApre induction of CREB-dependent neuroprotection relies on enhanced firing; chronic low-level NMDA receptor activity is not sufficient. a, Activation of CRE-dependent gene expression by NMDApre is AP dependent. Neurons were transfected with CRE-firefly luciferase plus Renilla transfection control and stimulated as indicated for 6 h. ∗p < 0.05; paired two-tailed t test (n = 4). b, The long-lasting CREB-dependent phase of NMDApre-dependent neuroprotection is dependent on AP firing. Neurons were exposed to the indicated concentrations of NMDA ± TTX for 12 h, after which all activity and NMDA receptors were blocked by TTX plus MK-801. Neurons were then exposed to the indicated apoptotic stimuli. An asterisk indicates significant neuroprotection as compared with control neurons not exposed to NMDA; p < 0.05; paired two-tailed t test (n = 4). c, NMDApre-induced neuroprotection against excitotoxic insults is dependent on firing activity. Neurons were exposed to the indicated level of NMDA ± TTX for 12 h before the toxic insult (Glutamate; 40 μm) for 1 h. Death was assessed 24 h later. In all panels, the mean ± SEM is shown of at least three independent experiments. An asterisk indicates significant neuroprotection as compared with control neurons not exposed to NMDA; p < 0.05; paired two-tailed t test. d, Induction of BDNF mRNA expression by NMDApre is AP dependent. RT-PCR was performed on RNA extracts from neurons treated for 4 h as indicated. The example shown is typical of three similar experiments. e, Analysis of all three experiments performed by digital scanning of gel photographs, with BDNF band intensity normalized to GAPDH; ∗p < 0.05, paired two-tailed t test. f, g, NMDApre induces TrkB activation that is AP dependent. Neurons were stimulated as indicated for 1 h, followed by Western blot analysis of phospho-TrkB. The example shown is typical of three similar experiments, the analysis of which is shown in g. ∗p < 0.05; paired two-tailed t test. h, BDNF supplementation is sufficient to reverse the inhibitory effect of TTX on NMDApre-induced neuroprotection. Neurons were subjected to trophic deprivation (72 h) and treated as indicated in the presence or absence of BDNF (25 ng/ml).
AP-dependent BDNF signaling is neuroprotective
A key CREB-regulated gene implicated in mediating the preconditioning effect of subtoxic NMDA is BDNF (Jiang et al., 2005). According to our results with the use of the CRE reporter, we would predict that NMDApre would induce transcription of the BDNF gene in a TTX-sensitive manner. We found that 10 μm NMDA did indeed cause an upregulation of BDNF mRNA levels after 4 h of stimulation. Furthermore, this was attenuated severely in the presence of TTX (Fig. 4d) (for analysis, see Fig. 4e). Therefore, induction of this CRE-regulated prosurvival gene expression by NMDApre is mediated mainly via its effect on enhancing firing rates.
NMDApre also is known to trigger the rapid release of existing BDNF, which contributes to the protecting effect (Jiang et al., 2005). Because synaptic activity is able to trigger release of BDNF (Kohara et al., 2001), we hypothesized that NMDApre-induced activation of the receptor of BDNF, TrkB, also would be AP-dependent. Thus we assayed the release of biologically active BDNF by looking for activation of the BDNF receptor TrkB with the use of an anti phospho-Tyr515 TrK antibody. NMDApre promoted TrkB phosphorylation, and, moreover, this activation of TrkB was blocked by TTX (Fig. 4f) (for analysis, see 4g). Thus the NMDApre-induced TrkB activation, as well as BDNF transcription, was reliant on the ability of NMDApre to enhance firing rates and could not be elicited by chronic low-level NMDA receptor activity alone. Furthermore, treatment of neurons with exogenous BDNF was sufficient to reverse the inhibitory effect of TTX on NMDApre-dependent neuroprotection (Fig. 4h), indicating that induction of BDNF transcription and BDNF release may be one of several protective pathways induced by enhanced AP firing that is important for neuroprotection by NMDApre. Consistent with this, the Trk receptor antagonist K252a impaired NMDApre-induced neuroprotection under conditions of trophic deprivation (data not shown).
