

Abstract
We tested the hypothesis that runoff of uranium-bearing particles from mining waste disposal areas was a significant mechanism for redistribution of uranium in the northeastern part of the Upper Puerco River watershed (New Mexico). However, our results were not consistent with this hypothesis. Analysis of >100 sediment and suspended sediment samples collected adjacent to and downstream from uranium source areas indicated that uranium levels in the majority of the samples were not elevated above background. Samples collected within 50 m of a known waste disposal site were subjected to detailed geochemical characterization. Uranium in these samples was found to be highly soluble; treatment with synthetic pore water for 24 h caused dissolution of 10–50% of total uranium in the samples. Equilibrium uranium concentrations in pore water were >4.0 mg/L and were sustained in repeated wetting events, effectively depleting soluble uranium from the solid phase. The dissolution rate of uranium appeared to be controlled by solid-phase diffusion of uranium from within uranium-bearing mineral particles. X-ray adsorption spectroscopy indicated the presence of a soluble uranyl silicate, and possibly a uranyl phosphate. These phases were exhausted in transported sediment suggesting that uranium was readily mobilized from sediments in the Upper Puerco watershed and transported in the dissolved load. These results could have significance for uranium risk assessment as well as mining waste management and cleanup efforts.
Introduction
The Uranium Procurement Program established by the Atomic Energy Commission in the late 1940s led to a mining boom on the Colorado Plateau that lasted until the late 1980s. Exploration activities, mine development, and ore processing–much of which occurred in the Grants uranium district in northwestern New Mexico (1,2)–resulted in over 75 production sites within ~4,660 km2 of the Navajo Nation (3). Many of these sites have been abandoned, leavingbehind a legacy of waste. Due to concerns over potential environmental and human health risks, some sites have been remediated; however, more than 90% of these sites remain unreclaimed (3). A complete assessment of human health risks at these sites requires a detailed understanding of uranium speciation, mineralogy, and kinetic controls on mobilization and transport.
In 1979, Church Rock, New Mexico, and downstream communities including Gallup, experienced the largest release of radioactive material in U.S. history when an earthen dam at a former uranium mill collapsed, releasing ~340,000 m3 of tailings liquor to the north fork of the Puerco River (4). During this time of active mining and processing, flow in the Puerco was perennial from mining-related releases. Concentrations of uranium and other contaminants were up to five times above background levels in groundwater, surface water, and suspended sediments (5), which presumably were stored in floodplains (6, 7).
Uranium geochemistry is well-studied in soils (8–11) and groundwater (12–15), but few studies have examined the effects of transient flow and episodic flooding on uranium transport and redistribution. Previous studies of surficial uranium transport in other mining communities have suggested particulate transport of uranium controlled by physical erosion processes (16–18). Thus, the objectives of this study were to (1) investigate the spatial distribution of uranium in the north fork of the Puerco River and its tributaries; (2) characterize the geochemistry and mineral associations of uranium in sediments associated with a mine waste pile; and (3) determine the rates of U solid–solution partitioning and its possible role in regulating U transport. To do this, we analyzed sediment, river water, and suspended sediments collected during both wet and dry weather, and performed batch experiments to determine uranium dissolution kinetics from sediments. In addition, X-ray adsorption spectroscopy was employed to directly probe the oxidation state, chemical form, and phase-associations of uranium in samples containing elevated uranium levels.
Materials And Methods
Site Description and Sample Selection
This study was conducted in the mine-rich, Upper Puerco watershed in the Church Rock, Pinedale, Coyote Canyon, and Nahodishgish chapters of the Navajo Nation (McKinley County, New Mexico, Figure SI-1; Supporting Information). The climate is semiarid to arid; precipitation averages <25 cm/y and falls primarily in August and September. The north fork of the Puerco River, which cuts through the study area, flows intermittently only after heavy precipitation. Depth to ground-water in the shallow alluvial aquifer is estimated at 5–6 m (19). Ores mined were primarily coffinite (U(SiO4)1−x(OH)4x) and uraninite (UO2) (20). To characterize the spatial extent of uranium in this watershed, sediment and water samples were taken in seven drainage areas (Figure SI-1) including: controls (no known anthropogenic input (areas 1 and 2), uranium-bearing-outcrop (area 3), downstream of abandoned mines or uranium processing facilities (areas 4–6), and waste pile (area 7). Sampling was designed to capture changes in uranium concentrations in a variety of geomorphic features including terraces, outwashes, and channel deposits of adjoining drainages. Sediment samples (n = 100) were collected with a soil auger to a maximum depth of 90 cm below ground surface (bgs). Three sediment samples from area 7 collected at 0–2, 20, and 90 cm (referred to as A7-0, A7-20, and A7-90) were subjected to detailed geochemical characterization. All samples were collected in August of 2006 except those from area 7 which were collected in August of 2005.
Although the Puerco is dry for much of the year, heavy rains permitted collection of suspended sediments. Samples were collected in 10-L jugs and kept on ice for 7 days to allow settling of particles in the 0.04–2000 μm range. Water was decanted and sediments were then preserved for elemental analysis. The suspended sediment samples were only used to estimate uranium mass in the particulate load. To address the potential for uranium loss (sorption, precipitation) or gain (dissolution) by the settling sediments, concentrations of uranium were measured in the aqueous phase before and after the 7-day settling period. The uranium concentration was ~2 μg/L at 0 and 7 days; thus, it does not appear that the sediments gained or lost significant uranium mass during the suspended sediment settling period. Samples of surface water from three stock ponds in areas 3 and 5, and a rainwater sample were also collected and analyzed for uranium content. All water samples were filtered immediately on site (Whatman, 25-mm diameter, 0.2-μm pore size, polypropylene syringe filter), acidified (0.1 mL of 10 N HCl), and refrigerated until analysis.
Digestion and Analysis of Solids
Samples (1.0 g) of sediment, thoroughly homogenized by hand, were acid-digested using concentrated HNO3, HCl, and HF (9:3:2 mL) in 30-mL Teflon containers. Samples were decomposed at 90 °C for 2 h and evaporated to dryness at 70 °C. Residues were extracted in 1 mL of HNO3 (70%) and diluted prior to analysis. Elemental compositions were determined using an inductively coupled plasma optical emission spectrometer (Thermo Iris Intrepid II). A soil standard was used to evaluate performance of the analytical method. Typical internal uncertainties of 3–4% for trace elements and 6–7% for uranium were achieved following internal standardization and correction for relevant spectral interferences.
Solids from area 7 were digested in 9 mL of trace mineral grade nitric acid for 2 h at 90 °C. Residues were extracted in 1 mL of HNO3 (70%) and diluted prior to analysis. Elemental compositions were determined for all extractions using an ELAN 6000 ICP-MS. Standard curves and quality control samples were analyzed every 5 samples. Laser scattering particle size analysis was performed on all sediment samples.
Dissolution Experiments
Batch experiments to determine the amount and kinetics of uranium dissolution were performed on A7-0, A7-20, and A7-90 as well as samples collected from areas 2 and 5. All sediment samples were thoroughly homogenized by hand before subsamples were taken. Samples (5.0 g) were placed in 100-mL glass beakers with 50 mL of synthetic pore water (5 mM NaHCO3 and 2 mM CaCl2) to maintain a pH between pH 8.1 and 8.4 and an ionic strength of 12 mM. Table SI-1 included in the Supporting Information shows Visual Minteq predictions of saturated and undersaturated mineral species in equilibrium with likely uranyl mineral phases in synthetic porewater. The same pore water solution was used throughout all the batch experiments. Samples were shaken (100 rpm) and pore water aliquots (2 mL) and sample pH were collected at 0.082, 0.5, 1.5, 6, 24, 48, and 96 h. The samples were then filtered (0.2 μm), acidified (20 μL of 12 N HCl), and refrigerated until analysis.
Solid-phase/dissolved-phase partitioning was measured by adding different volumes of pore water solution to known masses of solids from A7-0.A7-20, andA7-90. Ten 0.1-g splits of sediment were placed in centrifuge tubes along with 2–40 mL of pore water solution. The tubes were then capped and shaken at 75 rpm for 24 h, after which 2 mL of solution was decanted, filtered (0.2 μm), acidified (20 μL of concentrated HCl), and refrigerated until analysis. This experiment was repeated with 0.5-g splits of sedimentto vary the solid-solution mass ratio.
Experiments were also performed to determine the effects of repeated sediment wetting and drying on uranium dissolution. Splits (1.0 g) of A7-0, A7-20, and A7-90 were treated with 10 mL of synthetic pore water solution in 25-mL centrifuge tubes, which were capped and shaken at 75 rpm for 24 h. pH of the solution was then measured, and 2-mL aliquots of liquid were removed, filtered (0.2 μm), and acidified with 20 μL of concentrated HCl. Sediment samples were centrifuged (4000 rpm at 3600g) and the supernatant (~8 mL) was decanted. The sediments were then dried at 60 °C to a constant weight, and the experiment was repeated twice, for a total of three cycles. Residual sediment was preserved for elemental and spectroscopic analysis.
X-Ray Absorption Spectroscopy
X-ray absorption spectroscopy (XAS) was used to characterize uranyl mineral phases in A7-0, A7-20, and A7-90 both before and after extraction with synthetic pore water solution. X-ray absorption spectroscopy was performed at the Stanford Synchrotron Radiation Laboratory (SSRL) on beamline 10-2. Procedures for the collection and internal calibration of uranium X-ray absorption spectra are reported elsewhere (8). In preparation for X-ray absorption spectroscopy (XAS) ~1.0-g sediment samples were sealed in Teflon sample holders with low-permeability Kapton film. At least 6 spectra were collected for the majority of samples from −200 to +1000 eV about the uranium LIII-edge (17166 eV) and averaged. Spectral processing was performed using WinXAS 3.1 software (21). X-ray absorption near edge structure (XANES) spectra were fit using linear combinations of uranium dioxide (U(IV)) and uranyl (U(VI)) standards to determine the oxidation state and quantity of uranium species in each sample.
Extended X-ray absorption fine structure (EXAFS) spectra were converted to k-space using a Fermi Level of 17166 eV, fit with a seven-point cubic spline, and weighted by k3 (8). Spectra were Fourier-transformed to produce a radial structure-function from approximately 4–10 Å. Fits were simulated using Feff 8 (22) and structural data were compiled in Catalano andBrown (23). Linear combination fitting of EXAFS spectra was performed to determine relative quantities of uranyl-mineral phases including rutherfordine (8), zellerite (23), compreignacite (23), boltwoodite (23), α-uranophane (23), meta-autunite (24), and ankoleite (8).