Discussion
This investigation has revealed that neuroprotection induced by subtoxic levels of NMDA is mediated by enhancing the firing rate. NMDApre strongly promotes network activity, which in turn activates CREB-dependent gene expression, Akt signaling, and, subsequently, neuroprotection. In the absence of this firing activity the low-level chronic activation of NMDA receptors is unable to promote protection. The transition from a protecting dose of NMDA to a toxic dose is marked by striking qualitative changes in the intracellular Ca2+ signals evoked: from AP-dependent transients (protecting) to AP-independent sustained elevated levels (toxic). The effects of these different Ca2+ profiles on the mitochondria are equally striking, with only toxic levels of NMDA inducing the loss of Ψm or sustained accumulation of mitochondrial Ca2+.
Induction of excitability by protecting doses of NMDA
Our observation that low doses of NMDA promote firing in hippocampal neuronal cultures is consistent with observations that low NMDA can induce burst activity in hippocampal slices from developing and adult rats (Jensen and Wang, 1996). There are a number of known mechanisms by which NMDA could promote excitability, involving both presynaptic and postsynaptic mechanisms, and dependent on both Na+ and Ca2+ influx. NMDA receptor-dependent Na+ and Ca2+ influx will cause postsynaptic depolarization of neurons nearer to firing threshold and thus will make spontaneous EPSPs more likely to evoke an AP. In addition, presynaptic NMDA receptors can facilitate axon excitability in a mechanism dependent on Na+ entry (Suarez et al., 2005). Alternatively, the Ca2+ that NMDA receptors pass can induce classic LTP-like potentiation of currents mediated by non-NMDA ionotropic glutamate receptors (namely AMPA receptors) (Lisman, 2003; Malinow, 2003; Lynch, 2004). NMDA-induced potentiation also can be expressed at the presynaptic locus in the form of increased release probability (Malgaroli and Tsien, 1992). The overall effect of low doses of NMDA on network activity may be a cumulative result of these and other mechanisms.
Low NMDA causes preferential activation of synaptic over extrasynaptic NMDA receptors
We previously showed that synaptic NMDA receptor activity promotes long-lasting neuroprotection against apoptotic stimuli via the induction of CREB-dependent gene expression (Papadia et al., 2005). However, we also have shown that extrasynaptic NMDA receptor activity promotes CREB inactivation (Hardingham and Bading, 2002; Hardingham et al., 2002). The dominant nature of the CREB shut-off signal triggered by extrasynaptic NMDA receptor activation (Hardingham et al., 2002) at first glance would indicate that low levels of bath-applied NMDA would be poor activators of CREB-dependent gene expression. Therefore, initially we were surprised that NMDApre could activate CREB. However, this observation is explained in light of the fact that NMDApre results in the preferential activation of synaptic NMDA receptors.
Although all NMDA receptors (synaptic and extrasynaptic) will be activated directly by low doses of NMDA, the majority of Ca2+ influx is attributable to the NMDA-induced increase in firing (Fig. 2c). AP-induced intracellular Ca2+ influx is mediated mainly by synaptic NMDA receptors in our system of hippocampal neuronal cultures (Hardingham et al., 2001, 2002). Thus the majority of NMDA receptor-dependent Ca2+ influx induced by NMDApre is via the transsynaptic activation of synaptic NMDA receptors by glutamate. As such, synaptic NMDA receptors are activated far more strongly than extrasynaptic receptors, enabling synaptic signaling to dominate and activate CREB. The scenario is very different with toxic doses of NMDA; firing is suppressed and the Ca2+ influx is attributable to the direct activation of synaptic and extrasynaptic NMDA receptors. The dominant nature of the extrasynaptic NMDA receptor-dependent CREB shut-off signal means that CREB is induced poorly by the bath application of high doses of glutamate or NMDA (Bading et al., 1993; Hardingham and Bading, 1999; Sala et al., 2000; Hardingham et al., 2002), and activation of important CREB-regulated pro-survival genes such as BDNF is suppressed (Hardingham et al., 2002).