X-Ray Microprobe
X-ray microprobe analysis was used to confirm the elemental distribution and mineralogy of uranium within selected uranium-bearing samples at the grain scale. Data were collected at SSRL beamline 2–3, which is equipped with Kirkpatrick–Baez focusing optics to achieve a nominal spot size of 2 μm Fluorescence data was collected using an incident energy of 18.0 keV. Prior to data collection, dried samples were sieved to 2 mm and then mounted in a thin film on Kapton tape. Complete μXRF spectra were collected every 5 μm, with a dwell time of 500 μs at each point. Spectral processing and image analysis was achieved using Microprobe Toolkit (25). Regions of interest (ROIs) were defined for uranium–as well as K, P, S, Cl, Ca, Fe, Mn, Cu, Zn, and Pb–and integrated counts were used to estimate elemental abundance. Counts at each point were normalized by absorbance to account for samples of variable density and sample thickness. This was achieved by dividing the counts for a specific ROI by the ratio of incident and transmitted beam intensities (I0/I1).
Results and Discussion
Spatial Distribution of Uranium
Excluding samples taken from area 7, there was no statistical difference (p < 0.07) between mean uranium concentrations in samples from areas thought to be contaminated (5.1 ± 2.0 mg/kg) and control areas (4.2 ± 2.0 mg/kg; Table 1), both of which are similar to background U levels in nearby rock formations (26). Uranium concentrations measured in areas 1–6 are consistent with concentrations measured in sediment samples taken in Upper Puerco Watershed between 1976 and 1979 (range 0.62–7.8 mg/kg) (27). A significant correlation (p < 0.0001) was found between uranium concentration and mean particle size, with higher concentrations of uranium being associated with very fine sands (62.5–125 μm). The median concentration of U at area 7 (22.5 mg/kg) was 4–14 times higher than median concentrations in areas 1–6. Uranium concentrations did not vary significantly with depth in any of the seven areas sampled. (See SI Table SI-2 Elemental compositions of digestates for areas 1–6; and Table SI-3 Elemental compositions of A7-0, A7-20, A7-90).
TABLE 1.
Uranium in Sediment and Suspended Sediment from the Upper Puerco Watershed
| area | mean particle size μm | n | range U mg/kg | mean U ± SD mg/kg |
|---|---|---|---|---|
| 1 | 106.8 | 7 | 2.9–8.2 | 5.6 ± 1.8 |
| 2 | 260.6 | 16 | 1.7–7.7 | 3.6 ± 1.8 |
| 3 | 121.8 | 4 | 0.46–2.7 | 1.6 ± 0.99 |
| 4 | 283.0 | 21 | 1.7–7.3 | 4.3 ± 1.6 |
| 5 | 76.20 | 27 | 2.3–11 | 6.3 ± 2.0 |
| 6 | 103.1 | 22 | 1.5–7.8 | 4.9 ± 1.5 |
| 7a | 53.86 | 31 | 0.3–120 | |
| Puerco Riverb | 2.566 | 3 | 2.6–4.7 | 3.8 ± 1.1 |
| Stock Pondb | 1.971 | 2 | 4.7–4.9 | 4.8 ± 0.16 |
Concentrations of U from area 7 were not normally distributed; therefore mean ± SD are not reported. Median U was 22.5 mg/L.
Suspended sediment samples.
Uranium concentrations ranged from 2.6 to 4.7 mg/kg in suspended sediments (composed primarily of particle sizes <5 μm) in the Puerco River, and from 4.7 to 4.9 mg/kg in suspended sediment in the stock ponds. Concentrations of uranium ranged from < 1 μg/L (the detection limit) to 2 μg/L in all surface waters. Uranium concentrations were less than 1 μg/Lin rainwater.
Geochemical Characterization of Waste-Associated Sediment
Batch Dissolution
Dissolution of uranium from sediment samples (A7-0, A7-20, and A7-90) was rapid in the first 6 h of the experiment. After ~24 h nearly steady-state aqueous concentrations were achieved (Figure la). A7-90 (U(VI) = 86 mg/kg prior to batch dissolution) produced the highest dissolved uranium concentrations (1.78–4.15 mg/L), which represents ~50% of the uranium in the sediment. In contrast, dissolution of uranium from A7-20 (U(VI) = 91 mg/kg prior to batch dissolution) yielded lower concentrations of dissolved uranium (0.26–0.96 mg/L), and less solid-phase uranium dissolution (~20%). A7-0 (U(VI) = 117 mg/kg prior to batch dissolution) produced a range of uranium concentrations similar to that of A7-20 (0.37–0.88 mg/L), but the soluble uranium mass represented only 10% of the solid-phase uranium. The elevated uranium concentrations in solution suggest that discrete, soluble uranyl-bearing phases were present. Soluble uranium was not detected in identical experiments done on sediment samples from a control area (area 2) and downstream of another AUM (area 5).
Equilibrium modeling of uranyl phases was done using Visual Minteq and a modified version of the Lindsay thermodynamic database (28). Where important uranyl phases were absent (e.g., boltwoodite), kinetic data were entered to update the database. The pH and ionic strength of pore water was fixed to replicate experimental parameters. Concentrations of major ions and metals measured during dissolution experiments were used to the modify pore water solution to reflect the addition of sediment. Uranyl phases were added to the model one at a time to predict the equilibrium concentration of uranium in pore water (Table SI-4). In general, the modeled concentrations explain the experimental results. Gray lines in Figure 1(left) indicate the approximate equilibrium concentrations of boltwoodite, uranophane, and ankoleite. Experimental uranium concentrations measured for A7-90 appear to be approaching boltwoodite equilibrium, whereas A7-20 appears to be approaching equilibrium with uranophane.
FIGURE 1.
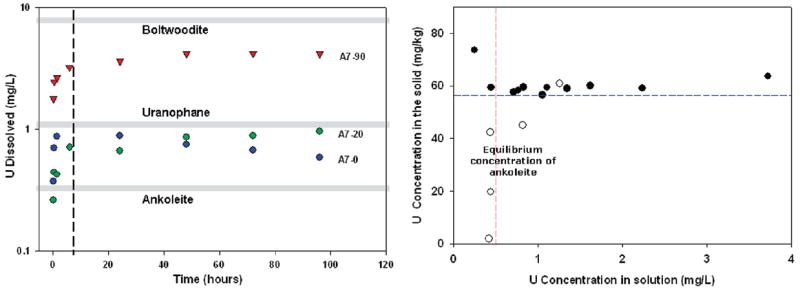
(Left panel) Results of uranium batch dissolution experiments. Dashed line represents apparent change in the rate of dissolution between 0–6 and 6–96 h. Gray lines represent equilibrium uranium concentrations of uranyl phases in synthetic porewater. (Right panel) Uranium dissolution after 24 h from fixed sediment masses of A7-90 (Black circles = 0.5 g; hollow circles = 0.1 g) in different volumes (2–40 ml) of synthetic pore water. Dashed red line represents the approximate solubility limit of one mineral phase (predicted as ankoleite using Visual Minteq), while the dashed blue line represents the approximate solid phase concentration of a second mineral phase(s).
Dissolution experiments were performed to determine the effect of pore water volume on uranium dissolution from fixed amounts of sediment sample A7-90. The results in Figure 1 (right) indicate that uranium dissolution was not well predicted by linear (Kd), Langmuir, or Freundlich isotherms. Figure 1 (right) contains two distinct regions. The first region (points above blue dashed line) exhibits roughly constant solid-phase uranium concentrations and high solution concentrations. This behavior is consistent with near complete dissolution of a soluble phase. The second region (represented by the points along the dashed red line), with lower solid-phase uranium levels and a uniform solution concentration of uranium of about 0.4–0.5 mg/L, represents the partial dissolution of this second mineral phase to a limit imposed by solubility. The predicted concentration for ankoleite in equilibrium with pore water solution was 0.52 mg/L. The apparent solubility control on dissolution (depicted by the red line in Figure 1 (right) is at approximately 0.5 mg/L, suggesting saturation of ankoleite. Dissolution of the most soluble phase decreases the solid phase concentration from 86 mg/kg total uranium to about 58 mg/kg. This implies that about 28 mg/kg U (about 33% of the total uranium) is associated with the more soluble phase, while 58 mg/kg U (about 67% of the total uranium) is associated with one or more less soluble phases.
The rates of uranium dissolution were rapid (Figure 1 (left)), and nearly complete within 6 h. The rates of uranium dissolution for A7-20 and A7-90 were best fit using the linear form of the parabolic diffusion equation with dissolution data (29)
| (1) |
Where r = radius of the grain; Qt = quantity of diffusing substance leaving at time t;Q∞, = corresponding quantity of diffusing substance after infinite time; and D = apparent diffusion coefficient. Particle size radii used for A7-20 and A7-90 were 0.0085 and 0.0056 cm, respectively. Diffusion coefficients from 0 to 6 h were calculated at 9.72 × 10−9 and 7.75 × 10−9 cm2/s for A7-20 and A7-90, consistent with diffusion of uranyl at the solid–water interface (30). For the remainder of the experiment, diffusion coefficients were slower for A7-20 and A7-90, at 6.92 × 10−10 and 3.11 × 10−10 cm2/s, suggesting the possibility of asecond diffusion domain. Differences in diffusivities may be explained by rapid dissolution from grain surfaces followed by slower diffusion of uranium from internal pores (31). Our results here are consistent with other studies showing diffusion-limited dissolution of uranyl-silicates (32).
Wet-Dry Cycle Dissolution
The results of experiments involving repeated sediment wetting and drying indicate that more soluble uranium was present in samples A7-20 and A7-90 than in A7-0 (Table SI-5). The concentrations of uranium resulting from dissolution of A7-20 and A7-90 after the first cycle were 3.03 and 2.05 mg/L, respectively, compared to 0.68 mg/L from A7-0. However, after the second cycle, the uranium concentrations resulting from dissolution of A7-20 (0.30 mg/L) and A7-90 (0.95 mg/L) were not significantly different than A7-0 (0.63 mg/L). The total mass of uranium removed during the three cycles from A7-20 (36.1 mg/kg) and A7-90 (37.6 mg/kg) was more than double the mass removed from A7-0 (17.8 mg/kg).
The difference in the mass of soluble uranium removed from the A7-20 and A7-90 between cycles 1 and 2 is taken as further evidence of the presence of amore soluble mineral phase (or phases) in these deeper samples. Less soluble uranyl phases likely dominate in the surficial sample (A7-0), resulting in lower solid phase depletion compared to A7-20 and A7-90.