A role for enhanced excitability and CREB activation in anoxic or ischemic preconditioning?
There is considerable evidence that ischemic tolerance brought about by a brief sublethal episode of ischemia requires NMDA receptor activity (Kato et al., 1992; Bond et al., 1999; Grabb and Choi, 1999). It remains to be seen to what extent enhanced firing is responsible for NMDA receptor-dependent hypoxic or ischemic preconditioning. Hypoxic/ischemic insults indeed can induce acute periods of hyperexcitability (Jensen et al., 1998), and increasing excitability is clearly neuroprotective against excitotoxic insults both in slices (Perez-Pinzon et al., 1999) and in cultures (Lee et al., 2005; Papadia et al., 2005) in which CREB is implicated. Brief episodes of hypoxia/ischemia-induced excitability therefore may contribute to the tolerance process via the activation of synaptic NMDA receptors.
The induction of CREB-dependent gene expression as a protecting response to ischemic episodes first was proposed by Dragunow and coworkers, who observed that neurons that survive stroke in vivo exhibited sustained CREB phosphorylation (Walton et al., 1996). In contrast, vulnerable neurons that are destined to die are characterized by exhibiting rapid CREB dephosphorylation (Walton and Dragunow, 2000). We propose that transsynaptic AP-dependent mechanisms may be responsible for sustained activation of CREB in response to a subtoxic-preconditioning ischemic episode. In contrast, CREB dephosphorylation will take place if the ischemic episode is too strong and the neurons depolarize, with sustained elevated levels of extracellular glutamate mediating CREB dephosphorylation via extrasynaptic NMDA receptor activity (Hardingham et al., 1999, 2002; Sala et al., 2000; Hardingham and Bading, 2002; Leveill et al., 2005)
A role for activity-dependent Akt activation in ischemic preconditioning
Although CREB activation is needed for the induction of long-lasting neuroprotection by synaptic NMDA receptor activity, during ongoing activity it is not required (Papadia et al., 2005). Under these conditions the activation of the PI3K-Akt pathway is both necessary and sufficient for neuroprotection. Our finding that induction of the PI3K-Akt pathway by NMDApre is reliant on AP firing leads us to predict that NMDA receptor-dependent preconditioning-induced Akt activity in vivo would be triggered by enhanced firing activity. Although the induction of Akt by a preconditioning episode in vivo recently has been shown to be NMDA receptor-dependent (Miao et al., 2005), its dependence on enhanced neuronal excitability/firing rates is unknown.
The induction of ischemic tolerance in the hippocampal CA1 subfield by a sublethal episode has been shown to require PI3K activity (Yano et al., 2001) and triggers an increase in Akt activity that persists for several days. Such a sustained level of activity is likely to rely on ongoing signaling, such as one that could be provided by an increase in local network excitability. In support of this, we have shown that enhancing firing rates by network disinhibition in vitro induces Akt activity via synaptic NMDA receptors that can be sustained for days (Papadia et al., 2005). In contrast, the toxic dose of glutamate used in this study (40 μm) induces a transient increase in Akt, followed by suppression of Akt activity below baseline within a matter of hours (S. Papadia and G. Hardingham, unpublished observation). Such a transient induction also is observed in vivo after a lethal ischemic episode (Yano et al., 2001; Kawano et al., 2002). Thus the transience of Akt induction is a good predictor of the toxicity of the insult, just as the transience of CREB phosphorylation is (see above).
Akt activation triggered by NMDApre targeted two important anti-apoptotic pathways: inhibition of GSK3β and nuclear export of FOXOs. In vivo studies have shown that GSK3β inhibition protects against an ischemic episode (Kelly et al., 2004), and FOXO activation is associated with lowered Akt activity after an ischemic episode (Kawano et al., 2002). Thus Akt-dependent GSK3β inhibition and FOXO export both may contribute to preconditioning in vivo, although Akt-dependent neuroprotection can be effected via other routes as well, such as p53 inhibition (Yamaguchi et al., 2001), BAD (Bcl-associated death promoter) phosphorylation (Datta et al., 1997), and JNK (Jun N-terminal kinase) inhibition (Miao et al., 2005). The activation of CREB by synaptic NMDA receptor activity in hippocampal neurons does not involve the PI3K-Akt pathway (Papadia et al., 2005).