Spectroscopy
X-ray Absorption Spectroscopy of Solid-Phase Minerals
XANES spectra revealed that solid-phase uranium exists primarily as U(VI) in samples A7-0, A7-20, and A7-90 (Figure 2). Linear combination fitting with U(IV) and U(VI) reference spectra found that more than 99% of the sample is U(VI); however, a small component of U(IV) cannot be ruled out (22). XANES spectra for uranium did not vary with sediment depth and did not change after the wet-dry cycles. The EXAFS spectra of A7-20 and A7-90 appeared to be distinct from A7-0 (Figure 3 (left)). The spectra share common fitting of the first shell, corresponding to U–O coordination of the axial and equatorial oxygens of the uranyl cation (UO22+) at 1.8 and 2.3–2.4 Å. Second-nearest neighbor shells are only well defined in A7-0, with silica shells present at 3.09 and 3.66 Å(Figure 3(right)). These distances are indicative of the bonding environment of uranyl in uranophane-group minerals (22). These and other more distant neighbor shells were not effectively fit with single shells in either A7-20 or A7-90, possibly due to the presence of multiple uranium-bearing minerals with interfering spectral components, the presence of a multiple scattering, or noise (8). The EXAFS spectra of low-uranium samples from control sites were not of sufficient quality to permit spectral analysis.
FIGURE 2.
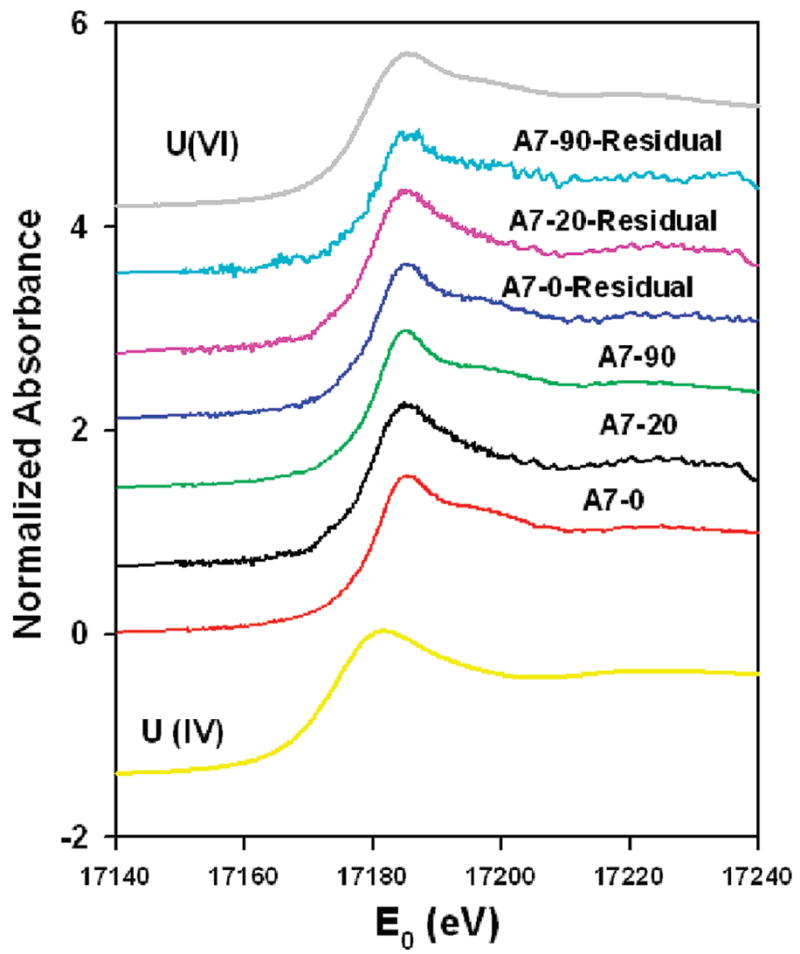
XANES spectra of A7-0, A7-20, and A7-90 prior to and after three cyclical extractions with synthetic pore water. (Absorbance was multiplied by an offset factor for each spectrum to prevent overlay of data).
FIGURE 3.
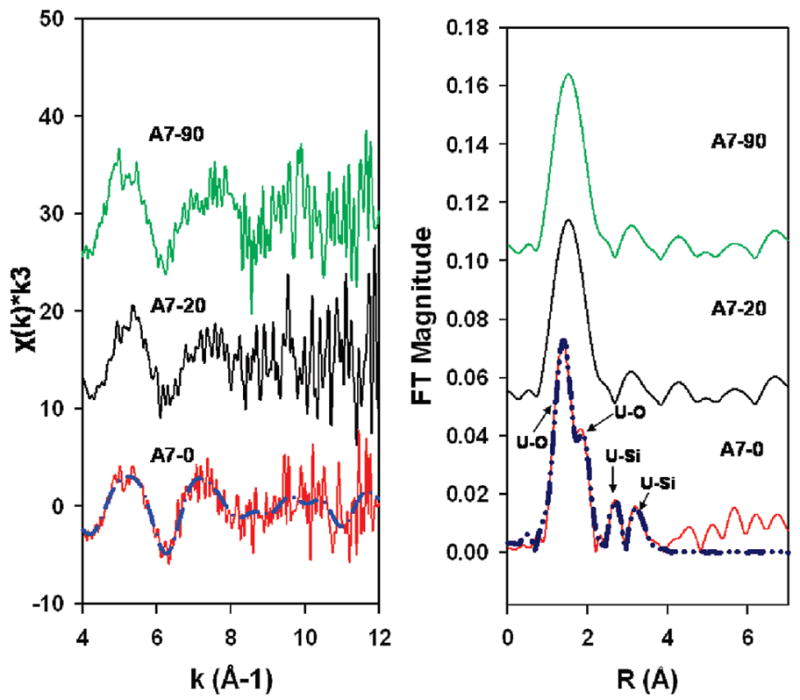
(Left) EXAFS spectra: Solid lines represent experimental data and dashed lines represent theoretical Feff fitting. (Right) Radial structure–functions of A7-0, A7-20, and A7-90. Solid lines represent experimental data and dashed lines represent theoretical shell fits. FT = Fourier transform and R = bond length.
Linear combination fitting of EXAFS spectra reveals that one or more uranophane-group minerals were the dominant soluble components of A7-0, A7-20, and A7-90 (Table SI-6). The second, less-soluble uranium-bearing mineral suggested by the dissolution data was more difficult to identify by linear combination fitting. Linear combination fitting of A7-0 required the presence of multiple phases, including a uranophane group mineral(s) (likely boltwoodite, K2 (UO2)2(SiO3OH)2·1.5H2O, or uranophane (Ca(UO2)2SiO3(OH)2·5H2O) and a uranyl phosphate (autunite, Na2(UO2)2(PO4)2·6H2O, or ankoleite, K2(UO2)2(PO4)2·6H2O). Both fits were of similar quality and thus hard to resolve. A7-20 and A7-90 spectra were best fit with a single uranophane-group mineral.
Microprobe X-ray Fluorescence
Spatial distributions (maps) of uranium, calcium, and potassium in the samples are presented in Figure 4(left). Uranium counts were elevated above background throughout the entire maps for A7-0 and A7-90; uranium-rich regions (hotspots) were seen primarily in association with potassium-rich minerals (e.g., feldspars). In contrast, uranium abundance was not highly correlated with calcium (nor was it correlated with Fe or Mn levels). The strong correlation between uranium and potassium (Figure 4(right)) suggests that boltwoodite is the primary uranium mineral rather than uranophane. The weak uranium–calcium correlation also implies that meta-autunite may not be a major component, although the presence of ankoleite cannot be ruled out. We attempted to differentiate between these possibilities based on XANES spectra, which are sensitive to bonding environment; however, spectra of U-rich hotspots were of insufficient quality to differentiate among boltwoodite, uranophane, or autunite.
FIGURE 4.
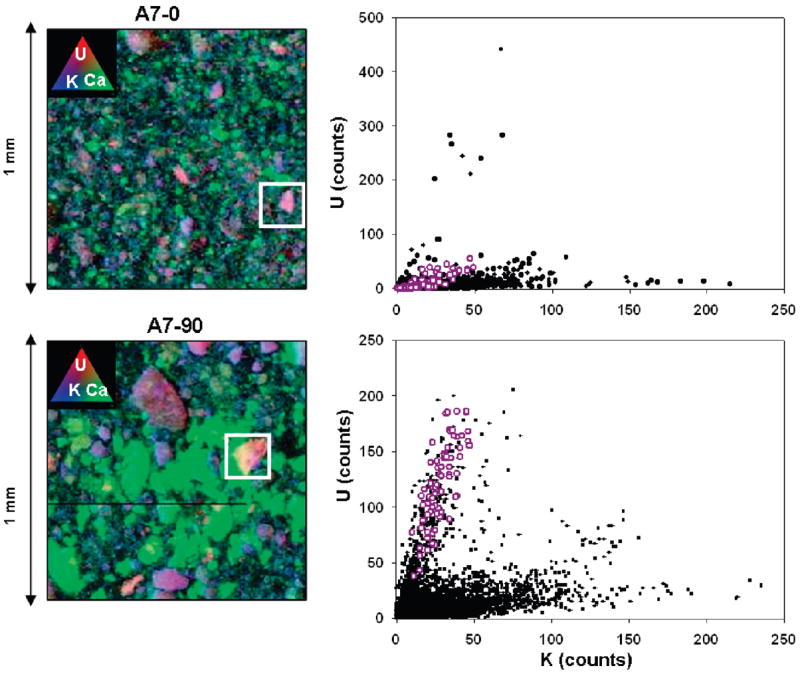
(Left) Tricolor X-ray fluorescence plots of U, Ca, and K in A7-0 and A7-90. Purple color represents the association between U and K. White boxes represent uranium “hot spots”. Green represents Ca. (Right) Correlation plots of U and K. Black dots represent all data (n = 25,000) where white circles represent U–K correlation at “hot-spot”.
XRF images were used to infer processes, including mineral dissolution, that have changed elemental composition and mineralogy in A7-0 andA7-90. Differences between the distribution of U in the A7-0 and A7-90 (Figure 4(left)) indicate that the uranium in each sample was associated with different mineral fractions. In A7-90, the bulk of uranium was associated with large (> 100 μm) mineral grains. In A7-0 there were few large mineral grains, and there were few small (<25 μm) uranium hotspots. Other K-bearing grains are also associated with uranium, but have much lower U:K ratios (Figure 4(right)). We attribute the difference in part to the leaching of uranium from K-rich uranyl silicate grains in surficial environments, consistent with the dissolution experiments examining uranium release.