Concluding remarks and model
This study shows that NMDA-induced pro-survival signaling is heavily reliant on AP-dependent activation of synaptic NMDA receptors, whereas pro-death signaling is provided most effectively by chronic AP-independent activation of NMDA receptors, including extrasynaptic ones. Thus the influence of extracellular NMDA or glutamate on neuronal fate is the net effect of these opposing signals. As can be seen in supplemental Figure S2 (available at www.jneurosci.org as supplemental material), pro-death AP-independent activation of extrasynaptic NMDA receptors will increase gradually with the concentration of extracellular ligand (curve 1). However, at low levels of NMDA the synaptic NMDA receptor activation rises steeply because of the dramatic increase in firing (curve 2). This allows prosurvival synaptic signaling to dominate at low doses. Once a dose of NMDA is reached that suppresses firing, pro-survival synaptic NMDA receptor signaling rises far more gradually, allowing extrasynaptic signaling to dominate. The simple smooth bell-shaped curve model (Lipton and Kater, 1989; Lipton and Nakanishi, 1999) used to describe neuronal survival/death as a function of NMDA/glutamate dose can be seen as a summation of these two curves (note that the “zero point” must be set to the pro-death side, because the absence of NMDA receptor activity is toxic).
It is becoming clear that the action of a global NMDA receptor antagonist in taking a neuron from one end of the bell-shaped curve (excitotoxic) to the other (pro-apoptotic) is of limited therapeutic benefit, perhaps contributing to the failure of NMDA receptor antagonists in clinical trials for stroke or traumatic brain injury (Ikonomidou and Turski, 2002). The supplemental figure S2 (available at www.jneurosci.org as supplemental material) illustrates the key pro-survival role that the NMDA receptor plays, demonstrating that, in developing therapies aimed at blocking NMDA receptor-dependent excitotoxic processes, the role of transsynaptic activation of synaptic NMDA receptors in promoting survival always must be considered and spared from inhibition.
Footnotes
*F.X.S. and S.P. contributed equally to this study.
This work is supported by the Wellcome Trust, the European Commission, the Royal Society, the Scottish Hospitals Endowment Research Trust, Tenovus Scotland, and the Alexander von Humboldt Foundation. We thank Domenico Accili and Grahame Mckenzie for providing plasmids.
References
- Adams SM, de Rivero Vaccari JC, Corriveau RA (2004). Pronounced cell death in the absence of NMDA receptors in the developing somatosensory thalamus. J Neurosci 24:9441–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden KC, Biggs WH III (2002). Regulation of the FOXO family of transcription factors by phosphatidylinositol-3 kinase-activated signaling. Arch Biochem Biophys 403:292–298. [DOI] [PubMed] [Google Scholar]
- Arnold FJ, Hofmann F, Bengtson CP, Wittmann M, Vanhoutte P, Bading H (2005). Microelectrode array recordings of cultured hippocampal networks reveal a simple model for transcription and protein synthesis-dependent plasticity. J Physiol (Lond) 564:3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H, Greenberg ME (1991). Stimulation of protein tyrosine phosphorylation by NMDA receptor activation. Science 253:912–914. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME (1993). Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260:181–186. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P (2003). The specificities of protein kinase inhibitors: an update. Biochem J 371:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balazs R, Hack N, Jorgensen OS (1988). Stimulation of the N-methyl-d-aspartate receptor has a trophic effect on differentiating cerebellar granule cells. Neurosci Lett 87:80–86. [DOI] [PubMed] [Google Scholar]
- Bambrick LL, Yarowsky PJ, Krueger BK (1995). Glutamate as a hippocampal neuron survival factor: an inherited defect in the trisomy 16 mouse. Proc Natl Acad Sci USA 92:9692–9696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond A, Lodge D, Hicks CA, Ward MA, O'Neill MJ (1999). NMDA receptor antagonism, but not AMPA receptor antagonism, attenuates induced ischaemic tolerance in the gerbil hippocampus. Eur J Pharmacol 380:91–99. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868. [DOI] [PubMed] [Google Scholar]
- Brunet A, Datta SR, Greenberg ME (2001). Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol 11:297–305. [DOI] [PubMed] [Google Scholar]
- Chuang DM, Gao XM, Paul SM (1992). N-methyl-d-aspartate exposure blocks glutamate toxicity in cultured cerebellar granule cells. Mol Pharmacol 42:210–216. [PubMed] [Google Scholar]
- Collins TJ, Lipp P, Berridge MJ, Bootman MD (2001). Mitochondrial Ca2+ uptake depends on the spatial and temporal profile of cytosolic Ca2+ signals. J Biol Chem 276:26411–26420. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS (2000). Glycogen synthase kinase-3β activity is critical for neuronal death caused by inhibiting phosphatidylinositol-3 kinase or Akt but not for death caused by nerve growth factor withdrawal. J Biol Chem 275:34266–34271. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997). Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91:231–241. [DOI] [PubMed] [Google Scholar]
- Dragunow M (2004). CREB and neurodegeneration. Front Biosci 9:100–103. [DOI] [PubMed] [Google Scholar]
- Friedman WJ (2000). Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci 20:6340–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa DG, Shinmei SS, Cai B (2000). Kainic acid-induced seizures produce necrotic, not apoptotic, neurons with internucleosomal DNA cleavage: implications for programmed cell death mechanisms. Neuroscience 98:41–53. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME (1994). Requirement for BDNF in activity-dependent survival of cortical neurons. Science 263:1618–1623. [DOI] [PubMed] [Google Scholar]
- Grabb MC, Choi DW (1999). Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J Neurosci 19:1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen HH, Briem T, Dzietko M, Sifringer M, Voss A, Rzeski W, Zdzisinska B, Thor F, Heumann R, Stepulak A, Bittigau P, Ikonomidou C (2004). Mechanisms leading to disseminated apoptosis following NMDA receptor blockade in the developing rat brain. Neurobiol Dis 16:440–453. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (1999). Calcium as a versatile second messenger in the control of gene expression. Microsc Res Tech 46:348–355. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2002). Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated. Biochim Biophys Acta 1600:148–153. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2003). The yin and yang of NMDA receptor signaling. Trends Neurosci 26:81–89. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Johnson CM, Bading H (1997). Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature 385:260–265. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Cruzalegui FH, Bading H (1999). Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron 22:789–798. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H (2001). Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci 4:261–267. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5:405–414. [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z (2000). Role of glycogen synthase kinase-3β in neuronal apoptosis induced by trophic withdrawal. J Neurosci 20:2567–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Qiao L, Wang S, Rong SB, Meuillet EJ, Berggren M, Gallegos A, Powis G, Kozikowski AP (2000). 3-(Hydroxymethyl)-bearing phosphatidylinositol ether lipid analogues and carbonate surrogates block PI3-K, Akt, and cancer cell growth. J Med Chem 43:3045–3051. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L (2002). Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol 1:383–386. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW (1999). Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283:70–74. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Stefovska V, Turski L (2000). Neuronal death enhanced by N-methyl-d-aspartate antagonists. Proc Natl Acad Sci USA 97:12885–12890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey SD, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH (2004). Defining the CREB regulon: a genome-wide analysis of CREB regulatory regions. Cell 119:1041–1054. [DOI] [PubMed] [Google Scholar]
- Ip NY, Li Y, Yancopoulos GD, Lindsay RM (1993). Cultured hippocampal neurons show responses to BDNF, NT-3, and NT-4, but not NGF. J Neurosci 13:3394–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen FE, Wang C (1996). Hypoxia-induced hyperexcitability in vivo and in vitro in the immature hippocampus. Epilepsy Res 26:131–140. [DOI] [PubMed] [Google Scholar]