Significance
The low concentrations of uranium in surficial sediment samples from areas 3–6 were not expected given the proximity of sampling sites to known uranium sources. Our results suggest that the low sediment uranium concentrations likely resulted from the dissolution and flushing of uranium during precipitation events. Surficial, weathered sediments are depleted of more soluble uranyl phases relative to deeper (>~20 cm) sediments. Our results further suggest that a uranophane group mineral (e.g., boltwoodite or uranophane) is the primary soluble phase in A7-0, A7-20, and A7-90, with a uranyl-phosphate (e.g., ankoleite) being a secondary, less soluble component. While we cannot clearly distinguish among these phases, available thermodynamic data indicate that these minerals are highly soluble (9). Equilibrium solubilities for pore water solutions in equilibrium with boltwoodite, uranophane, and ankoleite (calculated using Visual Minteq v2.52) are 9.0, 1.1, and 0.5 mg/L respectively, which are comparable to uranium concentrations reported in mine water discharges in Church Rock between 1969 and 1982 (0.7–3.71 mg/L) (33). The fate of uranium in the dissolved load is unknown; it is possible that soluble uranium is transported downward through the vadose zone with infiltrating precipitation, potentially impacting groundwater resources.
This study is among the first watershed-scale assessments of uranium geochemistry since the cessation of mining activities in the Upper Puerco watershed in the late 1980s. Our investigation points to the utility of geochemical characterization to support site-specific exposure concentrations and assumptions. This in turn may provide a more accurate foundation for human and ecological risk assessment and be useful in the development of risk management strategies. Results of this study maybe indicative of uranium mobilization in other areas in which geologically similar uranium ore was mined and brought to the surface for processing.
Acknowledgments
We are grateful to Chris Shuey of the Southwest Research and Information Center, Eastern Navajo Health Board, DiNEH project staff, and Mallery Downs and Miranda Cajero of the University of New Mexico Community Environmental Health Program (CEHP). Financial support was provided by a NIH Ruth L. Kirschstein National Research Service Award; Tisch College of Citizenship and Public Service; Tufts Institute of the Environment; the School of Engineering, the Graduate School of Arts and Sciences, and the Summer Scholars Program at Tufts; Church Rock Uranium Monitoring Project, and NIEHS grants (R25 ES013208, RO1 ES014565, and P30 ES012072) to UNM CEHP. Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory, a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences, with additional funding from the National Institute of Health.
Footnotes
Supporting Information Available
Map of the Upper Puerco Watershed study area (Figure SI-1); saturated and undersaturated mineral species in equilibrium with uranophane, boltwoodite, and ankoleite in synthetic pore water (Table SI-1); elemental compositions for digestates areas 1–6 (Table SI-2); elemental compositions of digestates for A7-0, A7-20, and A7-90 (Table SI-3); uranium concentrations in equilibrium with uranyl mineral phases in synthetic pore water solution (Table SI-4); Results of wet-dry cycle dissolution for A7-0, A7-20, and A7-90 (Table SI-5); linear combination best fit relative abundances of uranyl phases in A7-0, A7-20, andA7-90 (Table SI-6). This material is available free of charge via the Internet at http://pubs.acs.org
Literature Cited
- 1.Brugge D, Goble R. The History of Uranium Mining and the Navajo People. Am J Public Health. 2002;92:1410–1419. doi: 10.2105/ajph.92.9.1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ringholz RC. Uranium Frenzy: Boom and Bust on the Colorado Plateau. University of New Mexico Press; Albuquerque, NM: 1989. [Google Scholar]
- 3.United States Environmental Protection Agency. Eastern AUM Screening Assessment Report. U.S. EPA; San Fransico, CA: 2006. Abandoned Uranium Mines (AUM) on the Navajo Nation. [Google Scholar]
- 4.Shuey C, Taylor L, Siskin J. Church Rock Revisited: The Tailings Spill Three Years Later. Mine Talk. 1982 Summer–Fall [Google Scholar]
- 5.Graf JB, Gray JR, Wirt L. Streamflow transport of radio-nuclides and other chemical constituents in the Puerco and the Little Colorado River basins, Arizona and New Mexico, Water-Supply Paper 2459. U.S. Geological Survey; 1996. p. 89. [Google Scholar]
- 6.Van Metre PC, Gray JR. Effects of uranium mining discharges on water quality in the Puerco River basin, Arizona and New Mexico. Hydrol Sci J. 1992;37:463–480. [Google Scholar]
- 7.Miller JR, Wells SG. Types and processes of short-term sediment and uranium-tailings storage in arroyos. An example from the Rio Puerco of the West, New Mexico. In: Hadley RF, editor. Drainage Basin Sediment Delivery. International Association of Hydrological Sciences, International Commission on Continental Erosion; 1986. pp. 335–353. IAHS 159. [Google Scholar]
- 8.Bostick BC, Fendorf S, Barnett MO. Uranyl surface complexes formed on subsurface media from DOE facilities. Soil Sci Soc Am J. 2002;66:99–108. [Google Scholar]
- 9.Grenthe I, Fuger J, Konings RJM, Lemire RJ, Muller AB, Nguyen-Trung C, Wanner H. Chemical Thermodynamics of Uranium. North-Holland: Amsterdam; 1992. [Google Scholar]
- 10.Moyes LN, Parkman RH, Charnock JM. Uranium uptake from aqueous solution by interaction with goethite, lepidocrocite, muscovite, and mackinawite: An X-ray absorption spectroscopy study. Environ Sci Technol. 2000;34:1062–1068. doi: 10.1021/es010216g. [DOI] [PubMed] [Google Scholar]
- 11.Catalano JG, Brown GE. Uranyl adsorption onto mont-morillonite: Evaluation of binding sites and carbonate complexation. Geochim Cosmochim Acta. 2005;69:2995–3005. [Google Scholar]
- 12.Qafoku NP, Zachara JM, Liu C, Gassman PL, Qafoku OS, Smith SC. Kinetic Desorption and Sorption of U(VI) during Reactive Transport in a Contaminated Hanford Sediment. Environ Sci Technol. 2005;39:3157–3165. doi: 10.1021/es048462q. [DOI] [PubMed] [Google Scholar]
- 13.Barnett MO, Jardine PM, Brooks SC. Adsorption and transport of uranium (VI) in subsurface media. Soil Sci Soc Am J. 2000;64:908–917. [Google Scholar]
- 14.Dong W, Ball WP, Liu C, Wang Z, Stone AT, Bai J, Zachara JM. Influence of Calcite and Dissolved Calcium on Uranium(VI) Sorption to a Hanford Subsurface Sediment. Environ Sci Technol. 2005;39:7949–7955. doi: 10.1021/es0505088. [DOI] [PubMed] [Google Scholar]
- 15.Riley RG, Zachara JM, Wobber FJ. Chemical Contaminants on DOE Lands and Selection of Contaminant Mixtures for Subsurface Science Research. U.S. Department of Energy, Office of Energy Research; Washington, DC: 1992. DOE/ER-0547T. [Google Scholar]
- 16.Batson VL, Bertsch PM, Herbert BE. Transport of Anthropogenic Uranium from Sediments to Surface Waters During Episodic Storm Events. J Environ Qual. 1996;25:1129–1137. [Google Scholar]
- 17.Hancock GR, Grabham MK, Martin P. A methodology for the assessment of rehabilitation success of post mining landscapes - sediment and radionuclide transport at the former Nabarlek uranium mine, Northern Territory, Australia. Sci Total Environ. 2006;354:103–119. doi: 10.1016/j.scitotenv.2005.01.039. [DOI] [PubMed] [Google Scholar]
- 18.Lottermoser BG, Ashley PM, Costelloe MT. Contaminant dispersion at the rehabilitated Mary Kathleen uranium mine Australia. Environ Geol. 2005;48:748–761. [Google Scholar]
- 19.Davis GE, Hardt WP, Thompson LK, Cooley ME. Geohydrologicdata in the Navajo and Hopi Indian Reservations, Arizona, New Mexico and Utah, part I, Records of ground-water supplies. Arizona State Land Department Water Resources Phoenix; AZ: 1963. Report 12-A. [Google Scholar]
- 20.Baars DL. Navajo Country: A Geology and Natural History of the Four Corners Region. University of New Mexico Press; Albuquerque, NM: 1995. [Google Scholar]
- 21.Ressler T. WinXAS: a program for X-ray absorption spectroscopy data analysis under MS-Windows. J Synchrotron Radiat. 1998;5:118–122. doi: 10.1107/S0909049597019298. [DOI] [PubMed] [Google Scholar]
- 22.Ankudinov AL, Ravel B, Rehr JJ, Conradson SD. Realspace multiple-scattering calculation and interpretation of X-ray-absorption near-edge structure. Phys Rev B. 1998;58:7565–7576. [Google Scholar]
- 23.Catalano JG, Brown GE., Jr Analysis of uranyl-bearing phases by EXAFS spectroscopy: Interferences, multiple scattering, accuracy of structural parameters, and spectral differences. Am Mineral. 2004;89:1004–1021. [Google Scholar]
- 24.Wellman DM, Catalano JG, Icenhower JP, Gamerdinger AP. Synthesis and characterization of sodium meta-autunite, Na2[(UO2)(PO4)]2·3H2O. Radiochim Acta. 2005;93:393–399. [Google Scholar]
- 25.Sam Webb Analysis Software Microprobe Imaging Toolkit: SMAK. www.ssrl.slac.stanford.edu/~swebb/index.htm.
- 26.Agency for Toxic Substances and Disease Registry. Toxicological Profile for Uranium. ATSDR; Altanta, GA: 1999. [PubMed] [Google Scholar]
- 27.Smith SM. National Geochemical Database: Reformatted data from the National Uranium Resource Evaluation (NURE) Hydrogeochemical and Stream Sediment Reconnaissance (HSSR) Program, Version 140 (2006): U.S. Geological Survey Open-File Report 97-492,1997. [last accessed June 1, 2007]; WWW release only, http://pubs.usgs.gov/of/1997/ofr-97-0492/index.html.
- 28.Lindsay WL, Ajwa HA. Use of MINTEQA2 in teaching soil chemistry. In: Loeppert RH, Goldberg S, Schwab AP, editors. Chemical Equilibrium Reaction Models. American Society of Agronomy; Madison, WI: 1995. [Google Scholar]
- 29.Sparks DL, editor. Soil Physical Chemistry. CRC Press; Boca Raton, FL: 1999. pp. 145–148. [Google Scholar]
- 30.Thobodeaux LJ, editor. Environmental Chemodynamics: Movement of Chemicals in Air, Water, and Soil. Wiley and Sons; New York: 1996. [Google Scholar]
- 31.Liu CX, Zachara JM, Qafoku O. Dissolution of uranyl microprecipitates in subsurface sediments at Hanford site, USA. Geochim Cosmochim Acta. 2004;68:4519–4537. [Google Scholar]
- 32.Liu CX, Zachara JM, Yantasee W. Microscopic reactive diffusion of uranium in the contaminated sediments at Hanford, United States. Water Resour Res. 2006;42:13–15. [Google Scholar]
- 33.New Mexico Environmental Improvement Division. Water Quality Data for Discharges from New Mexico Uranium Mines and Mills. NMEID; 1980. [Google Scholar]