- Jensen FE, Wang C, Stafstrom CE, Liu Z, Geary C, Stevens MC (1998). Acute and chronic increases in excitability in rat hippocampal slices after perinatal hypoxia in vivo. J Neurophysiol 79:73–81. [DOI] [PubMed] [Google Scholar]
- Jiang X, Tian F, Mearow K, Okagaki P, Lipsky RH, Marini AM (2005). The excitoprotective effect of N-methyl-d-aspartate receptors is mediated by a brain-derived neurotrophic factor autocrine loop in cultured hippocampal neurons. J Neurochem 94:713–722. [DOI] [PubMed] [Google Scholar]
- Kato H, Liu Y, Araki T, Kogure K (1992). MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett 139:118–121. [DOI] [PubMed] [Google Scholar]
- Kawano T, Morioka M, Yano S, Hamada J, Ushio Y, Miyamoto E, Fukunaga K (2002). Decreased Akt activity is associated with activation of Forkhead transcription factor after transient forebrain ischemia in gerbil hippocampus. J Cereb Blood Flow Metab 22:926–934. [DOI] [PubMed] [Google Scholar]
- Keelan J, Vergun O, Duchen MR (1999). Excitotoxic mitochondrial depolarization requires both calcium and nitric oxide in rat hippocampal neurons. J Physiol (Lond) 520:797–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S, Zhao H, Hua Sun G, Cheng D, Qiao Y, Luo J, Martin K, Steinberg GK, Harrison SD, Yenari MA (2004). Glycogen synthase kinase 3β inhibitor Chir025 reduces neuronal death resulting from oxygen–glucose deprivation, glutamate excitotoxicity, and cerebral ischemia. Exp Neurol 188:378–386. [DOI] [PubMed] [Google Scholar]
- Kohara K, Kitamura A, Morishima M, Tsumoto T (2001). Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science 291:2419–2423. [DOI] [PubMed] [Google Scholar]
- Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM (1999). Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature 398:630–634. [DOI] [PubMed] [Google Scholar]
- Kovacs R, Kardos J, Heinemann U, Kann O (2005). Mitochondrial calcium ion and membrane potential transients follow the pattern of epileptiform discharges in hippocampal slice cultures. J Neurosci 25:4260–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon-Cazal M, Perez V, Bockaert J, Marin P (2002). Akt mediates the anti-apoptotic effect of NMDA, but not that induced by potassium depolarization in cultured cerebellar granule cells. Eur J Neurosci 16:575–583. [DOI] [PubMed] [Google Scholar]
- Lee B, Butcher GQ, Hoyt KR, Impey S, Obrietan K (2005). Activity-dependent neuroprotection and cAMP response element-binding protein (CREB): kinase coupling, stimulus intensity, and temporal regulation of CREB phosphorylation at serine 133. J Neurosci 25:1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveill F, Nicole O, Buisson A (2005). Cellular location of NMDA receptors influences their implication in excitotoxic injury. Soc Neurosci Abstr 31:946.2. [Google Scholar]
- Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA (2004). Glycogen synthase kinase-3β phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci 24:9993–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Kater SB (1989). Neurotransmitter regulation of neuronal outgrowth, plasticity, and survival. Trends Neurosci 12:265–270. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Nakanishi N (1999). Shakespeare in love—with NMDA receptors? Nat Med 5:270–271. [DOI] [PubMed] [Google Scholar]
- Lisman J (2003). Long-term potentiation: outstanding questions and attempted synthesis. Philos Trans R Soc Lond B Biol Sci 358:829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002). Function and regulation of CREB family transcription factors in the nervous system. Neuron 35:605–623. [DOI] [PubMed] [Google Scholar]
- Lynch MA (2004). Long-term potentiation and memory. Physiol Rev 84:87–136. [DOI] [PubMed] [Google Scholar]
- Malgaroli A, Tsien RW (1992). Glutamate-induced long-term potentiation of the frequency of miniature synaptic currents in cultured hippocampal neurons. Nature 357:134–139. [DOI] [PubMed] [Google Scholar]
- Malinow R (1991). Transmission between pairs of hippocampal slice neurons: quantal levels, oscillations, and LTP. Science 252:722–724. [DOI] [PubMed] [Google Scholar]
- Malinow R (2003). AMPA receptor trafficking and long-term potentiation. Philos Trans R Soc Lond B Biol Sci 358:707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M (2001). Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2:599–609. [DOI] [PubMed] [Google Scholar]
- Miao B, Yin XH, Pei DS, Zhang QG, Zhang GY (2005). Neuroprotective effects of preconditioning ischemia on ischemic brain injury through down-regulating activation of JNK1/2 via N-methyl-d-aspartate receptor-mediated Akt1 activation. J Biol Chem 280:21693–21699. [DOI] [PubMed] [Google Scholar]
- Monti B, Contestabile A (2000). Blockade of the NMDA receptor increases developmental apoptotic elimination of granule neurons and activates caspases in the rat cerebellum. Eur J Neurosci 12:3117–3123. [DOI] [PubMed] [Google Scholar]
- Monti B, Marri L, Contestabile A (2002). NMDA receptor-dependent CREB activation in survival of cerebellar granule cells during in vivo and in vitro development. Eur J Neurosci 16:1490–1498. [DOI] [PubMed] [Google Scholar]
- Nakae J, Barr V, Accili D (2000). Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the Forkhead transcription factor FKHR. EMBO J 19:989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG (2004). Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med 4:149–177. [DOI] [PubMed] [Google Scholar]
- Olney JW, Wozniak DF, Jevtovic-Todorovic V, Barber N, Bittigau P, Ikonomidou C (2002). Drug-induced apoptotic neurodegeneration in the developing brain. Brain Pathol 12:488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Stevenson P, Hardingham NR, Bading H, Hardingham GE (2005). Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J Neurosci 25:4279–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng TI, Greenamyre JT (1998). Privileged access to mitochondria of calcium influx through N-methyl-d-aspartate receptors. Mol Pharmacol 53:974–980. [PubMed] [Google Scholar]
- Peng TI, Jou MJ, Sheu SS, Greenamyre JT (1998). Visualization of NMDA receptor-induced mitochondrial calcium accumulation in striatal neurons. Exp Neurol 149:1–12. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Born JG, Centeno JM (1999). Calcium and increased excitability promote tolerance against anoxia in hippocampal slices. Brain Res 833:20–26. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA (2003). εPKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J Neurosci 23:384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha M, Martins RA, Linden R (1999). Activation of NMDA receptors protects against glutamate neurotoxicity in the retina: evidence for the involvement of neurotrophins. Brain Res 827:79–92. [DOI] [PubMed] [Google Scholar]
- Sala C, Rudolph-Correia S, Sheng M (2000). Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J Neurosci 20:3529–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A (1998). Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 20:727–740. [DOI] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ (1998). Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci 1:366–373. [DOI] [PubMed] [Google Scholar]
- Suarez LM, Suarez F, Del Olmo N, Ruiz M, Gonzalez-Escalada JR, Solis JM (2005). Presynaptic NMDA autoreceptors facilitate axon excitability: a new molecular target for the anticonvulsant gabapentin. Eur J Neurosci 21:197–209. [DOI] [PubMed] [Google Scholar]
- Sutton G, Chandler LJ (2002). Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J Neurochem 82:1097–1105. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME (1998). Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20:709–726. [DOI] [PubMed] [Google Scholar]
- Walton M, Dragunow M (2000). Is CREB a key to neuronal survival? Trends Neurosci 23:48–53. [DOI] [PubMed] [Google Scholar]
- Walton M, Sirimanne E, Williams C, Gluckman P, Dragunow M (1996). The role of the cyclic AMP-responsive element binding protein (CREB) in hypoxic-ischemic brain damage and repair. Brain Res Mol Brain Res 43:21–29. [DOI] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ (1996). Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci 16:5688–5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M (2001). Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem 276:5256–5264. [DOI] [PubMed] [Google Scholar]
- Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, Hamada J, Miyamoto E, Ushio Y (2001). Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J Cereb Blood Flow Metab 21:351–360. [DOI] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M (2005). Genome-wide analysis of cAMP response element-binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA 102:4459–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]